Introduction
Glucocorticoids (GCs) are frequently used to treat a
wide spectrum of chronic illnesses, including inflammation, cancer,
and autoimmune disorders (1).
However, prolonged and/or overdose administration of GCs has many
side-effects, such as hypertension, hyperglycemia, glaucoma,
osteonecrosis and osteoporosis (2–4). It is
clear that GC use increases bone loss and fracture risk (3–6). GCs can
induce apoptosis of osteoblasts and impair the function of these
cells. For instance, dexamethasone, a GC hormone, can upregulate
the expression of p53 in osteoblastic MC3T3-E1 cells, thus causing
cell apoptosis (7,8). Osteoblast apoptosis leads to decreased
bone formation. Oral use of bisphosphonates (alendronate and
risdronate) (9,10) has been introduced for the treatment
of GC-induced osteoporosis (GIOP). However, low adherence rates
limit the efficiency of oral bisphosphonates. Other medications
also have limitations such as inconvenient administration
[zoledronic acid (11)] and high
cost [PTH 1–34 (teriparatide) (12,13)].
Consequently, there is a need for the development of new treatment
options for GIOP.
Echinacoside (ECH) is a phenylethanoid glycoside
isolated from the traditional Chinese medicine Herba
Cistanches (14). ECH has
protective effects on the neuron system (15–20),
liver (21,22) and lung (23) through promoting cell proliferation,
and inhibiting inflammatory response, reactive oxygen species (ROS)
production and cell apoptosis. Although ECH can also promote
osteoblastic bone regeneration and has an extraordinary
antiosteoporotic activity in rat model (24,25),
there are no existing records describing the effects of ECH on
GC-induced osteoblastic cell apoptosis. Thus, we investigated the
influence that ECH exerts upon dexamethasone-induced osteoblastic
cell apoptosis and elucidated the preliminary mechanism in MC3T3-E1
cells.
Materials and methods
Cultivation of cells
The Cell Bank of the Chinese Academy of Sciences
(Shanghai, China) provided the murine osteoblastic MC3T3-E1 cells.
Growth of cells took place in α-minimal essential medium (α-MEM)
(Hyclone; GE Healthcare, Logan, UT, USA) containing 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc., Grand
Island, NY, USA) and penicillin/streptomycin (Beijing Solarbio
Science and Technology Co., Ltd., Beijing, China). Maintenance of
cells was conducted in an environment with 95% air and 5% carbon
dioxide at a temperature of 37°C.
The production of lentivirus
overexpressing p53
To create p53 expression construct, the protein
coding sequence (CDS) region of murine p53 gene was synthesized and
inserted into EcoRI/BamHI restriction sites of the
lentiviral expression vector pLVX-puro (Clontech Laboratories,
Inc., Palo Alto, CA, USA) by Genewiz, Inc. (Beijing, China). The
construct was verified by DNA sequencing. 293T cells (Shanghai
GeneChem Co., Ltd., Shanghai, China) were transfected with a
mixture of plasmids, including viral packaging plasmids and p53
expression plasmid (pLVX-p53) or control plasmid (pLVX) via
Lipofectamine 2000 (Invitrogen: Thermo Fisher Scientific, Inc.,
Carlsbad, CA, USA) based on the manufacturer's instructions. The
viral supernatant was collected at 48 h after transfection and used
to infect MC3T3-E1 cells. Evaluation of p53 expression was
conducted at 48 h after viral infection.
Quantitative polymerase chain reaction
(qPCR)
We used TRIzol reagent (Invitrogen: Thermo Fisher
Scientific, Inc.) to separate total RNA and MC3T3-E1 cells, and
RevertAid™ First Strand cDNA Synthesis kit (Thermo Fisher
Scientific, Inc., Rockford, IL, USA) according to the
manufacturer's instructions to reverse-transcribe total RNA. p53
mRNA level was detected via qPCR on ABI Prism 7300 Sequence
Detection System (Applied Biosystems: Thermo Fisher Scientific,
Inc., Foster City, CA, USA). The primers were as follows:
5′-CCCCTGTCATCTTTTGTCCCT-3′ and 5′-AGCTGGCAGAATAGCTTATTGAG-3′ for
p53, 5′-CTGCCCAGAACATCATCC-3′ and 5′-CTCAGATGCCTGCTTCAC-3′ for
GAPDH. Then, we assessed p53 mRNA levels via 2−ΔΔCT
method (26) with GAPDH as internal
control.
