Introduction
Vitiligo is a skin disease characterized by the lack
of pigmentation in the skin, it affects approximately 0.1 to 2% of
the world population. However, its prevalence varies considerably
among populations and ethnic groups: 0.14% in Russia, 1% in USA,
and 2.5% in Japan (1), although the
highest incidence has been described in Mexico (4%) and India
(8.8%) (1,2). The most common clinical variant of
vitiligo is vitiligo vulgaris (VV), in which the patient presents
asymptomatic, well-circumscribed, milky-white macules involving one
or multiple body regions or segments (3). The lack of pigmentation could be
attributed to two main causes: a) the absence of melanocytes, which
are dendritic cells derived from the neural crest that migrates to
the epidermis and then to the hair follicle during embryogenesis,
or b) the inability of these cells to produce and store melanin in
melanosomes in the process of melanogenesis (4). In this context, the pathological origin
of vitiligo has not yet been fully understood. Several hypotheses
and theories have been developed to explain these depigmentation
processes (5–8).
Although melanocyte is responsible for the
pigmentation process in vertebrates (9), the significance of the surrounding
environment has been neglected, e.g., keratinocytes (8,10).
Currently, it is known that the signaling mechanism that activates
the route of melanogenesis is controlled by genes, whose products
act as enzymes, structural proteins, transcriptional regulators,
transporters, receptors and growth factors related to the
melanogenesis process (11). Some of
the hormones and products from the hypothalamic-pituitary-adrenal
axis, and their respective receptors and negative regulators,
trigger a nuclear signaling cascade that leads to the activation or
repression of tyrosinase, a key enzyme in the pigmentation process,
including the final amount of melanin produced (12,13). A
major hormone regulator in melanin synthesis is the
α-melanocyte-stimulating hormone (α-MSH), produced by pituitary
gland, which interacts with a specific cell surface receptor
[Melanocortin 1 Receptor (MC1R)] to stimulate melanin synthesis and
other differentiated melanocyte functions (11).
Today, knowledge about genes and signaling pathways
potentially involved in the development of vitiligo is increasing
(14–17); for example, the expression profile of
approximately sixteen thousand genes, in an in vitro culture
of melanocytes obtained from five subjects with vitiligo, was
analyzed using microarrays (17),
describing five routes involved in the development of this disease:
i) Development of melanocytes; ii) intracellular processing and
vesicular trafficking of tyrosinase gene family protein; iii)
packaging and transport of melanosomes; iv) cell adhesion; and v)
processing and presentation of antigens (17).
In addition, previous reports of gene expression
involved in the melanocortin system (14) and melanogenesis signaling pathways
(15) showed modified expression
levels of proopiomelanocortin (POMC), melanocortin 1 receptor
(MC1R), melanocortin 4 receptor (MC4R), tyrosinase-related protein
1 (TYRP1) and dopachrome tautomerase (DCT), among other genes, in
tissue affected by vitiligo when compared with healthy tissue
samples from patients with vitiligo and normal skin from
controls.
To date, there are few studies that involve the
complete tissue analysis of patients with vitiligo. The
identification of the expression profile of genes potentially
involved in the development of the disease will be useful in
understanding the molecular mechanism of its development to select
potential therapeutic targets for its specific treatment.
In this study, we used microarray analysis to
characterize the transcriptional profile of the skin of patients
with VV and the identified transcripts were validated by the use of
high-throughput RNA sequencing in a larger set of patients to
determine the expression pattern that could play a role in the
pathogenesis of vitiligo and the clinical types of this
disease.
Materials and methods
Selection of participants
Fifty-five Northeastern Mexican patients from the
states of Coahuila, Nuevo Leon, San Luis Potosi, Tamaulipas, and
Zacatecas were recruited between November 2009 and May 2015 at the
Dermatology Department of the University Hospital-UANL, in
Monterrey, Nuevo Leon, Mexico. The Ethics and Research Committee of
the Faculty of Medicine-UANL approved and registered the protocol
and forms of informed consent under the code DE08-008 and DE13-001.
After signing their informed consent, the patients were interviewed
and evaluated to confirm the diagnosis of VV. Five healthy controls
were also included for the Microarray analysis. None of VV patients
had received any specific treatment in the previous six months to
recruitment. The stage of VV (active AVV/stable SVV) was determined
by intervals of time in the manifestation of new depigmented areas
(stable vitiligo with lesional stability of >1 year), or
enlargement of the already existing ones.
Sample collection and processing
Two skin biopsies of 4 mm were obtained from each
patient with vitiligo. The first biopsy was obtained from the
central part of the affected skin (called depigmented vitiligo
skin) and the second from the healthy areas of the skin of patients
with vitiligo (called pigmented vitiligo skin), generally 3 cm from
the affected skin. Only a skin biopsy was taken from healthy
control subjects (called control skin).
The skin tissue from the biopsies was immediately
suspended in 5 volumes of RNA Later Solution (Ambion; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) after collection, and stored at
4°C overnight. The next day, the supernatant was remove, and the
samples were stored at −80°C until the time for analysis.
Total RNA was isolated from the samples using the
RNeasy fibrous tissue mini kit (Qiagen, Inc., Valencia, CA, USA)
according to the manufacturer's instructions and quantified using a
NanoDrop® ND-8000 spectrophotometer (Thermo Fisher
Scientific, Inc., Wilmington, DE, USA) and Qubit® RNA BR
Assay kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
quality/integrity of the extracted RNA was evaluated by an Experion
Automated electrophoresis System (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Microarray assays
The biopsies taken from depigmented and pigmented
skin from ten vitiligo patients (5 AVV and 5 SVV) and a skin biopsy
of each of the five healthy controls were analyzed. For each
sample, 100 ng of total RNA was amplified, purified, fragmented and
labeled using the GeneChip® IVT Express kit,
(Affymetrix; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. 12.5 µg of fragmented and labeled
target were hybridized with Affymetrix GeneChip® Human
Gene u133 plus Array (Affymetrix; Thermo Fisher Scientific, Inc.),
at 45°C for 16 h, on a GeneChip® Hybridization Oven 640
(Affymetrix; Thermo Fisher Scientific, Inc.), according to the
manufacturer's recommendation. The hybridized arrays were washed
and stained on a GeneChip® Fluidics Station 450, scanned
on a GeneChip® Scanner 3000 7G (Affymetrix; Thermo
Fisher Scientific, Inc.), and then CEL image data files were
generated for each matrix.
