Introduction
Bullous pemphigoid (BP) is the most common
autoimmune bullous disease affecting the skin and mucosal
membranes, with antibodies directed against the 180-kDa BP antigen
(BP180) and the 230-kDa BP antigen (BP230) located in the basement
membrane zone (1). BP commonly
affects older patients and females are slightly more affected than
males (2).
Up to 20% of the patients with classical BP may have
at the onset of the disease a non-bullous phase of variable
duration with non-specific itchy lesions (eczematous patches,
urticarial plaques, polycyclic, targetoid, nodular, lichenoid or
vesicular lesions) (3).
Pemphigoid nodularis (PN) is a rare variant of BP
having clinical features of prurigo nodularis with an autoantibody
profile of BP (4). Hyperkeratotic,
excoriated and pruritic nodules on the extremities can be the first
sign of PN anticipating the blisters by weeks or months. In some
cases the blisters cannot be observed through-out the whole course
of the disease making diagnosis substantially difficult (3). The pathogenesis of PN is not well
understood. Skin physical trauma like persistent scratching exposes
hidden antigens of the basal membrane to the immune system, leading
to the production of autoantibodies in predisposed individuals
(5).
The aim of the study is to present a case of PN and
a systematic review of the published PN cases regarding clinical
aspects, histopathological features, laboratory findings and
treatment options.
Case report
Caucasian, 81-year female, with a history of
ischemic heart disease, chronic obstructive pulmonary disease and
bilateral gonarthrosis was admitted in to the hospital in July 2016
for a nodular-excoriated eruption on her trunk and extremities with
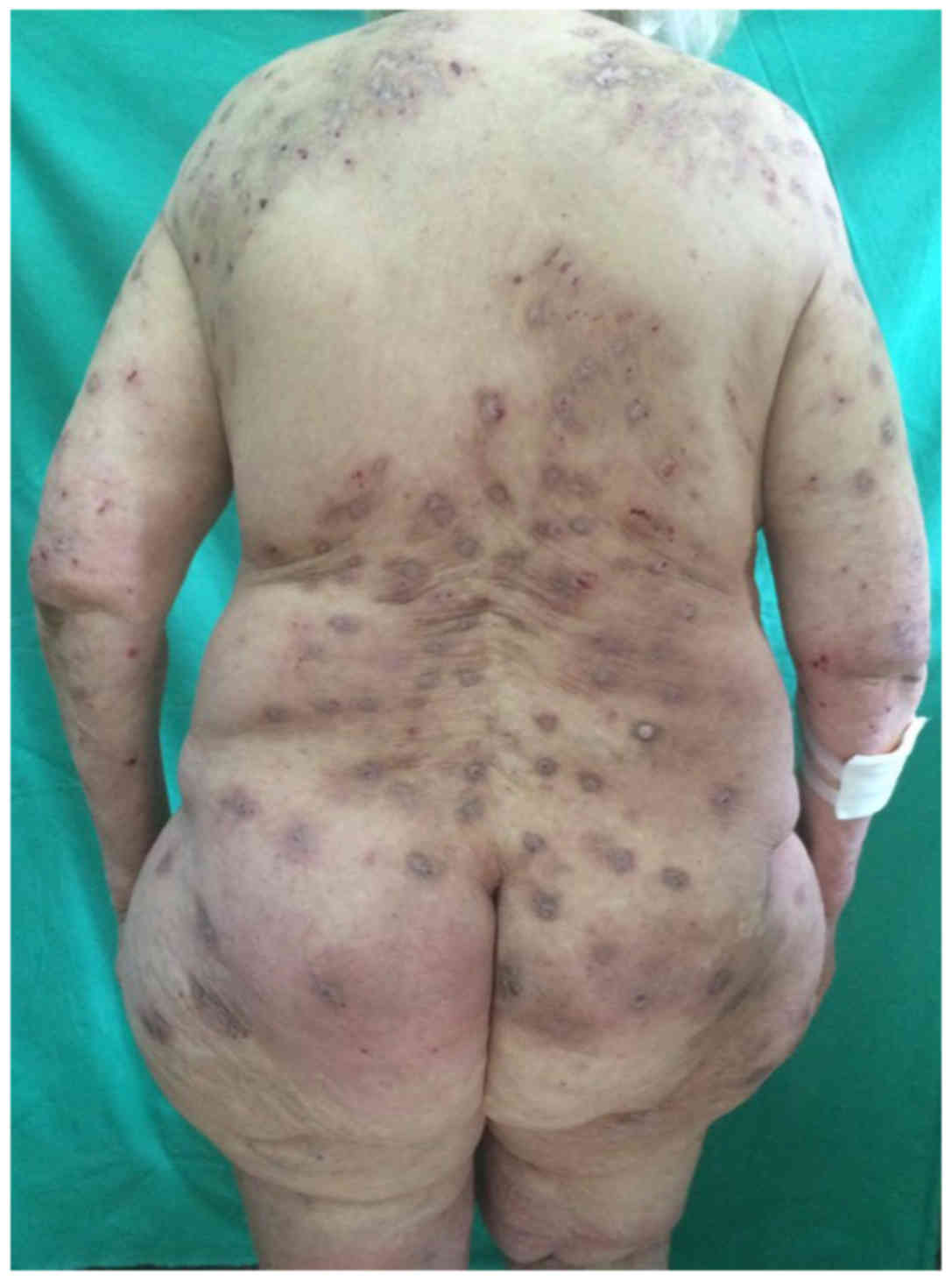
abrupt onset, ~2 months before admission. Clinical examination
revealed violaceous, highly pruritic nodular lesions, some with
excoriations, other with a pale and atrophic center (Fig. 1). Most lesions presented a
hyperpigmented, bluish halo. The lesions were located on the trunk
and limbs, with sparing of the face and interscapular region. All
the skin was dry and scaly. The emotional and psychosocial
background was stable. Complete blood count, liver and kidney
function were normal. Thyroid function was normal, but levels of
thyroid peroxidase and antithyroglobulin antibodies were increased.
