Introduction
Transforming growth factor-β (TGF-β) is a well-known
cytokine with pleiotropic biological functions. TGF-β has a pivotal
role in various physiological processes and pathological
conditions, including development, cancer, senescence, fibrosis,
wound healing and tissue regeneration (1–3). As an
immunosuppressive cytokine, TGF-β has a key role in tumor immune
evasion (4). The levels of TGF-β are
often observed to be elevated in the serum of patients with cancer
(5). Notably, TGF-β also influences
the biological characteristics of cancer stem cells. Research has
demonstrated that toll-like receptor 4/NANOG-dependent cancer stem
cells are defective in the TGF-β pathway (6). Perivascular TGF-β suppresses
proliferation and promotes invasion and heterogeneity in squamous
cell carcinoma stem cells (7).
Importantly, release from TGF-β-mediated inhibition restores
anti-tumor immunity (8).
TGF-β regulates numerous functions of epithelial
cells. TGF-β is a well-documented inducer of
epithelial-to-mesenchymal transition (EMT) during embryogenesis,
cancer progression and fibrosis (9).
Both exogenous TGF-β protein and TGF-β from other sources, such as
platelets, are able to induce EMT (10,11).
Several lines of evidence indicate that TGF-β is able to promote
cell proliferation of thyroid epithelial cells (12) and various tumor cells (13,14);
however, it also has antiproliferative effects on other cells
(15,16). Such conflicting findings suggest that
the effects of TGF-β are dependent on cell type and context.
The mammalian target of rapamycin (mTOR) signaling
pathway has a critical role in regulating basic cellular functions,
including cell proliferation, survival, mobility and angiogenesis
(17). Rapamycin has specific
antagonistic action on the function of mTOR. Rapamycin induces cell
cycle arrest in many cells (18,19);
however, the combinational effect of rapamycin and TGF-β on tumor
cells is unclear. In the present study, it was demonstrated that
TGF-β had a cytostatic effect on Michigan Cancer Foundation (MCF)-7
human breast cancer cells. TGF-β induced upregulation of the
cyclin-dependent kinase inhibitors (CKIs) p14ARF,
p15INK4b, p16INK4a and
p21WAF1/CIP1. Notably, it was demonstrated that
rapamycin enhanced the antiproliferative effect of TGF-β.
Materials and methods
Cell culture and reagents
Human breast cancer cell line MCF-7 was purchased
from American Type Culture Collection (Manassas, VA, USA). Tumor
cells were cultured and propagated in Dulbecco's modified Eagle
medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum at 37°C (5%
CO2). Rapamycin was obtained from Selleck Chemicals
(Houston, TX, USA) and dissolved in dimethyl sulfoxide. Recombinant
human TGF-β1 was purchased from HumanZyme, Inc. (Chicago, IL, USA)
and dissolved in 4 mM hydrochloric acid.
Cell viability assay
A total of 5×104 MCF-7 cells were plated
on 6-well plastic plates. Cells were treated with 5 and 10 ng/ml
TGF-β, in the presence or absence of 100 nM rapamycin, or an
equivalent volume of vehicles. Cell number was counted manually 120
h after treatment. An MTT assay was performed using 96-well plates
in order to determine cell viability. A total of 2.5×103
cells per well were seeded in the 96-well plates. Tumor cells were
exposed to 10 ng/ml TGF-β and/or 100 nM rapamycin on the following
day. Tumor cell viability was evaluated at 0, 24, 48, 72, 96 and
120 h after experiment initiation. For the MTT assay, 20 µl MTT
reagent (5 mg/ml) was added to each well and the plates were
incubated for an additional 4 h at 37°C. Subsequently, the formazan
precipitates in the cells were dissolved in 150 ml dimethyl
sulfoxide after removal of the supernatant. Absorbance was
determined at 570 nm and a growth curve was plotted.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from tumor tissues using an
RNA isolation kit (Axygen, Tewksbury, MA, USA). RNA was then
treated with DNase to remove genomic DNA and subsequently reverse
transcribed (PrimeScript RT reagent kit with gDNA Eraser; Takara
Biotechnology Co., Ltd., Dalian, China) according to the
manufacturer's instructions. qPCR was performed on a CFX 96
real-time PCR thermocycler (Bio-Rad Laboratories, Inc., Hercules,
CA, USA) using specific primers and SYBR Green supermix (Takara
Biotechnology Co., Ltd.). RT-qPCR experimental procedures were
performed according to the manufacturer's protocol with some
changes (SYBR Premix Ex Taq II; Takara Biotechnology Co., Ltd.).