Data analysis
CEL files were processed using the affy
package in R (https://cran.r-project.org). Data were pre-processed
using RMA and normalized using quantile-normalization (18). t-test and F-tests were used to
determine a P-value for differentially expressed genes between
groups. P-values were adjusted for multiple tests using the False
Discovery Rate (FDR) approach (19).
Principal component analysis (PCA) was performed in R.
Annotation and functional
analysis
Further information about the genes was obtained
from NetAffx™ Analysis Center (http://www.affymetrix.com) and NCBI databases
(http://www.ncbi.nlm.nih.gov).
Functional annotation was performed by uploading the
resulting gene list onto DAVID (20,21)
(Database for Annotation, Visualization and Integrated Discovery,
https://david.ncifcrf.gov/tools.jsp).
Genes were mapped to KEGG-pathways and scored according to P-values
(EASE Score, modified Fisher's exact test) and corrected for
multiple testing according to the Benjamini-Hochberg False
Discovery Rate correction provided by the DAVID tool.
TruSeq Targeted RNA Expression
analysis
Skin biopsies from 45 vitiligo patients (23 AVV and
22 SVV) were analyzed by TruSeq Targeted RNA Expression (Illumina,
Inc., San Diego, CA, USA). RNA was obtained and cDNA was prepared
with ProtoScript® Reverse Transcriptase (New England
Biolabs, Ipswich, MA, USA) according to the manufacturer's
instructions. RNA expression analysis was performed on an Illumina
MiSeq with MiSeq® Reagent Kit v3 (150 cycle) and
TruSeq® Targeted RNA Custom Panel Kit designed to detect
the expression profile of the calpain 3 (CAPN3), dopachrome
tautomerase (DCT), glycerol-3-phosphate dehydrogenase 1 (GPD1),
melan-A (MLANA) and tyrosinase-related protein 1 (TYRP1). RNA-Seq
data were analyzed within Base Space (https://basespace.illumina.com) using the tool TruSeq
Targeted RNA. Count data were exported and normalized per sample by
equalizing the total number of counts (multiplying by the average
of the total counts per sample then dividing by the accumulated
counts per sample).
Results
Microarray analysis
In the analysis performed on vitiligo samples using
microarray analysis, we first compared whether the unaffected skin
holds a higher similarity to healthy controls than to the affected
skin of the patients; one of the pigmented skin samples from a
vitiligo patient was discarded from the microarray analyses as it
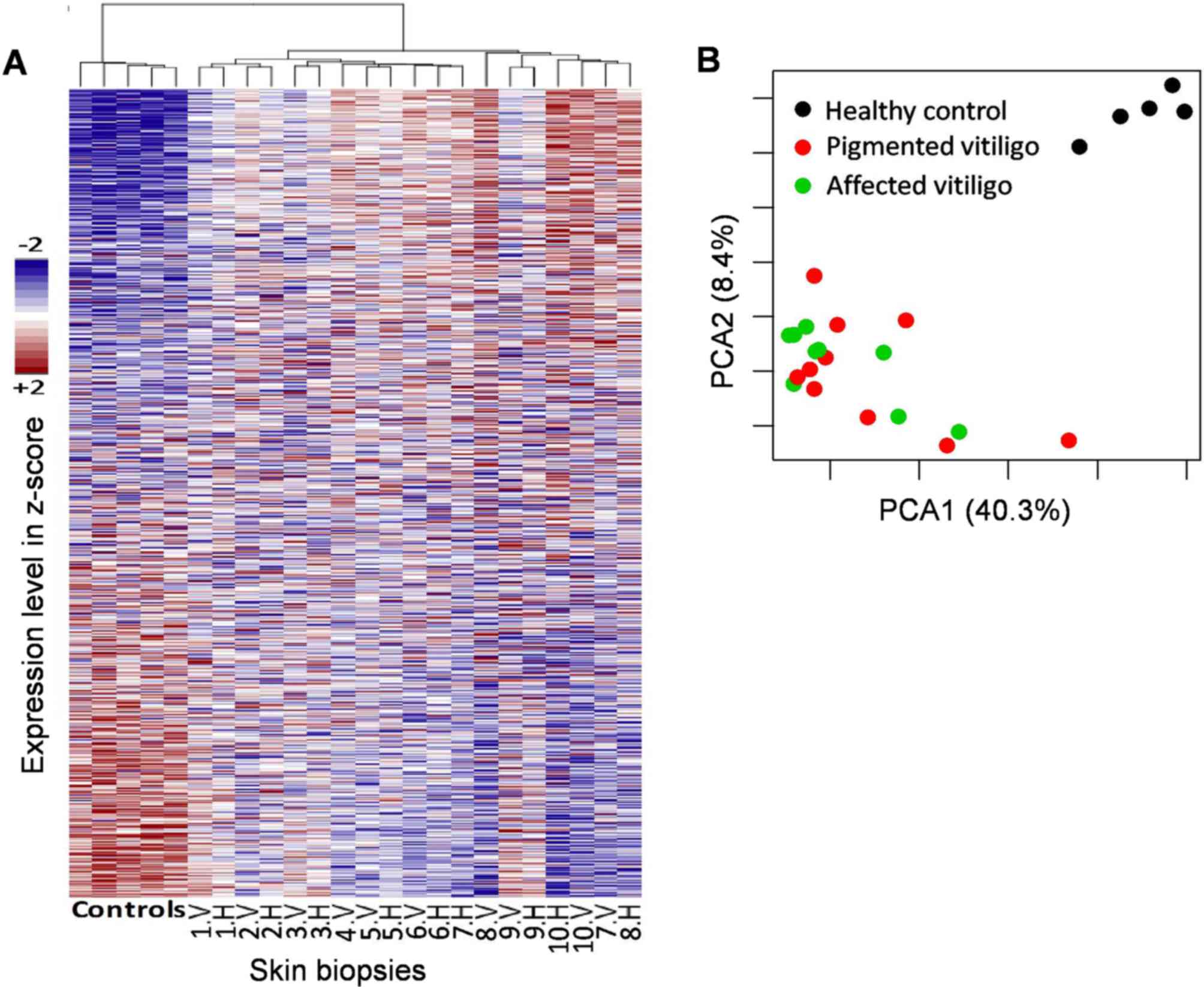
lacked sufficient quality to be used. A PCA and hierarchical
clustering shows that the unaffected skin is more similar to
vitiligo samples than to healthy controls (Fig. 1), suggesting that pigmented skin in
vitiligo patients is already affected regardless of its clinical
evidence.