IgE level in the blood was elevated (2002.9 UI/ml; normal <100.0
UI/ml). The first skin biopsy showed marked hyperkeratosis, focal
parakeratosis, and mild spongiosis; the dermis had marked mixed
inflammatory infiltrate (lymphocytes, plasmacytes, neutrophils,
eosinophils) with a diffuse and perivascular pattern. PAS stain
showed no mycotic colonies (Fig. 2).
Direct immunofluorescence (DIF) performed from the margin of the
same lesion was negative; ELISA for BP180 and BP230 were also
negative. Screening for an underlying malignancy was negative.
At this point, she was diagnosed with prurigo
nodularis, atopy, autoimmune thyroiditis and xerosis cutis. In
order to interrupt the cycle of itching-scratching a treatment with
topical corticosteroids and calcipotriol for 6 weeks was started,
with no improvement.
After 2 months urticarial plaques appeared with no
evidence of blister formation (Fig.
3). A new skin biopsy was performed from these new lesions
showing hyperkeratosis, focal parakeratosis, spongiosis,
subepidermal bulla and a lymphoplasmacytic and neutrophilic
infiltrate with diffuse and perivascular pattern (Fig. 4).
DIF revealed linear C3 deposits along the basal
membrane (Fig. 5). ELISA from serum
was positive for BP230 antibody (79.0 U/ml, normal <20.0 U/ml)
and was negative for BP180. The clinical and histologic aspect,
together with C3 linear distribution at the basal membrane and
positivity of BP230 antibodies were consistent with the rare
diagnosis of pemphigoid nodularis. She started a treatment with
prednisone (0.5 mg/kg/day) for 2 weeks which was gradually tapered
by 5 mg/week in the following 6 weeks. Topical therapy consisted in
one application of clobetasol propionate 0.05% in the evening. The
pruritus, nodules and urticaria-like eruption remitted under
corticotherapy. In order to minimize the risk of adverse reactions
and thus to discontinue the treatment with systemic
corticosteroids, we introduced fexofenadine hydrochloride (240
mg/day), montelukast sodium (10 mg/day) and continued topical
applications of clobetasol propionate. After 3 months, the nodular
lesions were improved, without any sign of recurrence.
Unfortunately, 2 weeks after the last visit the
patient died due to a hemorrhagic stroke, which, in our opinion,
was unrelated to the autoimmune bullous disease.
Materials and methods
We conducted a literature search in English and
French of the MEDLINE database on 18th May 2018 using the following
terms: ‘nodular pemphigoid’, ‘pemphigoid nodularis’, ‘pemphigoid’
and ‘nodularis’, without limitations on article type and restricted
the papers to those published in English or French. We collected
data on: age at diagnosis, sex, origin, associated diseases,
duration of symptoms before diagnosis, clinical presentation,
results of diagnostic tests, treatment options and outcome.
The study was approved by the Ethics Committee of
the Emergency County Hospital (Cluj-Napoca, Romania), and written
informed consent was provided by the patient for this study.
We defined PN patients as patients who presented
with a pruritic papulo-nodular eruption followed by a positive
analysis of either DIF, indirect immunofluorescence (IIF) or
immunoserology. Statistical analysis was performed using SPSS 20.0
(SPSS Inc., Chicago, IL, USA) software. Data were presented as mean
± SD.
