Introduction
Charcot-Marie-Tooth (CMT) neuropathies, a group of
peroneal progressive muscular atrophy disorders, comprise a
clinically and genetically heterogeneous group of monogenic
disorders affecting the peripheral nerves (1). More than 80 different genes are
genetically associated with CMT, leading to a prominent upgrade in
diagnostics and understanding of the intricate pathophysiological
mechanisms (2). CMT has been
categorized into two subsets following neuropathological and
electrophysiological criteria: The demyelinating type (CMT1), with
slow nerve conduction velocities (NCVs), and the axonal type
(CMT2), with milder NCVs (3). The
abnormalities in the axons of the peripheral nerves induce
hypotonia, foot deformities, distal weakness, sensory loss, and
optic atrophy (4).
CMT neuropathies frequently occur in the general
population with an estimated prevalence of 1 in 3,000 (2). CMT2 is divided into ≥20 subtypes
according to the disease-causing genes, including CMT2A, CMT2B and
CMT2D (5). Of these, CMT2A is the
most frequent axonal form of CMT allowing for 20–30% of diagnosed
cases. Mutations in kinesin family member 1B (KIF1B;
NM_015074) and mitofusin 2 (MFN2) are associated with
CMT2A. In contrast to the rare KIF1B gene mutation that
causes CMT2A1 (OMIM 118210), different forms of pathogenic
mutations in MFN2 most commonly account for autosomal
dominant CMT2A2A (OMIM 609260) and autosomal recessive CMT2A2B
(OMIM 617087) with differing severity (6). To date, over 170 missense mutations and
20 structural variations has been identified in MFN2, but
the mutational mechanism of variation remains unclear. Defected
mitofusin 2 can cause early onset severe forms or additional optic
atrophy symptoms and accounts for 18% of families with CMT2 in
mainland China (7).
In the present study, the phenotypes of affected
members were analyzed in a four generational pedigree and then
whole-exome sequencing (WES) was performed on the proband to screen
for the causative mutation. The pathogenicity of this mutation was
also verified.
Patients and methods
Patients
A four-generation family from Yancheng (China) with
autosomal dominant CMT was recruited in April 2007 at the
Department of Paediatric Orthopaedics, The Children's Hospital of
Soochow University (Suzhou, China) (Fig.
1). Blood samples were obtained from 7 affected individuals (3
females and 4 males) with CMT and 6 unaffected family members (II1,
II4, II5, II8, II9 and III1) after obtaining written consent. The
clinical features of the proband (III2) were analyzed
retrospectively. A detailed physical examination was performed on
all 7 patients. An electrophysiological examination was available
for 3 patients (III3, III4 and IV1). Written informed consent was
obtained from all participants and approval was obtained from the
Ethics Committee of the Chinese Academy of Medical Sciences
(Beijing, China).
WES
Genomic DNA was extracted from peripheral blood of
an affected family member (III2) using the QIAamp DNA Blood Mini
kit (Qiagen GmbH, Hilden, Germany). The DNA was broken into
fragments ranging from 180–280 bp using an ultrasonoscope (S2;
Covaris, Inc., Woburn, MA, USA) (8).
Adapter oligonucleotides from Illumina (single reads; Illumina
Inc., San Diego, CA, USA) were ligated to the ends and fragments
were amplified with the Paired-End Sequencing Library Prep kit
(Agilent Technologies, Inc., Santa Clara, CA, USA) according to the
manufacturer's protocol. Enrichment of coding exons was performed
with SureSelectXT Human All Exon V4 (Agilent Technologies, Inc.).
After hybridization of the sequencing primer, the bases were
incorporated using the Illumina HiSeq2000 platform (Illumina, Inc.)
for 90 cycles of sequencing per read to generate paired-end reads
including 90 bp at each end and 8 bp of the index tag (9). Image analysis and base calling were
performed using Illumina Pipeline (version 1.3.4; Illumina,
Inc.).