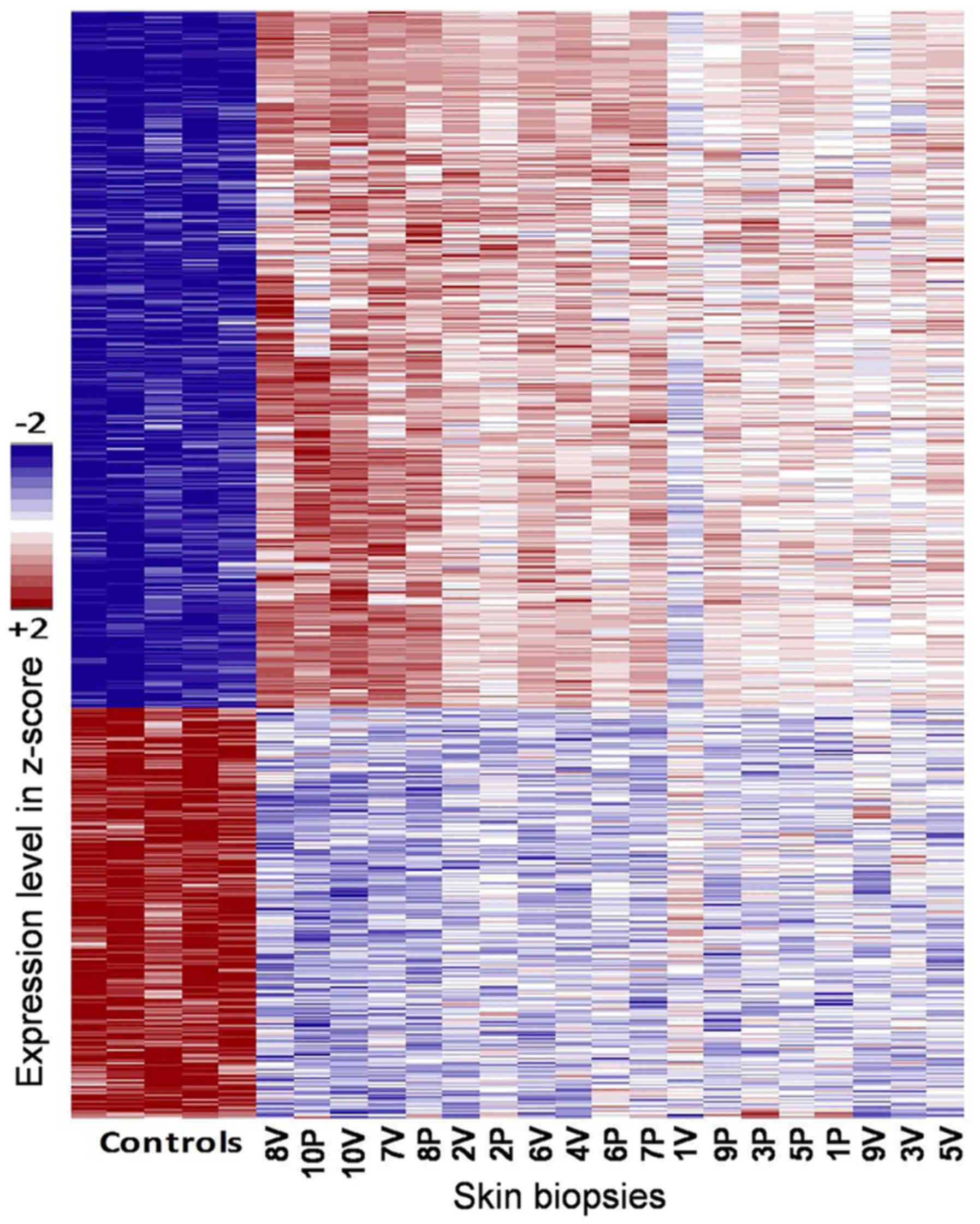
Further analysis comparing the healthy controls vs.
skin samples from vitiligo patients (de/pigmented tissue) confirmed
the existence of a large number of hits (1,927 probes at FDR
<0.1) (Fig. 2).
Then we compared different combinations of groups at
FDR <0.1, showing that there are differences between the three
groups, with altered expression patterns (over and under expressed)
when analyzing the more than 25,000 genetic targets present in
Affymetrix Gene Chip® Human Gene u133 plus Array.
Further, we observed several changes when comparing the healthy
controls and patients independent of sample type (pigmented or
depigmented skin of patients with vitiligo) that reached the 1,927
probes (Table I). When comparing the
pigmented tissue of vitiligo patients against healthy controls, the
difference decreases to 1,108 (Table
I). Surprisingly, the number decreases further to 722 when
comparing the depigmented vitiligo samples to healthy controls
(Table I). Finally, the differences
are modest when comparing the vitiligo depigmented samples against
pigmented asymptomatic tissue, consistent with the initial
observation that the asymptomatic tissue is already compromised.