Western blotting
Lysis of MC3T3-E1 cells took place in radio
immunoprecipitation assay buffer with protease inhibitors (Beijing
Solarbio Science & Technology Co., Ltd.), and centrifugation of
lysates continued for 15 min at a temperature of 4°C at 9,600 × g
so that precipitation can be removed. Subsequently, we assessed the
amount of protein with the BCA assay kit (Thermo Fisher Scientific,
Inc.). Equal amount of protein was added onto a 10 or 15% SDS-PAGE
and transferred to a nitrocellulose membrane (EMD Millipore,
Bredford, MA, USA). The membranes were blocked with 5% skim milk
and probed with primary antibodies. After washing 3 times with
phosphate-buffered saline (PBS), incubation of membranes with
horseradish peroxidase conjugated secondary antibody (1:1,000; cat
no. A0208; Beyotime Institute of Biotechnology, Shanghai, China)
was conducted. We conducted western blotting tests via an enhanced
chemiluminescence (ECL) kit (EMD Millipore). GAPDH was used as the
internal standard. The sources of primary antibodies were as
follows: Bax (Sc-493) and Bcl-2 (Sc-492) both from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA); p53 (no. 2524) and GAPDH
(no. 5174) both from Cell Signaling Technology, Inc. (Danvers, MA,
USA).
Experimental grouping
To determine the suitable dose of dexamethasone,
treatment of MC3T3-E1 cells was conducted with a series amounts of
dexamethasone (0, 1, 10, 100 and 1,000 nM; Sigma-Aldrich; Merck
KGaA, St. Louis, MO, USA). At 48 h after incubating cells, Cell
Counting Kit-8 (CCK-8) assays were conducted to detect cell
proliferation.
To select the appropriate dose of ECH, treatment of
MC3T3-E1 cells was conducted with 1,000 nM of dexamethasone and
various doses of ECH (0, 2.5, 5, 10, 20 and 40 mg/l; Shanghai
Aladdin Biochemical Technology Co., Ltd., Shanghai, China). Cells
grown under normal condition (mock) were served as control. CCK-8
assay was performed after 48 h of culture.
For all other experiments, the MC3T3-E1 cells were
split into five groups: group 1 (mock), cells were cultured under
normal condition; group 2, cells were transduced with pLVX
lentivirus; group 3, cells cultured with 10 mg/l of ECH; group 4,
cells with 20 µM of pifithrin-α (PFT-α; Selleck Chemicals, Houston,
TX, USA); group 5, cells transduced with pLVX-p53 lentivirus and
treated with 10 mg/l of ECH. Cells in groups 2–5 were treated with
1,000 nM dexamethasone. Cell apoptosis and protein expression were
measured after 48 h of culture.
Cell proliferation assay
We seeded MC3T3-E1 cells into 96-well plates at
3×103 cells/well and cultured at normal conditions
overnight. Cells were treated as indicated. After 48 h of culture,
CCK-8 (Signalway Antibody LLC, College Park, MD, USA) assay was
then performed according to the manufacturer's instructions.
Briefly, the culture medium in each well was replaced with 100 µl
of 10% CCK-8 solution in medium. After incubating the cells for a
duration of 1 h at a temperature of 37°C, we assessed the
absorbance at a wavelength of 450 nm with a microplate reader. The
relative cell viability (%) with connections to control wells was
calculated.
Cell apoptosis evaluation
We used Annexin V-FITC cell apoptosis kit (Beyotime
Institute of Biotechnology) for quantifying cells which are
undergoing early apoptosis following the manufacturer's
instructions. In brief, collection of treated cells was conducted
via trypsinization. The cells were washed via PBS, and resuspended
with Annexin binding buffer. Incubation of cells was conducted via
Annexin V-FITC for a duration of 15 min at a temperature of 4°C
followed by incubation with propidium iodide (PI) for 5 min at 4°C.
Cells in the lower right quadrant (Annexin V-FITC positive and PI
negative) were considered actively undergoing early apoptosis.