Error assessment and base calling were also
performed using Illumina Pipeline (version 1.3.4) to generate the
primary data. Clean reads of 90 bp length were mapped to the GRCh37
reference human genome from the National Center for Biotechnology
Information database (NCBI; http://www.ncbi.nlm.nih.gov/) using the Burrows
Wheeler Aligner Multi-Vision software package (version, 0.6.2;
Wellcome Sanger Institute, Cambridge, UK) (10). Single nucleotide variations (SNVs)
and indels were identified using SOAPsnp software (version, 2.04;
Beijing Genomics Institute, Shenzhen, China) and GATK Indel
Genotyper (version, 3.4–46; http://www.broadinstitute.org/gsa/wiki/index.php/),
respectively. Previously identified SNVs were filtered through the
NCBI dbVar (https://www.ncbi.nlm.nih.gov/dbvar/browse/) and ExAC
browser (http://exac.broadinstitute.org/). Candidate
disease-associated genes were obtained using Phenolyzer software
(http://phenolyzer.wglab.org/). The OMIM
database (https://omim.org) was searched using
‘hereditary motor and sensory neuropathy’ as key words and the
phenotypic series ‘Charcot-Marie-Tooth disease-PS118220’
(https://omim.org/phenotypicSeries/PS118220). Known
disease-causing mutations were recorded in the Human Gene Mutation
Database (HGMD) at the Institute of Medical Genetics in Cardiff, UK
(http://www.ghmd.cf.ac.uk/).
Candidate mutation confirmed by Sanger
sequencing
The genomic DNA of the family members was isolated
from peripheral blood using a standard SDS-proteinase
K-phenol/chloroform method (11).
The reference sequence of the candidate gene MFN2 was
obtained from the UCSC Genome Browser (http://genome.ucsc.edu), and the candidate variant was
confirmed by PCR-Sanger DNA sequencing. The PCR primers (forward,
5′-TGCTCCTCTGCTTAGTCA-3′; reverse, 5′-GAAACTGGCTGATCAAACGC-3′) were
designed with Primer 3.0 software (http://primer3.ut.ee/). The PCR reaction contained
20–100 ng genomic DNA, 0.5 µl each primer (10 µM), 4 µl dNTP (2.5
mM), 12.5 µl 2X GC buffer I, 2.5 U LA Taq DNA polymerase (Takara
Biotechnology Co., Ltd., Dalian, China) and deionized water to 25
µl. The PCR program was performed using the following conditions:
95°C for 3 min; followed by 38 cycles at 94°C for 30 sec, 58–60°C
for 30 sec; 72°C for 50 sec; and a final extension at 72°C for 8
min. The amplicons were run on an ABI 3730 Sequence Detection
System (Applied Biosystems; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The sequencing data were analyzed using
CodonCode Aligner (version 6.0.2.6; CodonCode Corporation,
Centerville, MA, USA).
Pathogenic validation by restriction
fragment length polymorphism analysis
PCR-restriction fragment length polymorphism
(PCR-RFLP) analysis was used to verify the candidate mutation in
all individuals of the pedigree and 120 healthy Chinese controls
(60 males and 60 females; age, 20–45 years) recruited between April
2016 and January 2018. Controls were from the Chinese Han
population with no family history of muscle diseases. The first PCR
was amplified longer fragments around the mutated site using the
procedure stated above. Nested-PCR was performed using a forward
(5′-TGCTCCTCTGCTTAGTCA-3′) and a mismatched reverse primer
5′-AATTTCAGTCGGTCTTCC-3′) using the thermocycling conditions stated
above. Amplicons from the nested-PCR were digested at 37°C for 15
min by MspI (Takara Biotechnology Co., Ltd.). An 8% neutral
polyacrylamide gel was used to distinguish alleles of the wild type
and mutant type by loss or gain of the restriction site.
Silver-staining (0.1% AgNO3) was used at room
temperature for 10 min prior to the final step of the chromogenic
reaction.
Pathogenic validation by
bioinformatics analysis
The protein sequences of human mitofusin 2
(NP_001121132.1) encoded by MFN2 and of the other homologous
proteins from nine different animals were obtained from the NCBI
Protein database (https://www.ncbi.nlm.nih.gov/protein/) in FASTA
format. Multiple sequence alignment and conservative analyses were
performed using MEGA software (version 7; Institute for Genomics
and Evolutionary Medicine, Temple University, Philadelphia, PA,
USA). The pathogenicity of several missense variants at the same
location including that found in the participant family were
respectively predicted using online tools PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/),
Scale-Invariant Feature Transform (SIFT; http://sift.jcvi.org/), MutationTaster (http://www.mutationtaster.org/) and M-CAP
(http://bejerano.stanford.edu/mcap/).