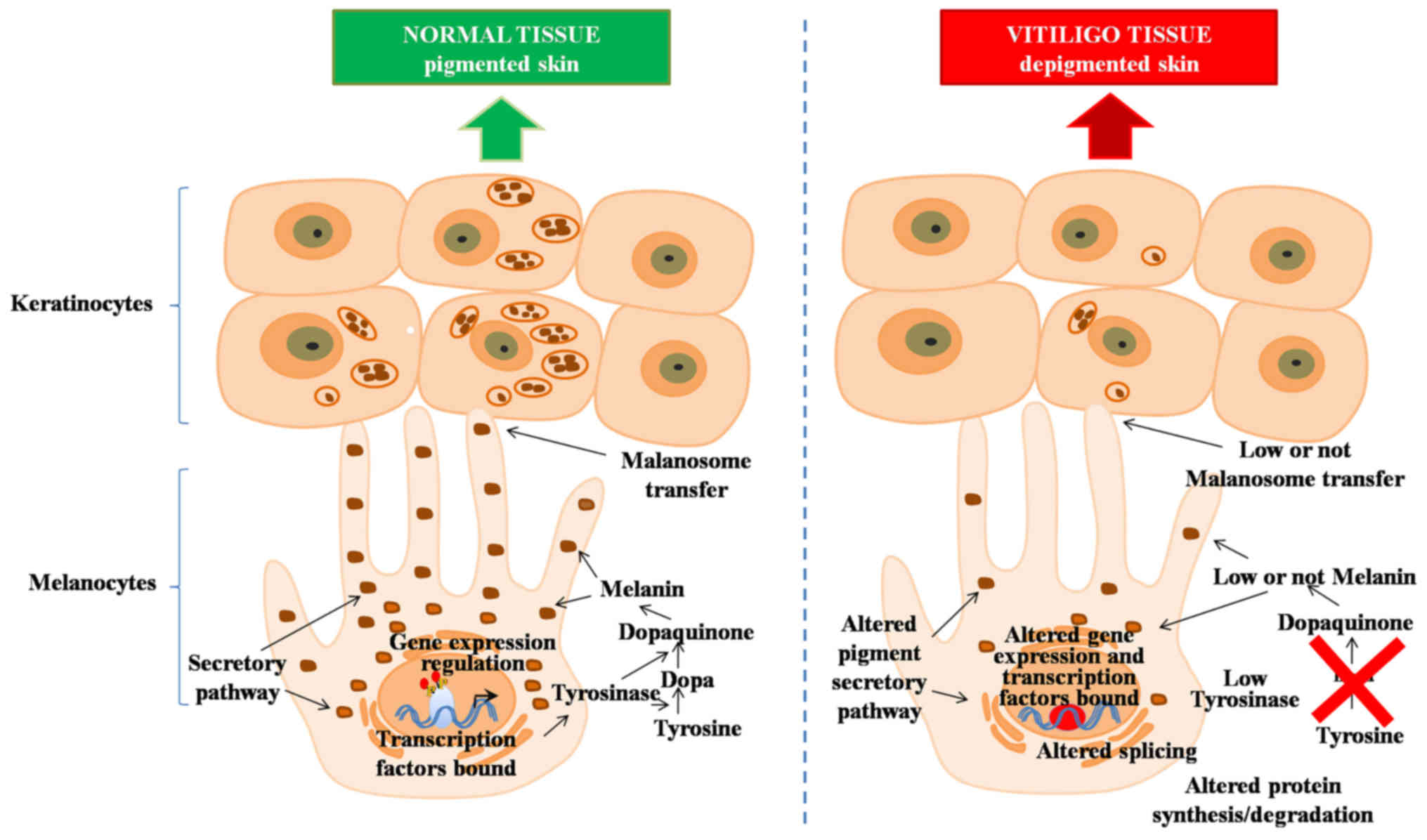
Therefore, we focused on these minor differences between vitiligo
samples and their healthy counterparts, observing the differences
in the important components of pigmentation (Fig. 3). Moreover, we noted that the
expression profile of these genes were similar to those from
healthy controls. On the other hand, no relationship was found
between expression pattern variations, gender, age, or type of
vitiligo.
 | Table I.Inference of pathways involved in
vitiligo, according to the probes that presented altered expression
patterns (over and sub expressed). |
Table I.
Inference of pathways involved in
vitiligo, according to the probes that presented altered expression
patterns (over and sub expressed).
| Comparison/N | Biological altered
pathways |
|---|
| Vitiligo
depigmented skin vs. vitiligo pigmented skin vs. controls
(N=1,927) | Alternative
splicing/Splice variant DNA-binding/Nucleic acid
binding/Zinc-finger/Transcription factor and transcription
regulation/Krueppel-associated box |
|
| Metal-binding |
|
| Phosphoprotein |
|
| Krueppel-associated
box |
|
| Coiled coil |
| Vitiligo pigmented
skin vs. controls (N=1,108) | Alternative
splicing/Splice variant |
|
| Intracellular |
|
| Krueppel-associated
box |
|
| Coiled coil |
| Vitiligo
depigmented skin vs. controls (N=722) | Keratin
filament |
|
| High sulphur
keratin-associated protein |
|
| Splice variant |
|
| Krueppel-associated
box |
| Vitiligo
depigmented skin vs. vitiligo pigmented skin (N=6) |
Melanosome/melanosome membrane |
|
| Uncharacterised
domain, di-copper centre |
|
| Tyrosinase/Tyrosine
metabolism |
|
| Topological domain:
Lumenal, melanosome |
|
| Melanin
biosynthesis/Melanogenesis |
Functional Annotation of observed
differences
Using the DAVID functional annotation tool, the
genes identified in each comparison were grouped into functional
categories such as alternative splicing processes, splice variants,
DNA and RNA binding domains, transcription, and expression
regulation, amongs others (Table I).
However, by focusing on the comparisons made between the pigmented
and depigmented skin of vitiligo patients it becomes apparent that
the mainly compromised genes are involved in the pigmentation
process, finding groups of genes responsible for pigment
production, granule and melanosome, melanogenesis, tyrosine
metabolism, melanin biosynthesis and metabolic process, all of
these involved in the development of this disease (Fig. 3).