The total PCR reaction volume was 25 µl, which contained 12.5 µl
supermix, 9.5 µl H2O, 1 µl cDNA, 1 µl forward primers
and 1 µl reverse primers. PCR conditions were as follows: Initial
denaturation for 30 sec at 95°C, 40 cycles at 95°C for 5 sec, 60°C
for 30 sec, 15 sec at 95°C and 5 sec at 65°C. Primers used for qPCR
have previously been described (20). The sequences were as follows: p14ARF,
forward 5′-TCCTCAGTAGCATCAGCACGAG-3′ and reverse
5′-AGAACATGGTGCGCAGGTTCTTG-3′; p15INK4b, forward
5′-GGGAGGGTAATGAAGCTGAG-3′ and reverse 5′-GGCCGTAAACTTAACGACACT-3′;
p16INK4a, forward 5′-GGGTCCCAGTCTGCAGTTA-3′ and reverse
5′-GGAGGGTCACCAAGAACCT-3′; p21 WAF1/CIP1, forward
5′-GCAGACCAGCATGACAGATTT-3′ and reverse
5′-GGATTAGGGCTTCCTCTTGGA-3′; and GAPDH, forward
5′-AGAAGGCTGGGGCTCATTTG-3′ and reverse 5′-AGGGGCCATCCACAGTCTTC-3′.
Relative gene expression levels were quantified using the
2−∆∆Cq method with GAPDH as reference (21). PCR was repeated in triplicate.
Flow cytometric analysis
A total of 5×104 MCF-7 cells were plated
on 6-well plastic plates and treated with 10 ng/ml TGF-β and/or 100
nM rapamycin, or an equivalent volume of vehicles. After 72 h,
cells were harvested and stained with annexin-V-FITC and propidium
iodide (PI). Apoptotic cells were quantified using a FACSCalibur
flow cytometer and analyzed using CellQuest software (version 6.0;
BD Biosciences, San Jose, CA, USA).
Statistical analysis
Statistical significance was determined using
Student's t-tests between two groups, and one-way analysis of
variance was used when comparing more than three groups followed by
least significant difference and Student-Newman-Keuls analysis.
SPSS version 13.0 software (SPSS Inc., Chicago, IL, USA) was used
for statistical analysis. Results were expressed as the mean ±
standard deviation. P<0.05 was considered to indicate a
statistically significant difference.
Results
TGF-β exhibits a cytostatic effect on
MCF-7 tumor cells
To investigate whether TGF-β influences the
proliferation of tumor cells, human breast cancer MCF-7 cells were
incubated with 5 or 10 ng/ml concentrations of human TGF-β. Results
demonstrated that TGF-β decreased cell numbers in a dose-dependent
manner. A dose of 10 ng/ml TGF-β significantly impaired tumor cell
proliferation compared with vehicle controls (P<0.05; Fig. 1A). A 5 ng/ml dose of TGF-β also
exhibited a significant cytostatic effect on MCF-7 cell
proliferation (P<0.05), although to a lesser extent when
compared with the 10 ng/ml dose (Fig.
1A). The MTT assay was adapted to assess the viability of tumor
cells in the presence of 10 ng/ml TGF-β. In the presence of TGF-β,
the viability of MCF-7 cells was significantly reduced compared
with the vehicle group from 48 h onwards (P<0.05; Fig. 1B). It was therefore concluded that
TGF-β exhibited a cytostatic effect on MCF-7 tumor cells.