Statistical analysis
Data are the means ± SD for all three independent
experiments. Tukey's multiple comparisons test and one-way analysis
of variance were conducted to analyze the differences between
groups via GraphPad Prism software (version 6.0; GraphPad Software,
Inc., San Diego, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Establishment of the suitable dose of
dexamethasone
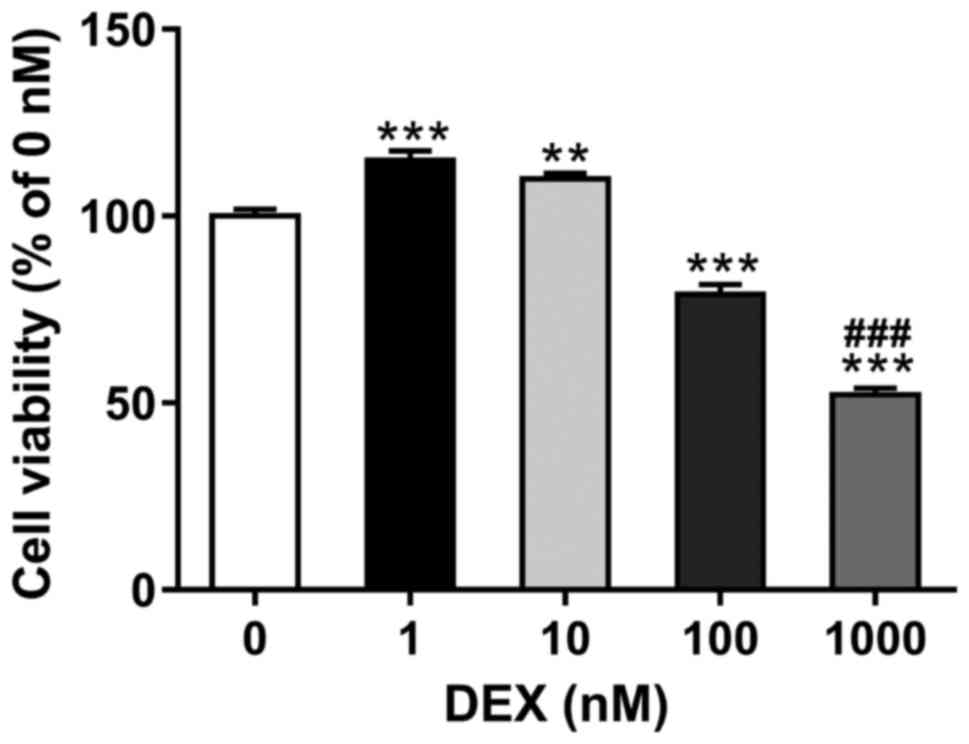
To determine the suitable dose of dexamethasone, the
MC3T3-E1 cells were exposed to increasing amounts of dexamethasone
(0, 1, 10, 100 and 1,000 nM) for 48 h. As shown in Fig. 1, relative cell viability as
determined by CCK-8 assay was increased in cells treated with low
concentrations of dexamethasone (1 and 10 nM), and significantly
reduced in cells exposed to high concentrations of dexamethasone
(100 and 1,000 nM). The most significant reduction was observed in
cells with 1,000 nM of dexamethasone. Thus, 1,000 nM of
dexamethasone was selected in the subsequent assays.
Effect of ECH on dexamethasone-induced
cell growth inhibition
We then explored the effect of ECH on
dexamethasone-induced cell growth inhibition. Treatment of MC3T3-E1
cells was conducted with 1,000 nM of dexamethasone and various
doses of ECH (0, 2.5, 5, 10, 20 and 40 mg/l; Fig. 2). At 48 h post-treatment, the
addition of ECH at doses of 5, 10, 20 and 40 mg/l notably decreased
dexamathasome-induced cell damage. ECH at a dose of 10 mg/l has
more obvious effect than that at a dose of 5 mg/l. Cell viability
was similar in cells treated with 10, 20 and 40 mg/l of ECH.
Therefore, 10 mg/l of ECH was used in the following
experiments.