TruSeq targeted RNA Expression
analysis expression validation patterns
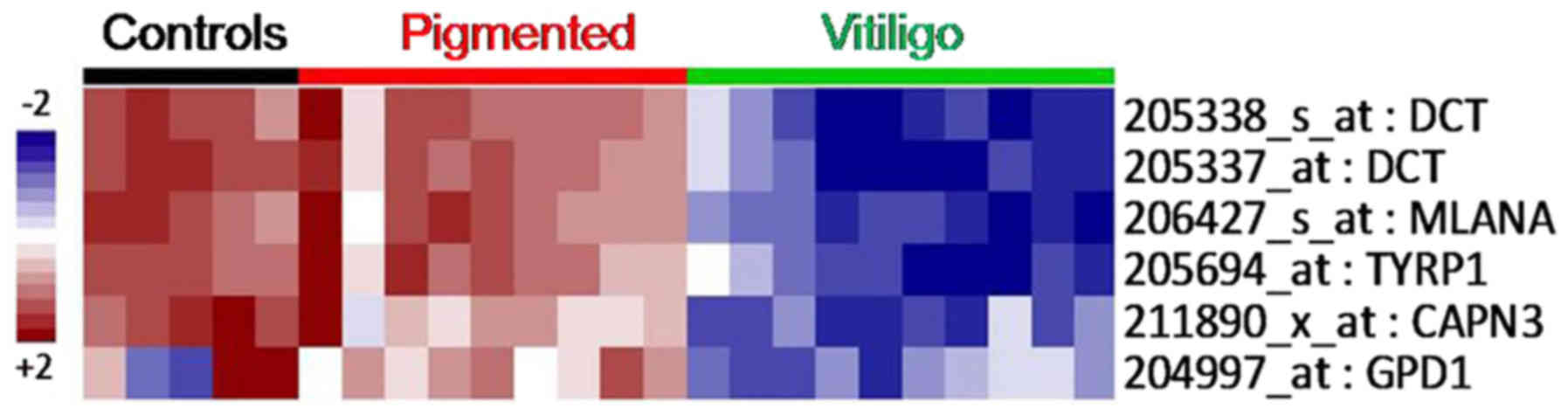
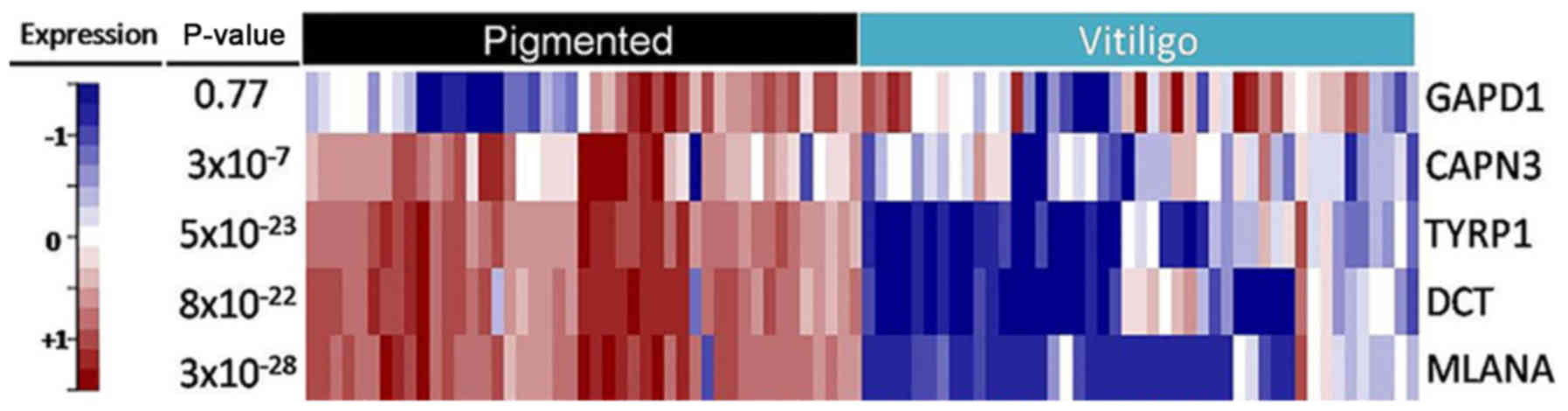
Surprisingly, the main differences between
asymptomatic and depigmented skin from vitiligo patients were
narrowed to only six probes (Fig.
4), five of which correspond to undrexpressed genes in vitiligo
tissue samples, i.e., dopachrome tautomerase (id. 205338_s_at,
205337_at DCT;), melan-A (id. 206427_s_at MLANA),
tyrosinase-related protein 1 (id. 205694_at TYRP1), calpain 3 (id.
211890_x_at CAPN3), and glycerol-3-phosphate dehydrogenase 1 (id.
204997_at: GPD1). These genes have been mostly related to
melanogenesis, tyrosine metabolism, and glycerophospholipid
metabolism. Therefore, these genes were selected for validation by
targeted RNA-Sequencing.
The expression analysis performed on skin biopsies
from 45 additional VV patients confirmed the existence of
significant differences in the expression pattern of three genes
involved in skin pigmentation: DCT, MLANA and TYRP1. In addition,
CAPN3 was also significant, although at a lower level. In addition
to microarray analysis, no relationship was identified between the
variants in the patterns of expression, gender, age, or type of
vitiligo. Moreover, in the case of the GAPD1 gene, no significant
difference in the expression pattern between asymptomatic skin and
vitiligo lesions was found by NGS when compared to previous
observations using microarray analysis (Fig. 5).
Discussion
Most vitiligo studies conducted in the Mexican
population are focused in demographics and clinical
characteristics. To date, the molecular etiology and genetic
factors interacting in the development of vitiligo within this
population have not been considered.
Up to date, there has been only two expression
studies of vitiligo using microarray technology. In the first
study, melanocyte culture in vitro from 5 samples of VV
patients were analyzed (17); in
addition to the small number of samples, it did not reflect the
contribution of the skin's environment as it only represents the
alterations of a single cellular type (melanocytes); further, the
expression pattern could be modified under cell culture conditions.
This report found altered an expression pattern of genes involved
in the development and function of melanocytes, proposing an
autoimmune response as a secondary event caused by the abnormal
function of melanocytes (17). In
our study, we also observed some similarities in the expression
pattern of genes involved in pigment synthesis, melanosome, granule
pigment, cytoplasmic vesicles (possibly melanosomas), plus redox
reaction processes related to diverse functions, including
cell-cell recognition, cell-surface receptors, muscle structure,
and the immune system. However, our results reflect the expression
profile of the whole skin.