TGF-β modulates cell cycle
regulators
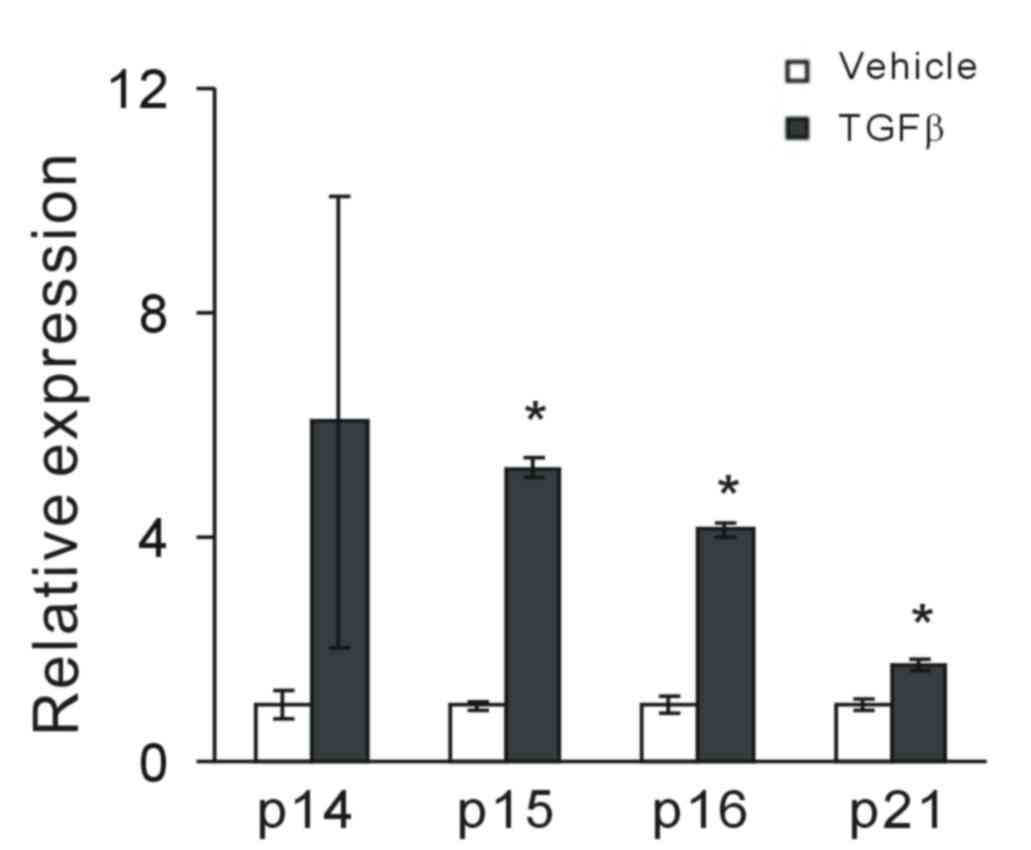
Considering that TGF-β had a cytostatic effect on
MCF-7 tumor cells, the effect of TGF-β on the induction of CKIs was
investigated. Following exposure to TGF-β, the expression of
cell-cycle inhibitors p14ARF, p15INK4b,
p16INK4a and p21WAF1/CIP1 was assessed.
RT-qPCR analysis demonstrated that TGF-β significantly increased
the expression levels of p15INK4b, p16INK4a
and p21WAF1/CIP1 in MCF-7 cells compared with vehicle
controls (P<0.05; Fig. 2); it
also increased p14ARF, though this was not statistically
significant. This suggests that TGF-β may have a potential
regulatory role in CKI expression.
Rapamycin enhances the
antiproliferative effect of TGF-β
The influence of rapamycin, a well-known mTOR
inhibitor, on the antiproliferative effect of TGF-β was
investigated. MCF-7 cells were exposed to 100 nM rapamycin and 5 or
10 ng/ml TGF-β. MTT assay demonstrated that rapamycin significantly
inhibited MCF-7 cell viability compared with vehicle controls after
48 h of experiment initiation. Unexpectedly, it was demonstrated
that in the presence of both rapamycin and TGF-β, MCF-7 cells
proliferated significantly slower compared with the single
treatments of rapamycin or TGF-β (P<0.05; Fig. 3). These results suggest that
rapamycin enhances the antiproliferative effect of TGF-β.