A total of 51 articles were found. We excluded from
our review: three articles because of language limitations, 6
articles due to incomplete data and 6 articles were not relevant to
the question.
A total of 36 articles included in the study
presented 47 cases of PN between 1981 and 2018 (Table I). All the characteristics of the
cases reported are summarized in Tables
II–V.
 | Table I.All included articles and cases
reported. |
Table I.
All included articles and cases
reported.
| Author (refs.) | No. of cases
reported | Publication
year |
|---|
| 36 articles | 47 cases | 1981–2018 |
| Yoneda et al
(22) | 1 | 2018 |
| Zhang et al
(23) | 1 | 2017 |
| Amber et al
(4) | 1 | 2017 |
| Yoshimoto et
al (24) | 1 | 2017 |
| Dangel et al
(25) | 1 | 2016 |
| Asahina et
al (15) | 1 | 2015 |
| Kwong and Lim
(26) | 1 | 2015 |
| Al-Salhi and
Alharithy (27) | 1 | 2015 |
| Das and
Bandyopadhyay (28) | 1 | 2014 |
| Mochizuki et
al (9) | 1 | 2013 |
| Matsudate et
al (29) | 2 | 2009 |
| Koga et al
(30) | 1 | 2009 |
| Aboumaria et
al (31) | 1 | 2008 |
| Teraki and Fukuda
(32) | 1 | 2008 |
| McGinnes et
al (16) | 1 | 2006 |
| Gach et al
(33) | 2 | 2005 |
| Tashiro et
al (34) | 2 | 2005 |
| Sakuma-Oyama et
al (35) | 1 | 2003 |
| Powell et al
(5) | 5 | 2002 |
| Schachter et
al (6) | 1 | 2001 |
| Gao et al
(36) | 1 | 2001 |
| Ameen et al
(37) | 1 | 2000 |
| Cliff and Golden
(38) | 3 | 1997 |
| Kossard (39) | 1 | 1996 |
| Tamada et al
(40) | 1 | 1995 |
| Bourke et al
(41) | 1 | 1994 |
| Ratnavel et
al (42) | 1 | 1994 |
| Gallo et al
(43) | 1 | 1993 |
| Borradori et
al (44) | 1 | 1992 |
| Ross et al
(45) | 3 | 1992 |
| Borradori et
al (46) | 1 | 1990 |
| Tani et al
(47) | 1 | 1989 |
| Shimizu et
al (48) | 1 | 1988 |
| Roenigk and Dahl
(49) | 1 | 1986 |
| Massa and Connolly
(50) | 1 | 1982 |
| Yung et al
(51) | 1 | 1981 |
| Table II.Demographic and personal history
characteristics. |
Table II.
Demographic and personal history
characteristics.
| Data | % (no. of pts) |
|---|
| Sex |
|
|
Male | 36.2 (17) |
|
Female | 63.8 |
|
Female:Male ratio | 1.8:1 (30) |
| Age (years; mean ±
SD) |
|
| 11–15
(3 pts) | 13.3±2.1; 14
years |
| 41–91
(44 pts) | 66.2±12.3; 70
years |
| Origin |
|
|
Caucasian | 25.5 (12) |
|
Non-Caucasian | 49.0 (23) |
| Not
mentioned | 25.5 (12) |
| Associated
diseases |
|
|
Autoimmune | 34.0 (16) |
|
Allergic | 4.2 (2) |
|
Inflammatory | 2.1 (1) |
|
Diabetes mellitus | 6.4 (3) |
| Table V.Treatment of reported cases. |
Table V.
Treatment of reported cases.