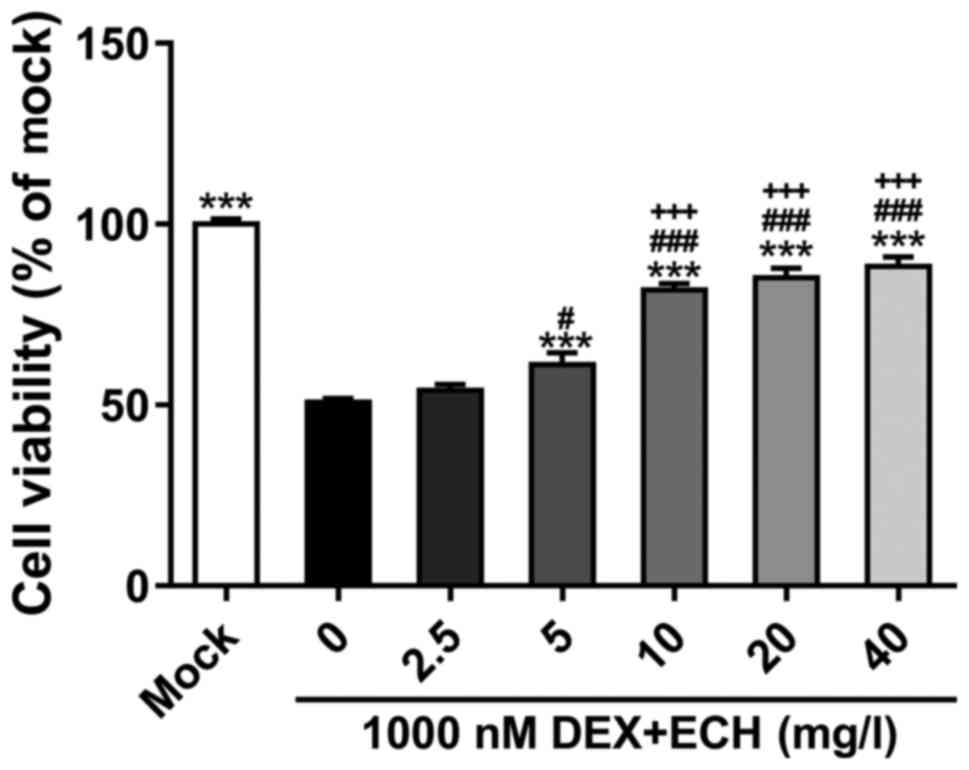 | Figure 2.Effect of ECH on DEX-induced cell
growth inhibition. MC3T3-E1 cell exposure to various amounts of ECH
(0, 2.5, 5, 10, 20 and 40 mg/l) in the presence of 1,000 nM of DEX
for 48 h, and CCK-8 was then performed. Cells grown under normal
condition (mock) served as negative control. ***P<0.001 vs.
1,000 nM DEX + 0 mg/l ECH; #P<0.05 and
###P<0.001 vs. 1,000 nM DEX + 2.5 mg/l ECH;
+++P<0.001 vs. DEX + 1 µM vs. 1,000 nM DEX + 5 mg/l
ECH. Data are the means ± SD for all three independent experiments,
each of which was repeated 3 times. ECH, echinacoside; DEX,
dexamethasone; CCK-8, Cell Counting Kit-8. |
Overexpression of p53 by constructing
lentivirus
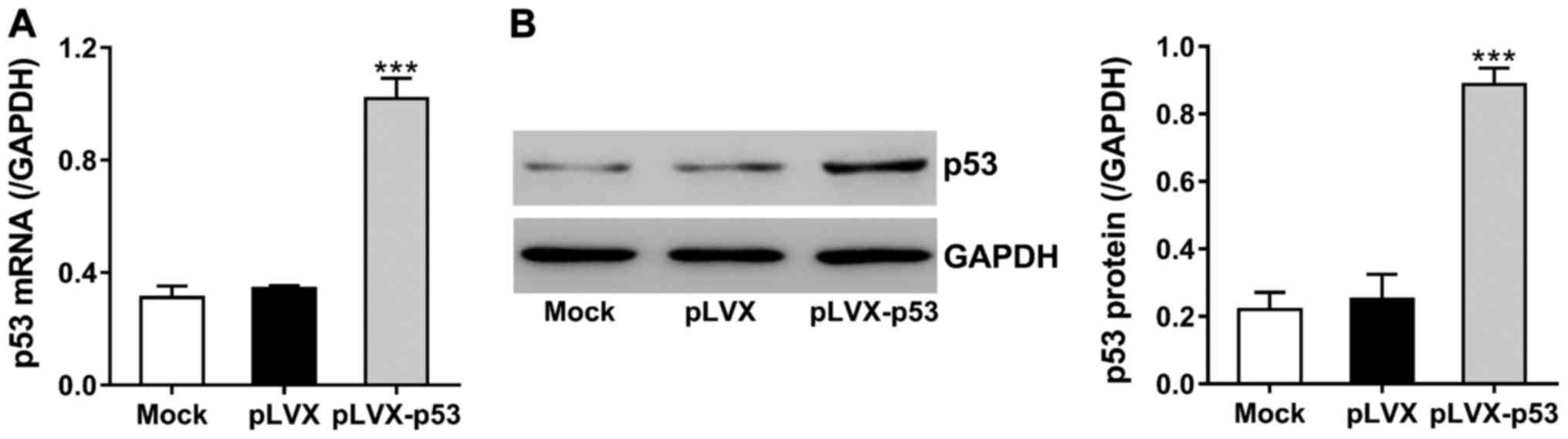
Dexamethasone can upregulate p53, thereby inducing
osteoblasts apoptosis (8). To study
the effects of ECH on dexamethasone-induced p53 expression, we
constructed lentivirus encoding p53 (pLVX-p53) and transduced the
virus into MC3T3-E1 cells. The expression of p53 was analyzed at 48
h after transduction. Fig. 3 shows
that p53 mRNA and protein levels were increased to >2 times in
pLVX-p53-tranduced cells as compared to cells with control virus
(pLVX).