The three-dimensional structures of normal and mutant mitofusin 2
were generated by homology modeling using SWISS-MODEL (http://swissmodel.expasy.org/) (12). The structure of mitofusin 2 was
examined with the crystal structure of the truncated mitofusin 1
structure (pdb 5GOM). The interactions between the mutant amino
acid (R397P) and the neighboring residues were exhibited and
simulated by PyMOL (Schrödinger, LLC, New York, NY, USA; http://www.pymol.org/). Homology modeling based on the
truncated mitofusin-1 (pdb 5GOM, unit 1A) solved with 2.8 Å
resolution.
Results
Clinical status of the patients
All 7 affected patients expressed manifestations of
bilateral finger contractures and a gait abnormality because of pes
cavus, lower limb weakness, foot drop and deformities. The proband
(III2) initially presented with symptoms at age 4 years (Fig. 2). Patient (II3) had similar pes
cavus, foot muscle atrophy, strephenopodia, and presented with
symptoms at age 7 years. Patient (II6) had more critical symptoms,
such as lower limb atrophy and limited walking ability after age 30
years with upper limb weakness. The present findings indicated that
the clinical manifestations and developmental progression of all
affected family members were similar, but the age of initial onset
was slightly different, with men aged 3–4 and women 7–8 years. In
addition, the male patients exhibited more severe symptoms than the
female patients, and they lost their ability to walk independently
after age 30. The electrophysiological examinations were performed
on 3 patients (III3, III4, and IV1). The results (recorded in the
case history without the electromyogram) exhibited milder NCV
changes. Based on the results of the physical and
electrophysiological examinations, the disease of this family was
diagnosed as CMT2.
WES identified the candidate mutation
in MFN2
The raw data were filtered into clean data and
>96.75% of reads had 99.9% accuracy to identify variations among
the patients. Following mapping to GRCh37, 99.95% of the yielded
reads were correctly matched, and average sequencing depth reached
at least 126-fold. In total, >23,250 exonic and 84,189 intronic
SNVs were identified by SAMtools, and highly frequent single
nucleotide polymorphism (SNPs) were excluded through international
criteria. A similar process was used to screen the 22,013 indels.
The candidate variations were produced through three processes: i)