|
|
|
|
| Clinical outcome
(no. of pts) |
|---|
|
|
|
|
|
|
|---|
| Therapy | Max dose | Min dose | Duration | Good | Partial | No response |
|---|
| Topical
corticosteroids | – | – | 18 weeks | 4 | 1 | 4 |
| Prednisone | 1 mg/kg/day | 0.5 mg/kg/day | 6–10 months | 9 | 1 | 0 |
| Prednisolone | 1 mg/kg/day | 0.1 mg/kg/day | 3 months | 7 | 9 | 1 |
| Betamethasone | 20 mg/day | 0.5 mg/day | NM | 2 | 0 | 0 |
|
Methylprednisolone | 0.4 mg/kg | 0.2 mg/kg | >6 months | 1 | 0 | 0 |
| Azathioprine | 50 mg tid | 50 mg/day | 12 months | 7 | 1 | 0 |
| Dapsone | 100 mg/day | 50 mg/day | NM | 3 | 1 | 0 |
| IVIg | 2 g/kg/day, 5
days | – | 10 months | 2 | 1 | 0 |
| Minocycline | 100 mg bid | 100 mg/day | NM | 2 | 1 | 2 |
| Mycophenolate
mofetil | 2 g/day | 1.5 g/day | NM | 1 | 1 | 0 |
|
Sulfamethoxyi-pyridazine | 500 mg tid | 500 mg/day | 2–4 years | 1 | 1 | 0 |
| Fexofenadine +
Montelukast | 240 mg bid +10
mg | – | 4 weeks | 1 | 1 | 1 |
| Niacinamide | 500 mg tid | – | 6 months | 1 | 0 | 2 |
| Suplataste
tosilate | 300 mg/day | – | NM | 1 | 0 | 0 |
| Rituximab | 375 mg/mp | – | NM | 1 | 0 | 0 |
| Triamcinolone
i.m. | 60 mg | – | NM | 0 | 1 | 0 |
| Cyclosporine | NM | NM | NM | 0 | 1 | 0 |
| Methotrexate | 15 mg/week | NM | 3 months | 0 | 0 | 1 |
|
Cyclophosphamide | NM | NM | NM | 0 | 0 | 1 |
| Oral
Antihistamines | NM | NM | NM | 0 | 0 | 6 |
The female to male ratio was 1.8:1. The mean age of
onset was 66.2 years, SD ± 12.3, median age 70 (for patients aged
41–91 years) and 13.3 years, SD ± 2.1, median age 14 (for patients
aged 11–15 years). Most of the patients were non-Caucasian [49.0%,
23 patients (pts)], 12 pts were Caucasian (25.5%) while for 12 pts
the origin was not mentioned. As comorbidities, we found autoimmune
diseases (rheumatoid arthritis, ulcerative colitis, autoimmune
thyroiditis, IPEX, bullous pemphigoid, dermatitis herpetiformis,
idiopathic chronic eosinophilic pneumonia) in 16 pts (34.0%),
inflammatory diseases (psoriasis) in 1 pts (2.1%), allergic
diseases (asthma and atopic diathesis) in 2 pts (4.2%) and diabetes
mellitus in 3 pts (6.4%) (Table
II).
For all patients the primary lesions were considered
as pruritic, hyperkeratotic or excoriated papules and/or nodules,
on the extremities and trunk, and 46.8% (22 pts) developed
secondary lesions (blisters, vesicles, urticarial plaques) before
or after the diagnosis of PN has been established. One case had
oral mucosa involvement. Data concerning the duration of the
disease (after onset of initial lesions) was found in 35 out of 47
pts (for 7 pts data was not recorded, and 5 pts had prior
blistering autoimmune disease). Mean duration was 23.6 months, SD ±
31.9, with the median value of 9 months. Histopathological
examination from a hyperkeratotic nodule was performed in 37 pts
(78.7%); spongiosis and/or subepidermal cleft was noted in 13 pts
(35.1%), which are not suggestive for prurigo nodularis, and dermal
eosinophils in 23 pts (62.2%), while one patient presented
non-specific features. High serum total IgE levels were found in 13
pts (27.7%) and eosinophilia in 5 pts (10.6%) (Table III).
| Table III.Clinical, histopathological and blood
tests findings. |
Table III.
Clinical, histopathological and blood
tests findings.
| Data | % (no. of pts) |
|---|
| Clinical
aspect |
|
| Primary
lesionsa and/or | 100 (47) |
|
secondary lesionsb | 46.8 (22) |
| Duration of
symptoms before diagnosis (months) |
|
| Mean ±
SD, 23.6±31.9 (range 1–132) | 74.5 (35) |
| Median
9 |
|
| Histopathology,
from a nodular lesion | 78.7% (37) |
|
Spongiosis and/or cleft | 35.1 (13) |
|
Eosinophilic infiltrate | 62.2 (23) |
| Blood tests |
|
|
Eosinophilia | 10.6 (5) |
| High
value of total IgE | 27.7 (13) |
Diagnosis was confirmed by immunofluorescence
analysis (DIF: linear deposits of IgG and/or C3 along the basement
membrane zone, IIF on monkey esophagus or 1M salt-split normal
human skin: linear deposit of IgG with epidermal binding >1:80)
and circulating autoantibody tests. Of 47 cases, DIF from pruritic
papule and/or nodule was carried out in 30 pts and was positive in
26 pts (86.7%). All 21 pts that developed secondary lesions
presented positive perilesional DIF (100%).
IIF was performed in 39 pts and was positive in 36
(92.3%). Immunoblot (western blotting) analysis was performed for
19 pts; it was positive in all cases: 4 pts for 180 kDa (21.1%), 11
pts (57.9%) for 230 kDa (220 kDa) and 4 pts (21.1%) for both.