p53 was involved in the effect of ECH
on dexamethasone-induced apoptosis
The MC3T3-E1 cells were treated with pLVX or
pLVX-p53, 10 mg/l of ECH, 20 µM of PFT-α (a p53 inhibitor), and
1,000 nM of dexamethasone, and then cell apoptosis was measured via
Annexin V-FITC apoptosis kit. As shown in Fig. 4A, dexamethasone treatment remarkably
induced cell apoptosis in comparison to the cells cultured under
normal condition (mock). ECH and PFT-α treatment significantly
protected the MC3T3-E1 cells against dexamethasone-induced
apoptosis. Additionally, p53 overexpression reversed the protective
effects of ECH.
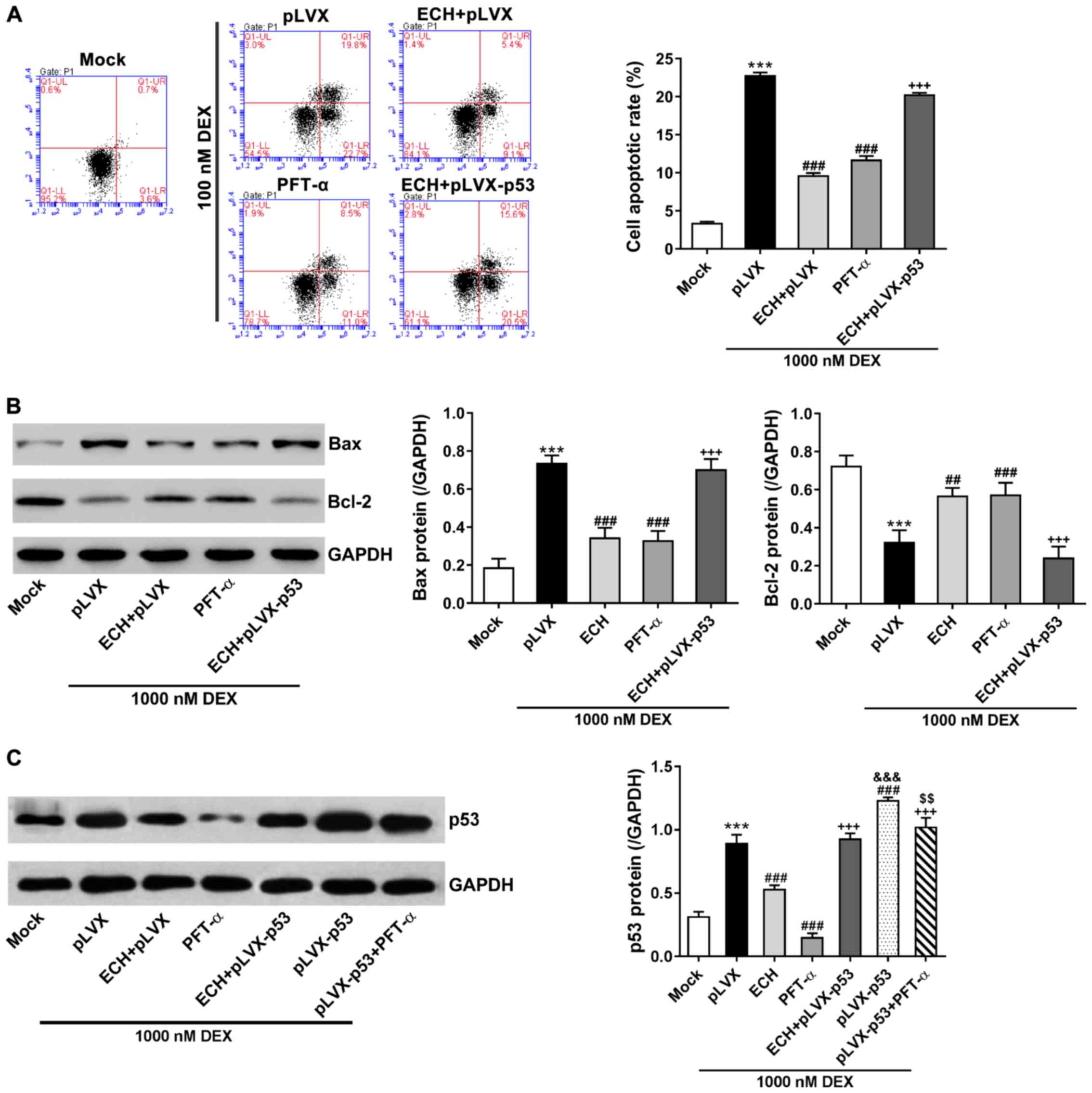 | Figure 4.p53 was involved in the effect of ECH
on DEX-induced apoptosis. The MC3T3-E1 cells were treated with pLVX
or pLVX-p53, 10 mg/l of ECH, 20 µM of PFT-α, and 1,000 nM of DEX as
indicated. Cells without any treatment (mock) served as negative
control. (A) At 48 h after treatment, we assessed cell apoptosis
via Annexin V-FITC apoptosis kit. (B and C) At 48 h after
treatment, we analyzed protein levels of p53, Bax and Bcl-2 via
western blotting with GAPDH as loading control. ***P<0.001 vs.
mock; ##P<0.01 and ###P<0.001 vs. pLVX
+ 1,000 nM DEX; +++P<0.001 vs. ECH;
&&&P<0.001 vs. ECH + pLVX-p53 + 1,000 nM
DEX; &&P<0.01 vs. PFT-α + 1,000 nM DEX. ECH,
echinacoside; DEX, dexamethasone; PFT-α, pifithrin-α. |
Bcl-2 and Bax, as important apoptosis regulators,
are known to be regulated by p53 (27). Western blotting indicated that the