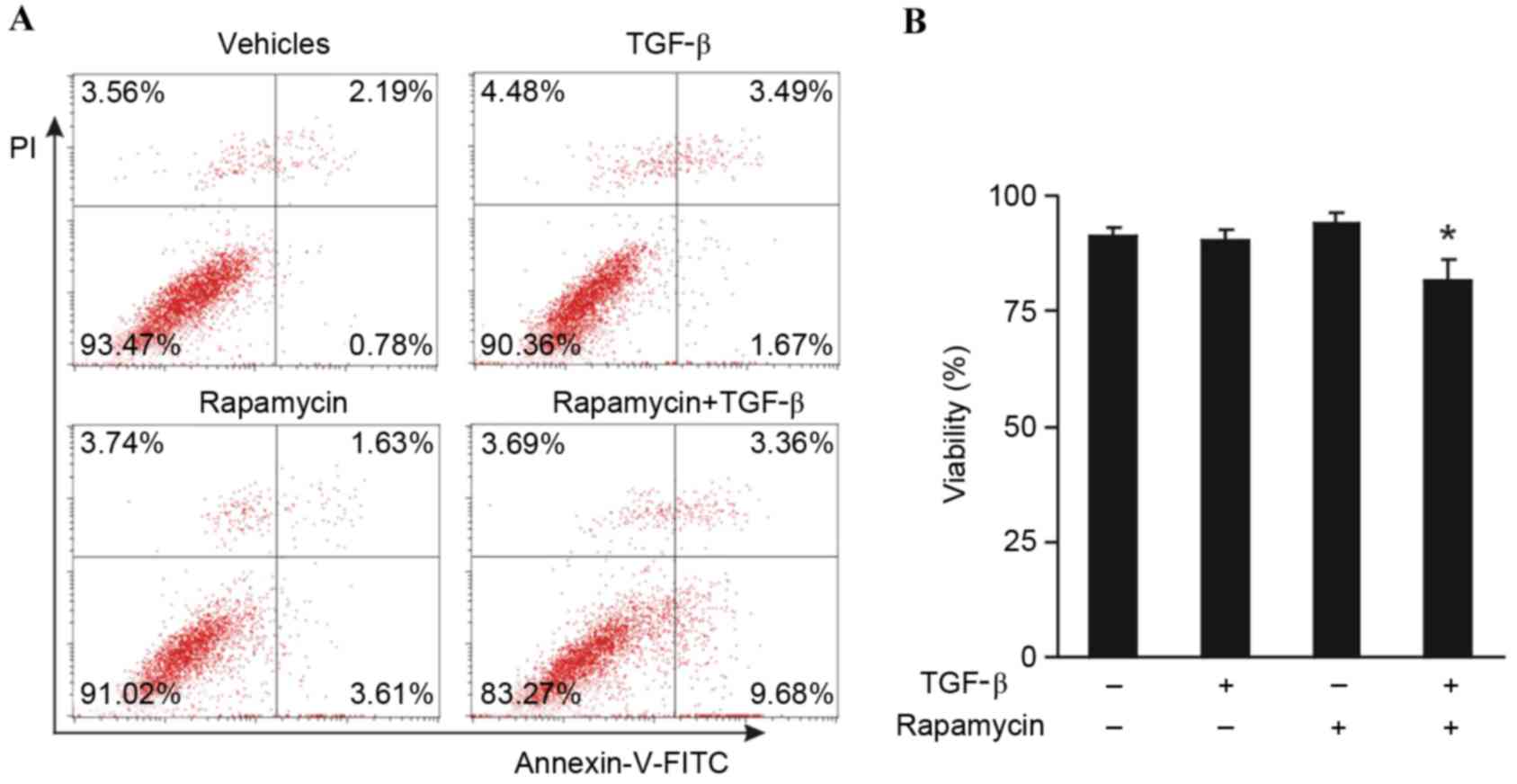
Combination treatment with rapamycin
and TGF-β induces apoptosis
An investigation was conducted in order to determine
whether the combination of rapamycin and TGF-β was able to induce
apoptosis of MCF-7 tumor cells. MCF-7 cells were treated with 100
nM rapamycin and 10 ng/ml TGF-β for 5 days, and subsequently the
cells were stained with annexin-V-FITC and PI. Enumeration of the
percentage of viable cells by flow cytometry demonstrated that
neither rapamycin nor TGF-β alone induced apoptosis; however, a
significant decrease in cell viability was observed in the presence
of both rapamycin and TGF-β compared with the vehicle control and
individual treatments of rapamycin or TGF-β (P<0.05; Fig. 4). These results suggest that the
combination of rapamycin and TGF-β induces MCF-7 tumor cell
apoptosis.