Nineteen patients were analyzed by ELISA and positive tests were
found in all patients; for BP180 in 12 pts (63.1%), for BP230 in 1
pts (5.3%) and for both (BP180+BP230) in 6 pts (31.6%) (Table IV).
| Table IV.Characteristics of immunofluorescence
microscopy and immunoserologic tests. |
Table IV.
Characteristics of immunofluorescence
microscopy and immunoserologic tests.
| Immunologic
tests | Positive (%) | Negative (%) | No. of pts |
|---|
| DIF, pruritic
papule and/or nodule | 26/30 (86.7) | 4/30 (13.3) | 46 |
| DIF, once the
secondary lesions appeareda | 21/21 (100) | 0 |
|
| IIF (monkey
esophagus/salt-split skin) | 36/39 (92.3) | 3/39 (7.9) | 39 |
| Immunoblot 180
kDa | 4/19 (21.1) | – | 19 |
| Immunoblot 230
kDa | 11/19 (57.9) | – |
|
| Immunoblot 180 kDa
and 230 kDa | 4/19 (21.1) | – |
|
| ELISA BP180 | 12/19 (63.1) | – | 19 |
| ELISA BP230 | 1/19 (5.3) | – |
|
| ELISA BP180 and
BP230 | 6/19 (31.6) | – |
|
Post-therapeutic clinical outcome of reported cases
was evaluated as ‘good’ (significant improvement of pruritus,
gradual resolution of skin lesions), ‘partial’ (persistence of some
skin lesions) and ‘no response’. We summarized available data as
the name of drugs, dosage (minimal and maximal), and duration of
administration in Table V. We found
that the best therapeutic options were systemic and topical
corticosteroids.
Discussion
Our systematic review summarized the reported
characteristics of PN, a rare variant of BP having clinical
features of prurigo nodularis with an autoantibody profile of BP.
The features of the present case report are similar to the ones
reported in the literature, highlighting the elderly patient group,
presenting initial papulo-nodular pruritic lesions and secondary
vesicular/ bullous lesions or urticarial plaques. This atypical
onset of PN partially explains the diagnosis delay, especially in
the absence of obvious clinical signs of BP. We consider that
clinicians should take into consideration DIF microscopy in elderly
patients with pruritic papulo-nodular eruption.
As for the pediatric patients, autoimmune blistering
diseases are rare and their prevalence unclear. Interestingly, in
the few cases of PN reported in children, most of them had previous
long-standing BP, suggesting a hyperproliferative integrin profile
(6).
Mucosal involvement was reported in 10–30% of
patients with BP (7). Our review
shows a lower incidence of 2.1%, suggesting that oral mucosa
involvement is less frequent in PN.
Drug-induced disease due to various medication
(antibiotics, NSAID, diuretics, anti-TNF-α, antidiabetics,
antiarrhythmics and antihypertensives) was reported in some cases
of BP (8). We found only one patient
who was treated for rheumatoid arthritis with etanercept
(anti-TNF-α drug) before the onset of PN, which imposed a
discontinuation of the drug with favorable effect (9).
Histopathologic findings in PN show features of
prurigo nodularis (orthohyperkeratosis, focal parakeratosis and
irregular epidermal hyperplasia) (10) and bullous pemphigoid (eosinophilic
spongiosis more than 50% and a subepidermal cleft 80%) (11). Skin nodule biopsies revealed
spongiosis and/or subepidermal split in some cases but, more
frequently, dermal eosinophilic infiltrate, indicating the
necessity of performing DIF microscopy and immunoserology assays in
addition to the histopathological examination.
DIF and IIF on salt-split skin microscopy have high
specificity, 98 and 100%, respectively (12) in BP and they are the most reported
positive diagnostic tests in nonbullous pemphigoid, 93.2 and 90.2%,
respectively (1). In the PN cases
that were reported, DIF microscopy performed from nodular lesions
was positive in most cases, while it was positive in all patients
when secondary lesions were clinically obvious. IIF was positive
similar to the previously reported data. As a result, a negative
DIF does not exclude PN.
The diagnosis must be completed with immunoserology
testing as ELISA or Immunoblot (western blotting). Since 2002 most
reported cases of PN were confirmed by ELISA and we found that all
were positive, either for BP180, BP230 or both. Moreover, ELISA
testing can be useful in follow-up, the levels of BP antibodies
decreasing as the clinical aspect improves under treatment.
A recent study indicated that BP patients can have
pathologic peripheral eosinophilia (50.2%) (13). This finding was significantly
correlated with the age of patients (older patients) and the
severity of the palmoplantar involvement. Moreover, peripheral
eosinophil count is significantly correlated with levels of both
anti-BP180 IgG and IgE (14). The
association between serum eosinophilia and BP or PN is still
unclear as Kridin found no correlation between atypical clinical
variants of BP (prurigo-like type) and serum eosinophilia (13), while our data revealed serum
eosinophilia in some patients, who also had high levels of serum
total IgE and anti-BP180 IgG antibody. Eosinophils play an
important role in the pathogenetic cascade of BP and PN as well as
in other autoimmune diseases (15,16).