changes of p53 and Bax protein expression were consistent with the
alterations of cell apoptosis, whereas Bcl-2 expression changed in
the opposite direction (Fig. 4B and
C). Notably, the protein levels of p53 were significantly
suppressed by ECH and PFT-α treatment in cells overexpressing p53
and treated with dexamethasone (Fig.
4C). These data suggest that ECH might exert protective effects
on dexamethasone-induced apoptosis via inhibiting p53
expression.
Discussion
The anti-apoptosis function of ECH has been
described in acute liver injury model induced by D-galactosamine
(D-GalN) and lipopolysaccharide (LPS), and in human neuroblastoma
(SH-SY5Y) cells induced by tumor necrosis factor-α or
1-methyl-4-phenylpyridinium ion (MPP+) (17,22,28). It
has been reported that dexamethasone treatment can inhibit the
proliferation and induce MC3T3-E1 cell apoptosis (8,29). In
the current study, relative cell viability was significantly
reduced in cells exposed to high concentrations of dexamethasone
(100 and 1,000 nM), which was consistent with a previous study
(8). Additional ECH treatment (5–40
mg/l) remarkably inhibited dexamethasone-suppressed cell viability.
Moreover, dexamethasone exposure (1,000 nM) stimulated MC3T3-E1
cell apoptosis, which the ECH treatment restrained to a significant
extent. The report gave a first depiction of how ECH is protected
against dexamethasone-induced osteoblast cell apoptosis and
suggested that ECH may have potential for clinical application in
GIOP.