Removing highly frequent SNPs, ii) reserving the variations from
exons and 10 bp around splicing sites, and iii) eliminating
synonymous mutations; eventually 2,223 variations remained. The
inheritance model was considered for further advanced analysis to
determine the potential disease-causing mutation. In this patient
(III2), potential SNVs and indels were separately identified in 541
and 178 candidate genes according to the obvious inheritance
pattern of autosomal dominance in this four-generation pedigree.
Following several filtering processes, the novel mutation was
ultimately identified as c.1190G>C in exon 10 of MFN2.
This mutation led to substitution of arginine for proline at amino
acid residue 397 (p.R397P).
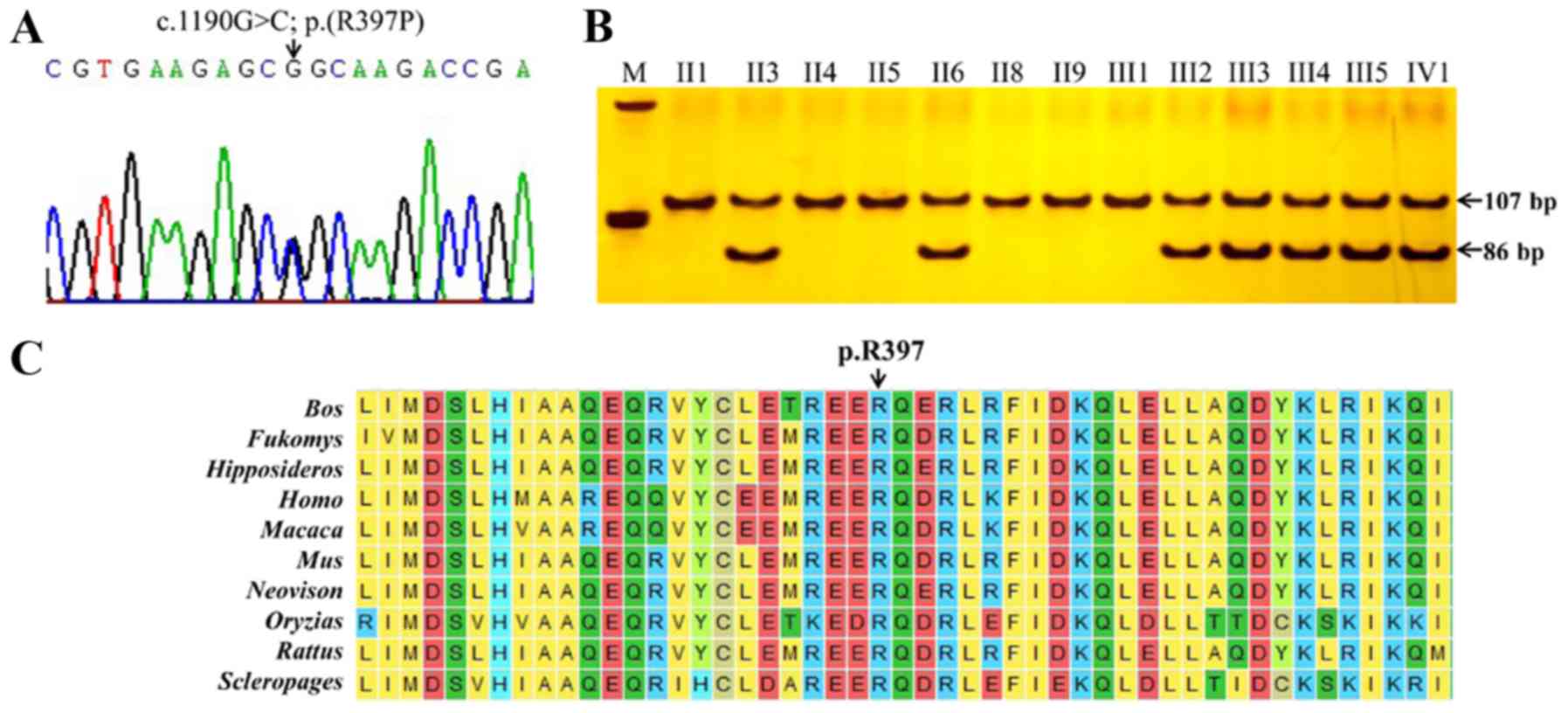
Mutation verification
The candidate mutation in the MFN2 gene was
verified by Sanger sequencing. As a result, the variant from Sanger
sequencing was completely consistent with that from the WES
analysis (Fig. 3A). PCR-RFLP
indicated that the wild-type allele failed to be digested by
MSPI and the mutant allele was successfully separated into
19 and 86 bp fragments. All affected individuals (II3, II6, III2,
III3, III4, III5, and IV1) carrying the heterozygous mutation
(c.1190G>C) presented the genotype of three 105, 86 and 19 bp
fragments; however, all normal individuals (II1, II4, II5, II8,
II9, and III1) and normal controls presented one fragment of 105 bp
(Fig. 3B). The close co-segregation
of the phenotype and genotype suggested that the R397P variant may
be the CMT causative mutation in the present family.
Bioinformatics analysis
A sequence conservation analysis of mitofusin 2
indicated that the amino acid located in position 397 was highly
conserved among 10 species (Fig.
3C). Substitution of the wild-type amino acid (arginine) with
the mutant amino acid (proline) may change its biological
function.
The c.1190G>C; p.(R397P) variation in MFN2
was not present in the HGMD database, indicating that this
variation is a rare variation in MFN2. Furthermore, the SIFT
PROVEAN, polyphen-2, Mutation Taster, and M-CAP programs were
respectively used to predict the pathogenicity of this variation
(Table I). The results from the
bioinformatics analysis suggested that c.1190G>C in MFN2
described a disease-causing mutation.
 | Table I.Prediction of the harmfulness of the
c.1190G>C; p. (R397P) mutation in MFN2. |
Table I.
Prediction of the harmfulness of the
c.1190G>C; p. (R397P) mutation in MFN2.
