Introduction
Familial hypercholesterolaemia (FH) is a rare
genetic disorder, which affects around 0.2% of the population
(1). The disease manifestations are
very high levels of serum low-density lipoprotein (LDL), xanthomas
and early coronary artery disease (CAD). The genetic mutations of
lipid handling genes in patients lead to hyperlipidemia that
usually develop into severe atherosclerosis and cause ischemia
heart diseases and heart failure. Due to technical challenging and
resource limitations, genetic screening is not possible to diagnose
patients with FH, especially in developing countries. Thus,
establishment of an easy and low cost diagnostic approach for FH is
needed.
In the present study, we reported a FH patient
admitted in our hospital with symptoms of heart failure. With
physical and laboratory tests, the patient showed typical FH
symptoms. Cardiac dysfunctions and coronary artery stenosis were
further determined by electrocardiogram, echocardiogram and CT
angiography. The patient was immediately treated with combination
of lipid lowering, anti-thrombosis and cardiac remodel improving
therapies. The patient was discharged when the heart failure
symptoms were successfully controlled.
Case report
A 13 years old female patient was admitted to
Children's Hospital of Chongqing Medical University (Chongqing,
China) due to symptoms of heart failure, including lower limb
swelling for 9 days, coughing and panic for 6 days. Upon arriving
hospital, the patient felt heart palpitations, breath shortness,
and fatigue after short walk. The lower extremity of the patient
had pitting edema, which had been aggravated with oliguria and
sweating for 9 days. In the course of the disease, there was a
paroxysmal cough accompanied with the yellow mucus phlegm and
bloody phlegm. None of the following symptoms was presented:
wheezing and dyspnea, eyelid edema, gross hematuria, foam urine,
chest tightness, chest pain, nausea, vomiting, abdominal
distension, diarrhea, dizziness, headache, syncope, or fever. Based
on her disease history, dermal yellow nodules were found right
after her birth, they developed into nodular xanthoma during her
growth.
Upon admitted in our hospital, physical examination
was performed: Her body temperature was 36.6°C, breath was 30 BPM,
heart rate was 142 BPM, and blood pressures were 110/58 mmHg. The
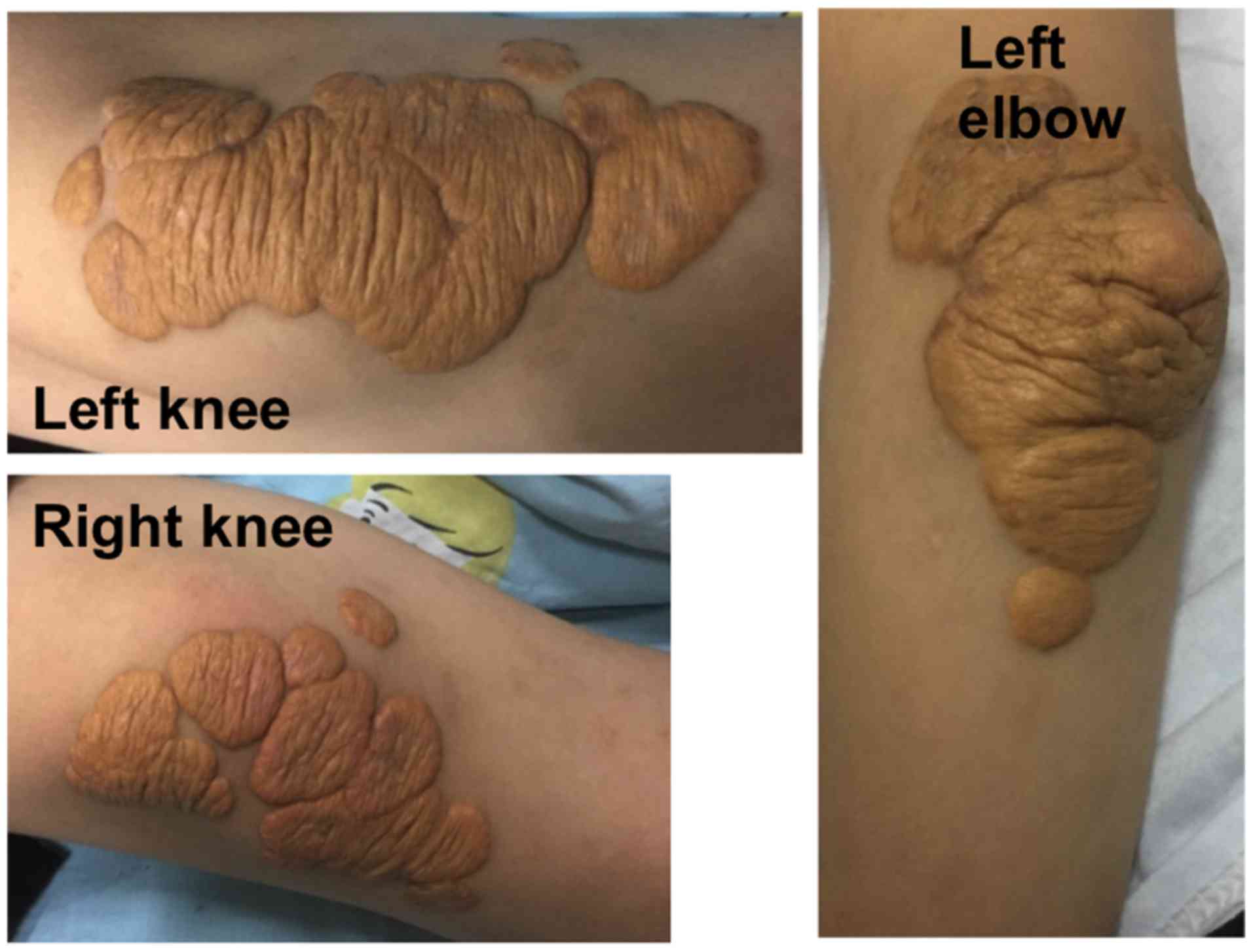
skin of double knees, left elbow, ankle, wrist, toe and coccygeal
joint had hemispherical yellow eminence subcutaneous nodules
(Fig. 1). The size of nodules ranged
from 1.0×2.0 to 5.0×6.0 cm. The boundary was clear, texture was
tough, and the surface was free from swelling and ulceration. The
apex beat was located at the 5th intercostal space about 3.0 cm
from left midclavicular line. No presentation of lifting type
pulsation and pericardial friction was detected. The heart beat
sounded slightly dull and arrhythmia could be detected.