Elevated IgE levels were reported in 70% of patients
with BP in 1974 (17). It was also
shown that IgE abnormalities and impaired B lymphocyte function is
correlated with serum levels of soluble CD23 (18). Although in the review of Saniklidou
et al on bullous pemphigoid no correlations were found
between IgE levels and the nodular form of the disease (19), our data showed high levels serum
total IgE in 13 patients (27.7%). Ten out of the later thirteen
patients had positive DIF performed from nodular lesions, positive
IIF and tissue eosinophilia.
Although literature data did not find an increased
risk for autoimmune disorders in patients with BP (20), we found one third of patients having
this association. These findings could be explained by a genetic
susceptibility to develop autoimmune diseases.
Treatment in PN remains difficult and challenging as
the condition is chronic, severely pruritic, occurring in elderly
patients with multiple comorbidities. Due to adverse effects of
immunosuppressive medication there is a need to find therapeutic
alternatives. Reported data regarding treatment are incomplete,
lacking details such as dosage, duration of administration.
The best therapeutic results were obtained using
systemic corticosteroids, azathioprine, dapsone, intravenous Ig,
minocycline. However, topical and systemic corticosteroids continue
to remain the gold standard in PN. Our review shows that the
therapeutic response is directly correlated with the dose and the
treatment duration.
In our case, after a significant improvement of the
pruritus and the aspect of the nodules under corticosteroid
treatment, we chose to continue due to comorbidities, with a
combination of montelukast sodium (leukotriene receptor inhibitor)
and fexofenadine hydrochloride (second generation antihistamine
antagonist) (21). Unfortunately, we
could not evaluate the therapeutic outcome on the long-term, due to
the death of the patient, which we believe was unrelated to PN.
Our review has a few limitations. The first is that
the results are based on case reports and small case series.
Moreover, data regarding the treatment is inconsistent in most of
the published articles. As strength, our review offers a clear
overview concerning the most important aspects of PN in cases
reported so far.
In conclusion, PN should be recognized as a rare
form of bullous pemphigoid and must be taken into consideration as
a differential diagnosis of pruritic papulo-nodular eruption in the
elderly. Refractory, chronic and unexplained pruritus in those
patients should lead to skin biopsy and DIF/IIF microscopy as well
as immunoserology analysis to detect PN.
Acknowledgements
Not applicable.
Funding
The publication of the manuscript was partially
supported by the Transylvanian Association of Dermatologists
(ADT).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CV, SCS, LU were responsible for the conception of
manuscript and the interpretation of data. ADP and RC contributed
to the interpretation and analysis of data. EC contributed to
drafting the manuscript and revising it critically for important
intellectual content. All authors contributed to the literature
search, read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
the Emergency County Hospital (Cluj-Napoca, Romania). The patient
provided written informed consent for the present study.
Patient consent for publication
The patient gave written consent for the
investigations that were performed (blood tests and biopsy) and for
the publication of medical data for scientific purposes.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
BP
|
bullous pemphigoid
|
|
BP180
|
180 kDa BP antigen
|
|
BP230
|
230 kDa BP antigen
|
|
DIF
|
direct immunofluorescence
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
|
Ig
|
immunoglobulin
|
|
IPEX
|
immune dysregulation,
polyendocrinopathy, enteropathy, X-linked syndrome
|
|
IIF
|
indirect immunofluorescence
|
|
NSAID
|
non-steroidal anti-inflammatory
drugs
|
|
PN
|
pemphigoid nodularis
|
|
Pts
|
patients
|
|
SD
|
standard deviation
|
|
TNF-α
|
tumor necrosis factor-α
|
References
|
1
|
Lamberts A, Meijer JM and Jonkman MF:
Nonbullous pemphigoid: A systematic review. J Am Acad Dermatol.
78:989–995. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Alpsoy E, Akman-Karakas A and Uzun S:
Geographic variations in epidemiology of two autoimmune bullous
diseases: Pemphigus and bullous pemphigoid. Arch Dermatol Res.
307:291–298. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cozzani E, Gasparini G, Burlando M, Drago
F and Parodi A: Atypical presentations of bullous pemphigoid:
Clinical and immunopathological aspects. Autoimmun Rev. 14:438–445.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Amber KT, Korta DZ, de Feraudy S and
Grando SA: Vesiculobullous eruption in a patient receiving psoralen
ultraviolet A (PUVA) treatment for prurigo nodules: A case of
PUVA-aggravated pemphigoid nodularis. Clin Exp Dermatol.