| Methods | Score | Prediction |
|---|
| SIFT | 0.122 | Tolerated |
| Polyphen-2 | 0.548 | Possibly
damaging |
| MutationTaster | – | Disease causing |
| M-CAP | 0.1 | Possibly
pathogenic |
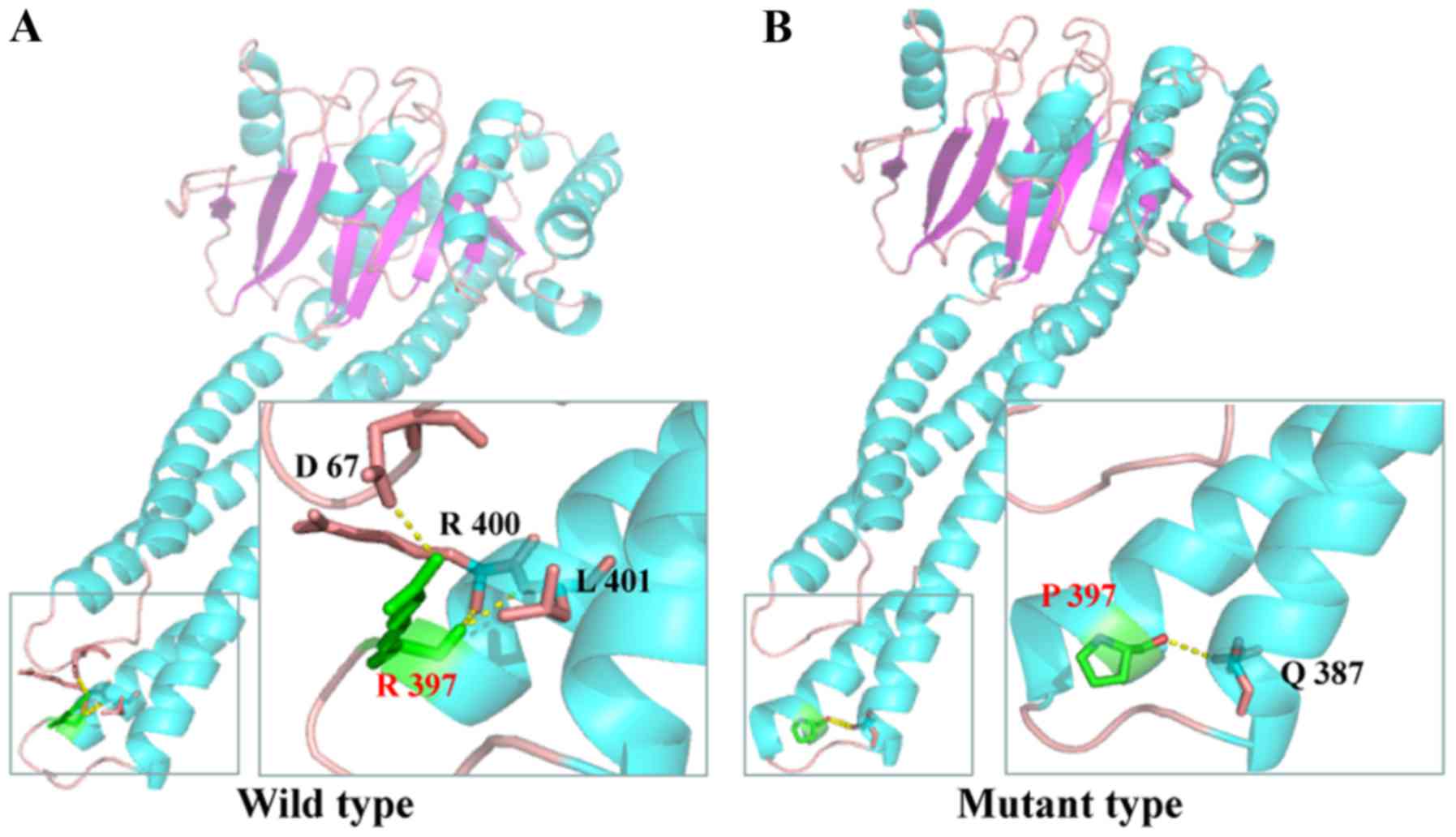
According to the SWISS-MODEL prediction, R397,
located in a critical position of the coiled-coil domain,
contributed to terminate the helix bundle (Fig. 4A). However, the change in R397P led
to extension of the helix bundle. R397 interacted with the R400,
L401 and D67 residues through H-bonds, whereas the more hydrophobic
residue proline resulted in the reformation of H-bonds with
glutamine at position 387 (Fig.
4B).
Discussion
CMT diseases are a group of clinically and
genetically heterogeneous neuropathies with easily confused
phenotypes, including neuropathy-associated features and systemic
impairment of the central nervous system (13). CMT is divided into two major types
based on electrophysiological criteria: Type 1 has slower NCVs
(<38 m/s), and type 2 has normal or slightly reduced NCVs
(14). The present patients
exhibited milder changes in the NCVs. As a result, the family was
tentatively classified as CMT2. As causative genes associated with
CMT are constantly being identified, type 2 has been categorized
into several subtypes, making it challenging to determine the
correct subtype for a patient with CMT (15).