Furthermore, we tried to explore the preliminary
mechanism of the protective effects of ECH. Dexamethasone can
enhance p53 transcription, thereby inducing osteoblasts apoptosis
and cell cycle arrest (8). p53 is
downregulated by ECH in SH-SY5Y cells (28). Here, PFT-α (a p53 inhibitor) had
similar protective effects as ECH, while p53 overexpression
reversed the protective effects of ECH. The changes of p53 protein
expression under different conditions were in agreement with the
alterations of cell apoptosis. These results indicate that ECH
might exert protective effects on dexamethasone-induced
osteoblastic cell apoptosis via inhibiting p53 expression. Bcl-2
family proteins, such as Bcl-2 and Bax, can alter the permeability
of mitochondrial membrane, activate effector caspases and cause
cell apoptosis (30). It is
well-known that p53 can regulate Bcl-2 and Bax (27). Here, treatment with PFT-α or ECH
attenuated the inhibitory effects of dexamethasone on
anti-apoptosis protein Bcl-2 levels as well as the stimulatory
effects dexamethasone has on pro-apoptosis protein Bax levels,
while p53 overexpression reversed the effects of ECH. These results
further demonstrated the involvement of p53 in the protective
function of ECH.
In summary, we demonstrated that ECH has a promising
protective effect on osteoblastic cell apoptosis induced by
dexamethasone, suggesting that ECH may have potential for clinical
application in the treatment of GIOP.
Acknowledgements
Not applicable.
Funding
This study was funded by the Key Discipline
Construction Project of Pudong Health Bureau of Shanghai
(PWZx2014-17), the Shanghai Pudong Commission of Health and Family
Planning (no. PWRq2015-11), the Shanghai Municipal Commission of
Health and Family Planning (201640389), and the Talents Training
Program of Seventh People's Hospital of Shanghai University of TCM
(grant no. QMX2016-05).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SL performed qPCR. SL and HJ were responsible for
western blotting. XG helped with cell proliferation and apoptosis
evaluation. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Buttgereit F, Burmester G-R and Lipworth
BJ: Optimised glucocorticoid therapy: The sharpening of an old
spear. Lancet. 365:801–803. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schäcke H, Döcke W-D and Asadullah K:
Mechanisms involved in the side effects of glucocorticoids.
Pharmacol Ther. 96:23–43. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fukushima W, Fujioka M, Kubo T, Tamakoshi
A, Nagai M and Hirota Y: Nationwide epidemiologic survey of
idiopathic osteonecrosis of the femoral head. Clin Orthop Relat
Res. 468:2715–2724. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Civitelli R and Ziambaras K: Epidemiology
of glucocorticoid-induced osteoporosis. J Endocrinol Invest.
31:(Suppl):. 2–6. 2008.PubMed/NCBI
|
|
5
|
Gudbjornsson B, Juliusson UI and
Gudjonsson FV: Prevalence of long term steroid treatment and the
frequency of decision making to prevent steroid induced
osteoporosis in daily clinical practice. Ann Rheum Dis. 61:32–36.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
van Staa TP, Geusens P, Pols HA, de Laet
C, Leufkens HG and Cooper C: A simple score for estimating the
long-term risk of fracture in patients using oral glucocorticoids.
QJM. 98:191–198. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rickard DJ, Sullivan TA, Shenker BJ, Leboy
PS and Kazhdan I: Induction of rapid osteoblast differentiation in
rat bone marrow stromal cell cultures by dexamethasone and BMP-2.
Dev Biol. 161:218–228. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li H, Qian W, Weng X, Wu Z, Li H, Zhuang
Q, Feng B and Bian Y: Glucocorticoid receptor and sequential P53
activation by dexamethasone mediates apoptosis and cell cycle
arrest of osteoblastic MC3T3-E1 cells. PLoS One. 7:e370302012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stoch SA, Saag KG, Greenwald M, Sebba AI,
Cohen S, Verbruggen N, Giezek H, West J and Schnitzer TJ:
Once-weekly oral alendronate 70 mg in patients with
glucocorticoid-induced bone loss: A 12-month randomized,
placebo-controlled clinical trial. J Rheumatol. 36:1705–1714. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wallach S, Cohen S, Reid DM, Hughes RA,
Hosking DJ, Laan RF, Doherty SM, Maricic M, Rosen C, Brown J, et
al: Effects of risedronate treatment on bone density and vertebral
fracture in patients on corticosteroid therapy. Calcif Tissue Int.