Discussion
The present study demonstrated that TGF-β exhibited
cytostatic effects on human breast adenocarcinoma MCF-7 cells,
which may be associated with the upregulation of CKIs, including
p14ARF, p15INK4b, p16INK4a and
p21WAF1/CIP1. It was also demonstrated that rapamycin
enhanced the antiproliferative effect of TGF-β. In addition,
rapamycin and TGF-β induced apoptosis of MCF-7 tumor cells.
TGF-β is involved in a variety of processes,
including proliferation, differentiation, apoptosis, adhesion, EMT,
and extracellular matrix deposition, which are essential for tissue
homeostasis (1). In the present
study, it was demonstrated that recombinant TGF-β inhibited the
proliferation of MCF-7 cells. This result was consistent with a
previous study by Mazars et al (22). Porcine TGF-β1 was used in the study
by Mazars et al (22),
whereas the present study used recombinant human TGF-β, which was
more suitable for the physiological conditions. CKIs have been
demonstrated to be causally associated with the inhibitory effect
of TGF-β; in ovarian cancer cells, TGF-β decreases cyclin-dependent
kinase 2 activity and induces p21WAF1/CIP1 (23). Other studies have demonstrated that
the effect of TGF-β on growth inhibition is mediated by Smad
complexes with forkhead box O factors, which activate
p15INK4b and p21CIP1 (24,25).
TGF-β has also been demonstrated to suppress transcription of the
Myc gene; in breast cancer cells that are insensitive to TGF-β, the
defective repression of Myc is frequently observed (26). TGF-β inhibits cell proliferation by
inhibiting c-Myc expression accompanied by the induction of p15 and
p21 expression (27,28). The transcription factor
CCAAT-enhancer binding protein β, essential for the induction of
p15INK4b and the repression of c-Myc, has been
demonstrated to be central to the cytostatic program initiated by
TGF-β (25). The results of the
present study demonstrated that, in MCF-7 tumor cells, TGF-β
induced the expression of p14ARF, p15INK4b,
p16INK4a and p21WAF1/CIP1, which may be
associated with the growth-arresting effects of TGF-β. Notably, it
has been reported that the response of fibroblasts and epithelial
cells to TGF-β differs, with TGF-β increasing the proliferation of
fibroblasts and inducing cell cycle arrest of epithelial cells
(29). TGF-β also functions to
maintain the pool of quiescent hematopoietic stem cells (30). Notably, TGF-β transcriptionally
activates p21, which stabilizes nuclear factor (erythroid-derived
2)-like 2, enhancing glutathione metabolism and diminishing the
effectiveness of anticancer therapeutics (7).