In the present study, the c.1190G>C; p.(R397P)
missense mutation in MFN2 was successfully detected by WES,
which indicates the power of WES to identify causative mutations
for such genetically heterogeneous disorders. Mitofusin 2 encoded
by MFN2 is associated with the mobility of mitochondria in
peripheral nerves (16–18). A mutation in this protein accounts
for ~90% of severe and early onset CMT2 cases (7). The patients of the current study
exhibited onset at a mean age of 5.5 years and expressed the
classical CMT2 phenotype, including pes cavus, foot drop and foot
muscle atrophy. Those clinical manifestations were highly
consistent with the phenotype induced by the MFN2 mutant
(5).
These findings suggest that the c.1190G>C
mutation identified in MFN2 is associated with the genetic
etiology of the disease in this family for the following reasons:
i) This mutation was co-segregated between genotype and phenotype
in the family, and absent from all normal controls observed. The
inheritance model was consistent with the previously reported
autosomal dominant inheritance of CMT2A2A (19). ii) This mutation has no recorded
population frequency described in the ExAC Browser and NCBI dbvar
database (https://www.ncbi.nlm.nih.gov/dbvar/), indicating that
this variation is a rare event in the human genome. iii) The p.R397
amino acid in the mitofusin 2 protein was highly evolutionarily
conserved through sequence alignment among 10 species. iv) The
substituted amino acid changed the hydrophobicity and charge
characteristics of the mitofusin 2 coiled-coiled domain, which may
disrupt normal physiological processes. In summary, the novel
mutation c.1190G>C; p.(R397P) contributed to the CMT2A phenotype
in this four-generation family.
In conclusion, a novel MFN2 mutation was
identified in a Chinese family with CMT2A through WES, which
expanded the mutational spectrum of CMT2A. Early prenatal
intervention is available with an accurate molecular diagnosis of
CMT. There are no known effective disease-modifying treatments for
MFN2-associated disorders, however, gene-based technologies offer
potential to develop treatment, including antisense
oligonucleotides, RNA interference to silence mutant expression and
induced pluripotent stem cells created in vitro to screen
novel drugs (20–22). Further investigation and in
vitro and in vivo studies are required to assess the
impact of MFN2 mutations on neuronal functions and to
further the development of novel therapeutic strategies (23).
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Key
Research and Development Program of China (grant nos.
2016YFE0128400 and 2016YFC0905100) and CAMS Innovation Fund for
Medical Sciences (CIFMS; grant no. 2016-I2M-3-003).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YY conducted experiments, data analysis and wrote
the manuscript. XDW provided pedigree information and performed
physical examinations. SL analyzed data. XLZ and XZ designed and
supervised the study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from all
participants, and approval was obtained from the Ethics Committee
of the Chinese Academy of Medical Sciences (Beijing, China).
Patient consent for publication
Written consent was obtained from the patients.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kazamel M and Boes CJ: Charcot marie tooth
disease (CMT): Historical perspectives and evolution. J Neurol.
262:801–805. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Timmerman V, Strickland AV and Züchner S:
Genetics of charcot-marie-tooth (CMT) disease within the frame of
the human genome project success. Genes (Basel). 5:13–32. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zuchner S, Mersiyanova IV, Muglia M,
Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E,
Patitucci A, Senderek J, et al: Mutations in the mitochondrial
GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A.
Nat Genet. 36:449–451. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tufano M, Cappuccio G, Terrone G,
Manganelli F, Pisciotta C, Geroldi A, Capponi S and Del Giudice E:
Early onset Charcot-Marie-Tooth neuropathy type 2A and severe
developmental delay: Expanding the clinical phenotype of
MFN2-related neuropathy. J Peripher Nerv Syst. 20:415–418. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stuppia G, Rizzo F, Riboldi G, Del Bo R,
Nizzardo M, Simone C, Comi GP, Bresolin N and Corti S: MFN2-related
neuropathies: Clinical features, molecular pathogenesis and
therapeutic perspectives. J Neurol Sci. 356:7–18. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Siskind CE, Panchal S, Smith CO, Feely SM,
Dalton JC, Schindler AB and Krajewski KM: A review of genetic
counseling for Charcot Marie Tooth disease (CMT). J Genet Couns.