42:833–835. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Powell AM, Albert S, Gratian MJ,
Bittencourt R, Bhogal BS and Black MM: Pemphigoid nodularis
(non-bullous): A clinicopathological study of five cases. Br J
Dermatol. 147:343–349. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schachter M, Brieva JC, Jones JC,
Zillikens D, Skrobek C and Chan LS: Pemphigoid nodularis associated
with autoantibodies to the NC16A domain of BP180 and a
hyperproliferative integrin profile. J Am Acad Dermatol.
45:747–754. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Di Zenzo G, Della Torre R, Zambruno G and
Borradori L: Bullous pemphigoid: From the clinic to the bench. Clin
Dermatol. 30:3–16. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stavropoulos PG, Soura E and Antoniou C:
Drug-induced pemphigoid: A review of the literature. J Eur Acad
Dermatol Venereol. 28:1133–1140. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mochizuki M, Fujine E, Tawada C, Kanoh H
and Seishima M: Pemphigoid nodularis possibly induced by
etanercept. J Dermatol. 40:578–579. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zeidler C, Yosipovitch G and Ständer S:
Prurigo nodularis and its management. Dermatol Clin. 36:189–197.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schmidt E and Zillikens D: Pemphigoid
diseases. Lancet. 381:320–332. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sárdy M, Kostaki D, Varga R, Peris K and
Ruzicka T: Comparative study of direct and indirect
immunofluorescence and of bullous pemphigoid 180 and 230
enzyme-linked immunosorbent assays for diagnosis of bullous
pemphigoid. J Am Acad Dermatol. 69:748–753. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kridin K: Peripheral eosinophilia in
bullous pemphigoid: Prevalence and influence on the clinical
manifestation. Br J Dermatol. Apr 16–2018.(Epub ahead of print).
View Article : Google Scholar
|
|
14
|
Messingham KN, Holahan HM, Frydman AS,
Fullenkamp C, Srikantha R and Fairley JA: Human eosinophils express
the high affinity IgE receptor, FcεRI, in bullous pemphigoid. PLoS
One. 9:e1077252014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Asahina A, Niizuma A, Ohzono A, Ishii N,
Koga H and Hashimoto T: Pemphigoid nodularis with diverse IgG, IgA
and IgE antibodies showing neutrophilic papillary microabscesses.
Acta Derm Venereol. 95:239–240. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
McGinness JL, Bivens MM, Greer KE,
Patterson JW and Saulsbury FT: Immune dysregulation,
polyendocrinopathy, enteropathy, X-linked syndrome (IPEX)
associated with pemphigoid nodularis: A case report and review of
the literature. J Am Acad Dermatol. 55:143–148. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Arbesman CE, Wypych JI, Reisman RE and
Beutner EH: IgE levels in sera of patients with pemphigus or
bullous pemphigoid. Arch Dermatol. 110:378–381. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schmidt E, Bröcker EB and Zillikens D:
High levels of soluble CD23 in blister fluid of patients with
bullous pemphigoid. Arch Dermatol. 131:966–967. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Saniklidou AH, Tighe PJ, Fairclough LC and
Todd I: IgE autoantibodies and their association with the disease
activity and phenotype in bullous pemphigoid: A systematic review.
Arch Dermatol Res. 310:11–28. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Taylor G, Venning V, Wojnarowska F and
Welch K: Bullous pemphigoid and autoimmunity. J Am Acad Dermatol.
29:181–184. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shintani T, Ohata C, Koga H, Ohyama B,
Hamada T, Nakama T, Furumura M, Tsuruta D, Ishii N and Hashimoto T:
Combination therapy of fexofenadine and montelukast is effective in
prurigo nodularis and pemphigoid nodularis. Dermatol Ther
(Heidelb). 27:135–139. 2014. View Article : Google Scholar
|
|
22
|
Yoneda K, Ishii N, Nakai K, Kubota Y and
Hashimoto T: Localized nodular pemphigoid. Int J Dermatol.