mTOR complex 1 is a critical regulator of Gap 1 (G1)
cell cycle progression and rapamycin is able to induce G1 cell
cycle arrest in MDA-MB-231 breast cancer cells (19). The present study demonstrated that
100 nM rapamycin alone was able to inhibit the proliferation of
MCF-7 cells. This effect may be associated with the relatively low
levels of phospholipase D activity in MCF-7 cells (31). The present study demonstrated, for
the first time, that rapamycin enhances the growth-arresting effect
of TGF-β on MCF-7 tumor cells. Therefore, it is now evident that
low doses of rapamycin are sufficient for activating TGF-β
signaling (18). Rapamycin-induced
G1 cell cycle arrest employs both TGF-β and retinoblastoma pathways
(19), which may partly explain the
combinational action of TGF-β and rapamycin. Neither 100 nM
rapamycin nor 10 ng/ml TGF-β alone induced apoptosis in MCF-7
cells; however, the combination of both rapamycin and TGF-β
resulted in significant apoptosis. The induction of apoptosis by
TGF-β has been demonstrated to be cell type-dependent. TGF-β
induces apoptosis in thyrocytes, and p27kip1 reduction
is a key event in this process (16). Further investigation is required to
clarify the mechanism of the combinational effect of TGF-β and
rapamycin on apoptosis.
In conclusion, the present study demonstrated that
rapamycin enhances the antiproliferative effect of TGF-β on human
MCF-7 tumor cells. These findings advance the current understanding
of the biological effects of TGF-β and rapamycin.
Acknowledgements
This research was supported by the National Natural
Science Foundation of China (grant no. 81501609) and the China
Postdoctoral Science Foundation (grant no. 2015M582553).
References
|
1
|
Massagué J: TGFbeta in cancer. Cell.
134:215–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cipriano R, Kan CE, Graham J, Danielpour
D, Stampfer M and Jackson MW: TGF-β signaling engages an
ATM-CHK2-p53-independent RAS-induced senescence and prevents
malignant transformation in human mammary epithelial cells. Proc
Natl Acad Sci USA. 108:pp. 8668–8673. 2011; View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schiller M, Javelaud D and Mauviel A:
TGF-beta-induced SMAD signaling and gene regulation: Consequences
for extracellular matrix remodeling and wound healing. J Dermatol
Sci. 35:83–92. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Flavell RA, Sanjabi S, Wrzesinski SH and
Licona-Limón P: The polarization of immune cells in the tumour
environment by TGFbeta. Nat Rev Immunol. 10:554–567. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee JC, Lee KM, Kim DW and Heo DS:
Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies
impaired NK cytotoxicity in cancer patients. J Immunol.
172:7335–7340. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen CL, Tsukamoto H, Liu JC, Kashiwabara
C, Feldman D, Sher L, Dooley S, French SW, Mishra L, Petrovic L, et
al: Reciprocal regulation by TLR4 and TGF-β in tumor-initiating
stem-like cells. J Clin Invest. 123:2832–2849. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Oshimori N, Oristian D and Fuchs E: TGF-β
promotes heterogeneity and drug resistance in squamous cell
carcinoma. Cell. 160:963–976. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gorelik L and Flavell RA: Immune-mediated
eradication of tumors through the blockade of transforming growth
factor-beta signaling in T cells. Nat Med. 7:1118–1122. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shiota M, Zardan A, Takeuchi A, Kumano M,
Beraldi E, Naito S, Zoubeidi A and Gleave ME: Clusterin Mediates
TGF-β-induced epithelial-mesenchymal transition and metastasis via
twist1 in prostate cancer cells. Cancer Res. 72:5261–5272. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Labelle M, Begum S and Hynes RO: Direct
signaling between platelets and cancer cells induces an
epithelial-mesenchymal-like transition and promotes metastasis.
Cancer Cell. 20:576–590. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fang Y, Yu S and Braley-Mullen H: TGF-β
promotes proliferation of thyroid epithelial cells in IFN-γ(−/−)
mice by down-regulation of p21 and p27 via AKT pathway. Am J
Pathol. 180:650–660. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lu SL, Reh D, Li AG, Woods J, Corless CL,
Kulesz-Martin M and Wang XJ: Overexpression of transforming growth
factor beta1 in head and neck epithelia results in inflammation,
angiogenesis, and epithelial hyperproliferation. Cancer Res.