In a second report, an expression study using the
Smyth line from chicken, an avian model for human autoimmune
vitiligo, the microarray analysis results support the
multifactorial etiology of vitiligo, where inflammatory/innate
immune activity and oxidative stress, and the adaptive immune
response play a predominant role in melanocyte loss. The microarray
analysis results provided comprehensive information at
transcriptome level, supporting the multifactorial etiology of
vitiligo, where, along with the apparent inflammatory/innate immune
activity and oxidative stress, the adaptive immune response plays a
predominant role in melanocyte loss (22).
In the biological context of the skin, the
melanocyte represents less than ten percent of epidermal cells
(23,24). The functional clustering analysis
made using the DAVID database points towards the involvement of an
altered functional group composed by genes involved in diverse
biological processes, such as transcription and transcription
regulation, transcriptional repression, alternative splicing, and
keratin filament. The observation that melanocytes are the main
cell type affected in vitiligo does not rule out the participation
of other cellular components of the skin in the development of this
disease, as supported by the major affected pathways of pigment
synthesis, packaging and transport of pigment, when the depigmented
vitiligo tissue was compared with the pigmented skin of vitiligo.
For this reason, and considering that keratinocytes are the
predominant cell type in this tissue (25), their role could involve an important
function in skin pigmentation (10,26).
Even when the differences in expression patterns between a)
pigmented skin and b) affected skin vs. control skin the same
metabolic routes are affected, in the first case a greater number
of genes are involved (Table I),
these could be explained by compensatory mechanisms (14,27) in
which the skin of the vitiligo patient tends to stimulate
pigmentation by regulating the expression factors involved in
pigmentation, increasing or decreasing their expression to maintain
skin homeostasis.
After analyzing the genes showing a different
expression pattern, a group of 5 genes, involved in the melanogenic
routes, intracellular cysteine proteases, and oxidative stress,
were selected for validation by TruSeq Targeted RNA Expression
analysis. The validation assays showed a differential expression
tendency in four genes: CAPN3, DCT, MLANA and TYRP1. However, GPD1
was not found differentially expressed by NGS. These results are
consistent with Regazzetti et al (28), whom validated a group of genes in
skin biopsies from patients with vitiligo, finding low expression
levels of MLANA, DCT, and TYRP1 in vitiligo skin lesions when
compared to skin around the injured and uninjured
(pigmented/asymptomatic) tissue.
The CAPN3 gene was under expressed in the skin of
patients with vitiligo; previously, Stromberg et al reported
that this gene was up regulated in melanocyte cultures obtained
from vitiligo lesions (17). This
gene encodes a large subunit of neutral calpain of protease 3
activated by muscle calcium, a heterodimer consisting of a large
subunit and a small subunit is a major intracellular protease that
is ubiquitously expressed in human tissues and other and other
tissue-specific isoforms. Its function on the skin is not clear,
but it has been observed that CAPN3 variants can play a
pro-apoptotic role in melanoma cells and its down regulation, as
observed in highly aggressive melanomas, could contribute to their
progression (29). In melanocytic
cells, CAPN3 expression may be regulated by MITF (30) which regulates a broad variety of
genes whose functions range from pigment production (such as DCT,
MC1R, MLANA, TYR and TYP1) to the cell-cycle regulation, migration
and survival; moreover, MITF-mediated up regulation of CAPN3 has
been reported in melanoma (30).
The GPD1 gene encodes a member of the NAD-dependent
glycerol-3-phosphate dehydrogenase protein family, playing a
critical role in carbohydrate and lipid metabolism (31). Along with GPD2, the mitochondrial
isoform, it constitutes a glycerol phosphate shuttle that
facilitates the transfer of reducing equivalents from the cytosol
to the mitochondria, playing a crucial role in osmoregulation and
redox balance (32). In our study,
the microarray analysis showed the under expression of this gene in
tissue from patients with vitiligo lesions. As a result of
expression profile validation, made by RNA-Seq in a number of
samples, no significant differences were observed between the
expression profiles of affected pigmented skin in vitiligo
patients. However, we observed a higher expression in some of the
samples that possibly participate in the oxidative stress response
experienced by the skin cells of these patients, a mechanism that
has been described in this disease. So far, the participation of
this gene in vitiligo has not been explored.
In addition, NGS provides a better approach to gene
expression profile analysis, as this technology enables high
resolution research for all of the RNA present in a sample,
including mRNA sequence and abundance (33). Its usefulness has been compared with
microarray technology and Real-Time PCR in the analysis of
expression profiles (34–36); therefore, it has been used in
transcriptome analysis in clinical research (36–39).
The analysis of expression by directed sequencing of
RNA (RNAseq) has been helpful in the efficient evaluation of
expression profiles of multiple genes in human pathological and
non-pathological conditions (40–43). In
particular, RNAseq using NGS, not only shows the expression levels
of a transcript, but the presence of isoforms in the samples
analyzed (44). In patients affected
by dermatological diseases such as psoriasis, systemic lupus
erythematosus (45–47), and vitiligo, these tools are an
excellent alternative inidentifying modified gene expression
patterns in key routes of skin homeostasis, pigmentation, and cell
survival at a low cost and reduced time in comparison to
RT-qPCR.
In conclusion, we found alterations in the
expression pattern of depigmented vitiligo lesions when compared to
pigmented asymptomatic skin of vitiligo patients and against
healthy control samples. These results reflect that even the
clinically unaffected (pigmented) skin shows an impaired
melanogesis process in a patient with vitiligo; the observed
changes are related to processes regulating gene expression and
splicing that eventually will end up in skin depigmentation by
altering the levels of proteins involved in this process. Up
regulated expression profiles detected by microarray analysis for
CAPN3, DCT, MLAN-A and TYRP1 in VV patients was validated using NGS
TruSeq Targeted RNA Expression analysis, and whose altered
functions on pigment production, and possibly in melanocyte cell
survival, affect skin pigmentation, thus suggesting that these
tools are an excellent alternative in identifying the expression
pattern of genes involved in the development of this disease. On
the other hand, we did not find any differences in the gene
expression profiles of active or stable VV.