22:422–436. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xie Y, Li X, Liu L, Hu Z, Huang S, Zhan Y,
Zi X, Xia K, Tang B and Zhang R: MFN2-related genetic and clinical
features in a cohort of Chinese CMT2 patients. J Peripher Nerv
Syst. 21:38–44. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wei X, Ju X, Yi X, Zhu Q, Qu N, Liu T,
Chen Y, Jiang H, Yang G, Zhen R, et al: Identification of sequence
variants in genetic disease-causing genes using targeted
next-generation sequencing. PLoS One. 6:e295002011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang B, Zheng Z, Wang Z, Zhang X, Yang H,
Cai H and Fu Q: A novel missense mutation of TNNI2 in a Chinese
family cause distal arthrogryposis type 1. Am J Med Genet A.
170A:135–141. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jiang Y, Pan J, Guo D, Zhang W, Xie J,
Fang Z, Guo C, Fang Q, Jiang W and Guo Y: Two novel mutations in
the PPIB gene cause a rare pedigree of osteogenesis imperfecta type
IX. Clin Chim Acta. 469:111–118. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jiang M, Zhao X, Han W, Bian C, Li X, Wang
G, Ao Y, Li Y, Yi D, Zhe Y, et al: A novel deletion in TNNI2 causes
distal arthrogryposis in a large Chinese family with marked
variability of expression. Hum Genet. 120:238–242. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cao YL, Meng S, Chen Y, Feng JX, Gu DD, Yu
B, Li YJ, Yang JY, Liao S, Chan DC and Gao S: MFN1 structures
reveal nucleotide-triggered dimerization critical for mitochondrial
fusion. Nature. 542:372–376. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Harel T and Lupski JR: Charcot-Marie-Tooth
disease and pathways to molecular based therapies. Clin Genet.
86:422–431. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zimoń M, Battaloğlu E, Parman Y, Erdem S,
Baets J, De Vriendt E, Atkinson D, Almeida-Souza L, Deconinck T,
Ozes B, et al: Unraveling the genetic landscape of autosomal
recessive Charcot-Marie-Tooth neuropathies using a homozygosity
mapping approach. Neurogenetics. 16:33–42. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kostera-Pruszczyk A, Kosinska J, Pollak A,
Stawinski P, Walczak A, Wasilewska K, Potulska-Chromik A, Szczudlik
P, Kaminska A and Ploski R: Exome sequencing reveals mutations in
MFN2 and GDAP1 in severe Charcot-Marie-Tooth disease. J Peripher
Nerv Syst. 19:242–245. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guo X, Chen KH, Guo Y, Liao H, Tang J and
Xiao RP: Mitofusin 2 triggers vascular smooth muscle cell apoptosis
via mitochondrial death pathway. Circ Res. 101:1113–1122. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Santel A and Fuller MT: Control of
mitochondrial morphology by a human mitofusin. J Cell Sci.
114:867–874. 2001.PubMed/NCBI
|
|
18
|
Schon K, Spasic-Boskovic O, Brugger K,
Graves TD, Abbs S, Park SM, Ambegaonkar G and Armstrong R:
Mosaicism for a pathogenic MFN2 mutation causes minimal clinical
features of CMT2A in the parent of a severely affected child.
Neurogenetics. 18:49–55. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Helbig I, Hodge SE and Ottman R: Familial
cosegregation of rare genetic variants with disease in complex
disorders. Eur J Hum Genet. 21:444–450. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Deng Y, Wang CC, Choy KW, Du Q, Chen J,
Wang Q, Li L, Chung TK and Tang T: Therapeutic potentials of gene
silencing by RNA interference: Principles, challenges, and new
strategies. Gene. 538:217–227. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kanasty R, Dorkin JR, Vegas A and Anderson
D: Delivery materials for siRNA therapeutics. Nat Mater.
12:967–977. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nizzardo M, Simone C, Falcone M, Locatelli
F, Riboldi G, Comi GP and Corti S: Human motor neuron generation
from embryonic stem cells and induced pluripotent stem cells. Cell
Mol Life Sci. 67:3837–3847. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ekins S, Litterman NK, Arnold RJ, Burgess
RW, Freundlich JS, Gray SJ, Higgins JJ, Langley B, Willis DE,
Notterpek L, et al: A brief review of recent Charcot-Marie-Tooth
research and priorities. F1000 Res. 4:532015.
|