64:4405–4410. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tang B, Vu M, Booker T, Santner SJ, Miller
FR, Anver MR and Wakefield LM: TGF-beta switches from tumor
suppressor to prometastatic factor in a model of breast cancer
progression. J Clin Invest. 112:1116–1124. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lyons RM and Moses HL: Transforming growth
factors and the regulation of cell proliferation. Eur J Biochem.
187:467–473. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bravo SB, Pampin S, Cameselle-Teijeiro J,
Carneiro C, Domínguez F, Barreiro F and Alvarez CV: TGF-beta
induced apoptosis in human thyrocytes is mediated by p27kip1
reduction and is overridden in neoplastic thyrocytes by NF-kappaB
activation. Oncogene. 22:7819–7830. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chan S: Targeting the mammalian target of
rapamycin (mTOR): A new approach to treating cancer. Br J Cancer.
91:1420–1424. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yellen P, Saqcena M, Salloum D, Feng J,
Preda A, Xu L, Rodrik-Outmezguine V and Foster DA: High-dose
rapamycin induces apoptosis in human cancer cells by dissociating
mTOR complex 1 and suppressing phosphorylation of 4E-BP1. Cell
Cycle. 10:3948–3956. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chatterjee A, Mukhopadhyay S, Tung K,
Patel D and Foster DA: Rapamycin-induced G1 cell cycle arrest
employs both TGF-β and Rb pathways. Cancer Lett. 360:134–140. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wan G, Mathur R, Hu X, Liu Y, Zhang X,
Peng G and Lu X: Long non-coding RNA ANRIL (CDKN2B-AS) is induced
by the ATM-E2F1 signaling pathway. Cell Signal. 25:1086–1095. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mazars P, Barboule N, Baldin V, Vidal S,
Ducommun B and Valette A: Effects of TGF-beta 1 (transforming
growth factor-beta 1) on the cell cycle regulation of human breast
adenocarcinoma (MCF-7) cells. FEBS Lett. 362:295–300. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Elbendary A, Berchuck A, Davis P,
Havrilesky L, Bast RC Jr, Iglehart JD and Marks JR: Transforming
growth factor beta 1 can induce CIP1/WAF1 expression independent of
the p53 pathway in ovarian cancer cells. Cell Growth Diff.
5:1301–1307. 1994.PubMed/NCBI
|
|
24
|
Seoane J, Le HV, Shen L, Anderson SA and
Massagué J.: Integration of Smad and forkhead pathways in the
control of neuroepithelial and glioblastoma cell proliferation.
Cell. 117:211–223. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gomis RR, Alarcón C, He W, Wang Q, Seoane
J, Lash A and Massagué J: A FoxO-Smad synexpression group in human
keratinocytes. Proc Natl Acad Sci USA. 103:pp. 12747–12752. 2006;
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen CR, Kang Y and Massagué J: Defective
repression of c-myc in breast cancer cells: A loss at the core of
the transforming growth factor beta growth arrest program. Proc
Natl Acad Sci USA. 98:pp. 992–999. 2001; View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y
and Wang XF: Transforming growth factor beta induces the
cyclin-dependent kinase inhibitor p21 through a p53-independent
mechanism. Proc Natl Acad Sci USA. 92:pp. 5545–5549. 1995;
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li JM, Nichols MA, Chandrasekharan S,
Xiong Y and Wang XF: Transforming growth factor beta activates the
promoter of cyclin-dependent kinase inhibitor p15INK4B through an
Sp1 consensus site. J Biol Chem. 270:26750–26753. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hosobuchi M and Stampfer MR: Effects of
transforming growth factor beta on growth of human mammary
epithelial cells in culture. In Vitro Cell Dev Biol. 25:705–713.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mo AD, Joseph H and Nolta JA: Molecular
mechanism of transforming growth factor beta-mediated cell-cycle
modulation in primary human CD34(+) progenitors. Blood. 99:499–506.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen Y, Zheng Y and Foster DA:
Phospholipase D confers rapamycin resistance in human breast cancer
cells. Oncogene. 22:3937–3942. 2003. View Article : Google Scholar : PubMed/NCBI
|