Acknowledgements
Thanks to all participants and collaborators in this
study. Thanks, also, to the personnel of the Department of
Dermatology of the University's Hospital and the Molecular Biology,
Genomics and Sequencing Unit Center for Research and Development in
Health Sciences, UANL. We appreciate the kindness and help of the
personnel in the Unit of Molecular Diagnosis-Department of
Molecular Medicine, Faculty of Medicine-UANL, for their support
during the development of this study. The participating students
were supported by the CONACyT (grant no. 220719). The authors also
wish to thank Dr Daniel Díaz, Ph.D. for his kind assistance in
proofreading this manuscript.
References
|
1
|
Steiner D, Bedin V, Moraes MB, Tadeu R and
Steiner VT: Vitiligo. An Bras Dermatol. 79:335–351. 2004.
View Article : Google Scholar
|
|
2
|
Parsad D, Dogra S and Kanwar AJ: Quality
of life in patients with vitiligo. Health Qual Life Outcomes.
1:582003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ezzedine K, Lim HW, Suzuki T, Katayama I,
Hamzavi I, Lan CC, Goh BK, Anbar T, Silva de Castro C, Lee AY, et
al: Revised classification/nomenclature of vitiligo and related
issues: The vitiligo global issues consensus conference. Pigment
Cell Melanoma Res. 25:E1–E13. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sehgal VN and Srivastava G: Vitiligo:
Compendium of clinico-epidemiological features. Indian J Dermatol
Venereol Leprol. 73:149–156. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Spritz RA: The genetics of generalized
vitiligo and associated autoimmune diseases. J Dermatol Sci.
41:3–10. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Spritz RA: The genetics of generalized
vitiligo and associated autoimmune diseases. Pigment Cell Res.
20:271–278. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tazi-Ahnini R, McDonagh AJ, Wengraf DA,
Lovewell TR, Vasilopoulos Y, Messenger AG, Cork MJ and Gawkrodger
DJ: The autoimmune regulator gene (AIRE) is strongly associated
with vitiligo. Br J Dermatol. 159:591–596. 2008.PubMed/NCBI
|
|
8
|
Mohammed GF, Gomaa AH and Al-Dhubaibi MS:
Highlights in pathogenesis of vitiligo. World J Clin Cases.
3:221–230. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
McCurdy HM: Enzyme localization during
melanogenesis. J Cell Biol. 43:220–228. 1969. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee AY: Role of keratinocytes in the
development of vitiligo. Ann Dermatol. 24:115–125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Slominski A, Ermak G and Wortsman J:
Modification of melanogenesis in cultured human melanoma cells. In
Vitro Cell Dev Biol Anim. 35:564–565. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gillbro JM, Marles LK, Hibberts NA and
Schallreuter KU: Autocrine catecholamine biosynthesis and the
beta-adrenoceptor signal promote pigmentation in human epidermal
melanocytes. J Invest Dermatol. 123:346–353. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rousseau K, Kauser S, Pritchard LE,
Warhurst A, Oliver RL, Slominski A, Wei ET, Thody AJ, Tobin DJ and
White A: Proopiomelanocortin (POMC), the ACTH/melanocortin
precursor, is secreted by human epidermal keratinocytes and
melanocytes and stimulates melanogenesis. FASEB J. 21:1844–1856.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kingo K, Aunin E, Karelson M, Philips MA,
Rätsep R, Silm H, Vasar E, Soomets U and Kõks S: Gene expression
analysis of melanocortin system in vitiligo. J Dermatol Sci.
48:113–122. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kingo K, Aunin E, Karelson M, Rätsep R,
Silm H, Vasar E and Kõks S: Expressional changes in the
intracellular melanogenesis pathways and their possible role in the
pathogenesis of vitiligo. J Dermatol Sci. 52:39–46. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nagui NA, Mahmoud SB, Abdel Hay RM,
Hassieb MM and Rashed LA: Assessment of gene expression levels of
proopiomelanocortin (POMC) and melanocortin-1 receptor (MC1R) in
vitiligo. Australas J Dermatol. 58:e36–e39. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stromberg S, Bjorklund MG, Asplund A,
Rimini R, Lundeberg J, Nilsson P, Pontén F and Olsson MJ:
Transcriptional profiling of melanocytes from patients with
vitiligo vulgaris. Pigment Cell Melanoma Res. 21:162–171. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Irizarry RA, Hobbs B, Collin F,
Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP:
Exploration, normalization and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate: A practical and powerful approach to
multiple testing. J Royal Stat Soc. 57:289–300. 1995.
|
|
20
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang da W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shi F, Kong BW, Song JJ, Lee JY,
Dienglewicz RL and Erf GF: Understanding mechanisms of vitiligo
development in Smyth line of chickens by transcriptomic microarray
analysis of evolving autoimmune lesions. BMC Immunol. 13:182012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jimbow K, Quevedo WC Jr, Fitzpatrick TB
and Szabo G: Some aspects of melanin biology: 1950–1975. J Invest
Dermatol. 67:72–89. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Feinmesser M, Tsabari C, Fichman S, Hodak
E, Sulkes J and Okon E: Differential expression of proliferation-
and apoptosis-related markers in lentigo maligna and solar
keratosis keratinocytes. Am J Dermatopathol. 25:300–307. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Baroni A, Buommino E, De Gregorio V,
Ruocco E, Ruocco V and Wolf R: Structure and function of the
epidermis related to barrier properties. Clin Dermatol. 30:257–262.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yoshida Y, Hachiya A, Sriwiriyanont P,
Ohuchi A, Kitahara T, Takema Y, Visscher MO and Boissy RE:
Functional analysis of keratinocytes in skin color using a human
skin substitute model composed of cells derived from different skin
pigmentation types. FASEB J. 21:2829–2839. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Moretti S, Fabbri P, Baroni G, Berti S,
Bani D, Berti E, Nassini R, Lotti T and Massi D: Keratinocyte
dysfunction in vitiligo epidermis: Cytokine microenvironment and
correlation to keratinocyte apoptosis. Histol Histopathol.