57:587–589. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang W, Liu Y and Li C: Generalised
nodules in pemphigoid nodularis. Lancet. 389:19302017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yoshimoto N, Ujiie H, Hirata Y, Izumi K,
Nishie W and Shimizu H: Bullous pemphigoid developed in a patient
with prurigo nodularis. J Eur Acad Dermatol Venereol. 31:e187–e189.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dangel B, Kofler L and Metzler G: Nodular
subtype of bullous pemphigoid. J Cutan Med Surg. 20:570–572. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kwong HL and Lim SP: Pemphigoid nodularis
mimicking nodular prurigo in an immune-suppressed patient with
rheumatoid arthritis. Acta Derm Venereol. 95:237–238. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Al-Salhi W and Alharithy R: Pemphigoid
nodularis. J Cutan Med Surg. 19:153–155. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Das D and Bandyopadhyay D: Juvenile
pemphigoid nodularis: Report of a rare case. Indian Dermatol Online
J. 5:189–192. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Matsudate Y, Ansai SI, Hirose K, Kubo Y
and Arase S: Pemphigoid nodularis: The importance of ELISA for
diagnosis. Eur J Dermatol. 19:83–84. 2009.PubMed/NCBI
|
|
30
|
Koga H, Hamada T, Ohyama B, Nakama T,
Yasumoto S and Hashimoto T: An association of idiopathic chronic
eosinophilic pneumonia with pemphigoid nodularis: A rare variant of
bullous pemphigoid. Arch Dermatol. 145:1339–1340. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aboumaria A, Pelletier F, Aubin F, Algros
MP, Afifi Y and Humbert P: Pemphigoid nodularis. Ann Dermatol
Venereol. 135:251–252. 2008.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Teraki Y and Fukuda T: Pemphigoid
nodularis associated with psoriatic erythroderma: Successful
treatment with suplatast tosilate. Br J Dermatol. 158:424–426.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gach JE, Wilson NJ, Wojnarowska F and
Ilchyshyn A: Sulfamethoxypyridazine-responsive pemphigoid
nodularis: A report of two cases. J Am Acad Dermatol. 53 Suppl
1:S101–S104. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tashiro H, Arai H, Hashimoto T, Takezaki S
and Kawana S: Pemphigoid nodularis: two case studies and analysis
of autoantibodies before and after the development of generalized
blistering. J Nippon Med Sch. 72:60–65. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sakuma-Oyama Y, Powell AM, Albert S, Oyama
N, Bhogal BS and Black MM: Lichen planus pemphigoides evolving into
pemphigoid nodularis. Clin Exp Dermatol. 28:613–616. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gao XH, Lin J, Yang C, Ma L, Wang G, Wang
Y and Chen HD: A case of Kaposi's sarcoma associated with
pemphigoid nodularis. J Dermatol. 28:388–392. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ameen M, Harman KE and Black MM:
Pemphigoid nodularis associated with nifedipine. Br J Dermatol.
142:575–577. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cliff S and Holden CA: Pemphigoid
nodularis: A report of three cases and review of the literature. Br
J Dermatol. 136:398–401. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kossard S: Prurigo papules with blisters.
Australas J Dermatol. 37:104–105. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tamada Y, Yokochi K, Oshitani Y, Nitta Y,
Ikeya T, Hara K and Owaribe K: Pemphigoid nodularis: A case with
230 kDa hemidesmosomes antigen associated with bullous pemphigoid
antigen. J Dermatol. 22:201–204. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bourke JF, Berth-Jones J, Gawkrodger DJ
and Burns DA: Pemphigoid nodularis: A report of two cases. Clin Exp
Dermatol. 19:496–499. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ratnavel RC, Shanks AJ, Grant JW and
Norris PG: Juvenile pemphigoid nodularis. Br J Dermatol.
130:125–126. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gallo R, Parodi A and Rebora A: Pemphigoid
nodularis. Br J Dermatol. 129:744–745. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Borradori L, Prost C, Wolkenstein P,
Bernard P, Baccard M and Morel P: Localized pretibial pemphigoid
and pemphigoid nodularis. J Am Acad Dermatol. 27:863–867. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ross JS, McKee PH, Smith NP, Shimizu H,
Griffiths WA, Bhogal BS and Black MM: Unusual variants of
pemphigoid: From pruritus to pemphigoid nodularis. J Cutan Pathol.
19:212–216. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Borradori L, Rybojad M, Verola O, Flageul
B, Puissant A and Morel P: Pemphigoid nodularis. Arch Dermatol.
126:1522–1523. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tani M, Murata Y and Masaki H: Pemphigoid
nodularis. J Am Acad Dermatol. 21:1099–1104. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shimizu H, Hayakawa K and Nishikawa T: A
comparative immunoelectron microscopic study of typical and
atypical cases of pemphigoid. Br J Dermatol. 119:717–722. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Roenigk RK and Dahl MV: Bullous pemphigoid
and prurigo nodularis. J Am Acad Dermatol. 14:944–947. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Massa MC and Connolly SM: Bullous
pemphigoid with features of prurigo nodularis. Arch Dermatol.
118:937–939. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yung CW, Soltani K and Lorincz AL:
Pemphigoid nodularis. J Am Acad Dermatol. 5:54–60. 1981. View Article : Google Scholar : PubMed/NCBI
|