67:277–285. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Reid DM, Devogelaer J-P, Saag K, Roux C,
Lau CS, Reginster JY, Papanastasiou P, Ferreira A, Hartl F, Fashola
T, et al: HORIZON investigators: Zoledronic acid and risedronate in
the prevention and treatment of glucocorticoid-induced osteoporosis
(HORIZON): A multicentre, double-blind, double-dummy, randomised
controlled trial. Lancet. 373:1253–1263. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Langdahl BL, Rajzbaum G, Jakob F, Karras
D, Ljunggren O, Lems WF, Fahrleitner-Pammer A, Walsh JB, Barker C,
Kutahov A, et al: Reduction in fracture rate and back pain and
increased quality of life in postmenopausal women treated with
teriparatide: 18-Month data from the European Forsteo Observational
Study (EFOS). Calcif Tissue Int. 85:484–493. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Saag KG, Zanchetta JR, Devogelaer JP,
Adler RA, Eastell R, See K, Krege JH, Krohn K and Warner MR:
Effects of teriparatide versus alendronate for treating
glucocorticoid-induced osteoporosis: Thirty-six-month results of a
randomized, double-blind, controlled trial. Arthritis Rheum.
60:3346–3355. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jia Y, Guan Q, Guo Y and Du C:
Echinacoside stimulates cell proliferation and prevents cell
apoptosis in intestinal epithelial MODE-K cells by up-regulation of
transforming growth factor-β1 expression. J Pharmacol Sci.
118:99–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kuang R, Sun Y, Yuan W, Lei L and Zheng X:
Protective effects of echinacoside, one of the phenylethanoid
glycosides, on H(2)O(2)-induced cytotoxicity in PC12 cells. Planta
Med. 75:1499–1504. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kuang R, Sun Y and Zheng X: Suppression of
nitric oxide implicated in the protective effect of echinacoside on
H2O2-induced PC12 cell injury. Nat Prod
Commun. 5:571–574. 2010.PubMed/NCBI
|
|
17
|
Deng M, Zhao JY, Tu PF, Jiang Y, Li ZB and
Wang YH: Echinacoside rescues the SHSY5Y neuronal cells from
TNFalpha-induced apoptosis. Eur J Pharmacol. 505:11–18. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Geng X, Tian X, Tu P and Pu X:
Neuroprotective effects of echinacoside in the mouse MPTP model of
Parkinson's disease. Eur J Pharmacol. 564:66–74. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao Q, Gao J, Li W and Cai D:
Neurotrophic and neurorescue effects of echinacoside in the
subacute MPTP mouse model of Parkinson's disease. Brain Res.
1346:224–236. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu M, Lu C and Li W: Transient exposure
to echinacoside is sufficient to activate Trk signaling and protect
neuronal cells from rotenone. J Neurochem. 124:571–580. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu Y, Li L, Wen T and Li YQ: Protective
effects of echinacoside on carbon tetrachloride-induced
hepatotoxicity in rats. Toxicology. 232:50–56. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li X, Gou C, Yang H, Qiu J, Gu T and Wen
T: Echinacoside ameliorates D-galactosamine plus
lipopolysaccharide-induced acute liver injury in mice via
inhibition of apoptosis and inflammation. Scand J Gastroenterol.
49:993–1000. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Y, Xing J, Ai T, Wen T, Guan L and
Zhao J: Protection of echinacoside against acute lung injury caused
by oleic acid in rats. Free Radic Res. 41:798–805. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li F, Yang Y, Zhu P, Chen W, Qi D, Shi X,
Zhang C, Yang Z and Li P: Echinacoside promotes bone regeneration
by increasing OPG/RANKL ratio in MC3T3-E1 cells. Fitoterapia.
83:1443–1450. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li F, Yang X, Yang Y, Guo C, Zhang C, Yang
Z and Li P: Antiosteoporotic activity of echinacoside in
ovariectomized rats. Phytomedicine. 20:549–557. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Miyashita T, Krajewski S, Krajewska M,
Wang HG, Lin HK, Liebermann DA, Hoffman B and Reed JC: Tumor
suppressor p53 is a regulator of bcl-2 and bax gene expression in
vitro and in vivo. Oncogene. 9:1799–1805. 1994.PubMed/NCBI
|
|
28
|
Zhao Q, Yang X, Cai D, Ye L, Hou Y, Zhang
L, Cheng J, Shen Y, Wang K and Bai Y: Echinacoside protects against
MPP+-induced neuronal apoptosis via
ros/atf3/chop pathway regulation. Neurosci Bull.
32:349–362. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chua CC, Chua BH, Chen Z, Landy C and
Hamdy RC: Dexamethasone induces caspase activation in murine
osteoblastic MC3T3-E1 cells. Biochim Biophys Acta. 1642:79–85.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|