24:849–857. 2009.PubMed/NCBI
|
|
28
|
Regazzetti C, Joly F, Marty C, Rivier M,
Mehul B, Reiniche P, Mounier C, Rival Y, Piwnica D, Cavalié M, et
al: Transcriptional analysis of vitiligo skin reveals the
alteration of wnt pathway: A promising target for repigmenting
vitiligo patients. J Invest Dermatol. 135:3105–3114. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Moretti D, Del Bello B, Cosci E, Biagioli
M, Miracco C and Maellaro E: Novel variants of muscle calpain 3
identified in human melanoma cells: Cisplatin-induced changes in
vitro and differential expression in melanocytic lesions.
Carcinogenesis. 30:960–967. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hoek KS, Schlegel NC, Eichhoff OM, Widmer
DS, Praetorius C, Einarsson SO, Valgeirsdottir S, Bergsteinsdottir
K, Schepsky A, Dummer R and Steingrimsson E: Novel MITF targets
identified using a two-step DNA microarray strategy. Pigment Cell
Melanoma Res. 21:665–676. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guindalini C, Lee KS, Andersen ML,
Santos-Silva R, Bittencourt LR and Tufik S: The influence of
obstructive sleep apnea on the expression of glycerol-3-phosphate
dehydrogenase 1 gene. Exp Biol Med (Maywood). 235:52–56. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hubmann G, Guillouet S and Nevoigt E: Gpd1
and Gpd2 fine-tuning for sustainable reduction of glycerol
formation in Saccharomyces cerevisiae. Appl Environ Microbiol.
77:5857–5867. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Finotello F and Di Camillo B: Measuring
differential gene expression with RNA-seq: Challenges and
strategies for data analysis. Brief Funct Genomics. 14:130–142.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hurd PJ and Nelson CJ: Advantages of
next-generation sequencing versus the microarray in epigenetic
research. Brief Funct Genomic Proteomic. 8:174–183. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Git A, Dvinge H, Salmon-Divon M, Osborne
M, Kutter C, Hadfield J, Bertone P and Caldas C: Systematic
comparison of microarray profiling, real-time PCR and
next-generation sequencing technologies for measuring differential
microRNA expression. RNA. 16:991–1006. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kloster MB, Bilgrau AE, Rodrigo-Domingo M,
Bergkvist KS, Schmitz A, Sønderkær M, Bødker JS, Falgreen S,
Nyegaard M, Johnsen HE, et al: A model system for assessing and
comparing the ability of exon microarray and tag sequencing to
detect genes specific for malignant B-cells. BMC Genomics.
13:5962012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mastrokolias A, den Dunnen JT, van Ommen
GB, 't Hoen PA and van Roon-Mom WM: Increased sensitivity of next
generation sequencing-based expression profiling after globin
reduction in human blood RNA. BMC Genomics. 13:282012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gonorazky H, Liang M, Cummings B, Lek M,
Micallef J, Hawkins C, Basran R, Cohn R, Wilson MD, MacArthur D,
Marshall CR, et al: RNAseq analysis for the diagnosis of muscular
dystrophy. Ann Clin Transl Neurol. 3:55–60. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lesluyes T, Pérot G, Largeau MR, Brulard
C, Lagarde P, Dapremont V, Lucchesi C, Neuville A, Terrier P,
Vince-Ranchère D, et al: RNA sequencing validation of the
Complexity INdex in SARComas prognostic signature. Eur J Cancer.
57:104–111. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chandrasekharappa SC, Lach FP, Kimble DC,
Kamat A, Teer JK, Donovan FX, Flynn E, Sen SK, Thongthip S, Sanborn
E, et al: Massively parallel sequencing, aCGH and RNA-Seq
technologies provide a comprehensive molecular diagnosis of Fanconi
anemia. Blood. 121:e138–e148. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
O'Hurley G, Busch C, Fagerberg L,
Hallström BM, Stadler C, Tolf A, Lundberg E, Schwenk JM, Jirström
K, Bjartell A, et al: Analysis of the human prostate-specific
proteome defined by transcriptomics and antibody-based profiling
identifies TMEM79 and ACOXL as two putative, diagnostic markers in
prostate cancer. PLoS One. 10:e01334492015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Endsley MP, Moyle-Heyrman G, Karthikeyan
S, Lantvit DD, Davis DA, Wei JJ and Burdette JE: Spontaneous
transformation of murine oviductal epithelial cells: A model system
to investigate the onset of fallopian-derived tumors. Front Oncol.
5:1542015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Katayama S, Skoog T, Jouhilahti EM,
Siitonen HA, Nuutila K, Tervaniemi MH, Vuola J, Johnsson A,
Lönnerberg P, Linnarsson S, et al: Gene expression analysis of skin
grafts and cultured keratinocytes using synthetic RNA normalization
reveals insights into differentiation and growth control. BMC
Genomics. 16:4762015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang Z, Gerstein M and Snyder M: RNA-Seq:
A revolutionary tool for transcriptomics. Nat Rev Genet. 10:57–63.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Swindell WR, Remmer HA, Sarkar MK, Xing X,
Barnes DH, Wolterink L, Voorhees JJ, Nair RP, Johnston A, Elder JT
and Gudjonsson JE: Proteogenomic analysis of psoriasis reveals
discordant and concordant changes in mRNA and protein abundance.
Genome Med. 7:862015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Stone RC, Du P, Feng D, Dhawan K, Rönnblom
L, Eloranta ML, Donnelly R and Barnes BJ: RNA-Seq for enrichment
and analysis of IRF5 transcript expression in SLE. PLoS One.
8:e544872013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shi L, Zhang Z, Yu AM, Wang W, Wei Z,
Akhter E, Maurer K, Costa Reis P, Song L, Petri M and Sullivan KE:
The SLE transcriptome exhibits evidence of chronic endotoxin
exposure and has widespread dysregulation of non-coding and coding
RNAs. PLoS One. 9:e938462014. View Article : Google Scholar : PubMed/NCBI
|