Laboratory tests were performed: blood lipids were
extremely high [total cholesterol (TC) 14.73 mmol/l, high-density
lipoprotein (HDL) 0.59 mmol/l, LDL 13.44 mmol/l], B type
natriuretic peptide was 800 pg/ml, lactic acid was 4.04 mmol/l,
blood amine was 61.7 mol/l, CRP was 59 mg/l, PTC was 1.138 ng/ml.
In addition, the following tests were performed and showed normal:
Blood routine, urine routine, fecal routine, liver and kidney
function, electrolytes, myocardial markers, ESR, immunization,
autoantibodies, blood glucose and blood gas tandem mass
spectrometry.
Because the patients showed symptoms of heart
failure, cardiac related tests were performed. First,
electrocardiogram of the patient showed sinus tachycardia, QT
lengthening, ST-T change (Fig. 2).
Dynamic electrocardiogram test revealed that ventricular premature
beat presented 2 times/24 h, atrial premature beat presented
onceper 24 h, the average heart rate was 97 beats per minute, and
the heart rate variability was poor. Furthermore, cardiac
morphology and function were determined by echocardiography
(Fig. 3). Multiple pathological
changes have been observed, including enlarged left ventricle,
enlargement of left and right atrium, and right ventricle, decrease
of ventricular septum and left ventricular posterior wall motion,
and aortic sinus tube junction stenosis with hyperechoic structure
with moderate regurgitation, moderate to severe mitral valve
regurgitation, mild to moderate tricuspid valve regurgitation,
decreased left ventricular systolic function, slightly widened
inferior vena cava. In addition, abdominal B-ultrasound test
revealed a slight swelling of the liver. In order to examine the
cause of ischemic heart disease, chest CT angiography was performed
(Fig. 4): There were multiple points
of stenosis in the ascending aorta, left and right coronary artery
and descending aorta, while CT of double lungs showed normal. The
bifurcation of the left coronary artery was stenosis, the degree of
stenosis was 93%, the left anterior descending coronary artery had
stenosis after diagonal branches, the degree of stenosis was 68%,
the upper right coronary artery (from the start 27.9 mm) also had
stenosis, the degree of stenosis was 42%.
Except for the patients, no skin xanthoma and
premature coronary heart disease were found in the family.
Based on the patient's medical history, clinical
manifestation, blood lipid and cardiac test results. Diagnosis was
made: the patient showed typical FH with coronary atherosclerotic
heart disease. She had grad II cardiac insufficiency, nodular
xanthoma, atherosclerosis, and vavular calcification. In order to
confirm our diagnosis, a genetic test was prescribed to the
patient. Unfortunately, the parents of the patient refused to
perform the genetic test due to the financial difficulties.
Based on guidance from the 2014 European
Atherosclerotic Association and the 2016 British HoFH management,
the following treatments were prescribed to the patient: Sodium
phosphocreatine 1.0 g IV, qd, 11 days, VitC 3.0 g IV, qd, 11 days,
combined coenzyme needle 1 branches IV, qd, 11 days to nourish the
myocardium. Milrinone 50 µg/kg, slow intravenous injection for 10
min later, 0.5 µg/kg/min, 6 days, and maintain dobutamine 5
µg/kg/min IV continued 2 h, q8 h, 5 days, digoxin 0.125 mg/time,
po, qd for 6 days were used to enhance myocardial contractility.
Oral Atorvastatin 20 mg/time, qN for 6 days and diet control were
used to lower blood lipid. Ezetimibe Tablets: 10 mg/time, po, bid
for 6 days were administrated to stablize plaque. Benazepril 10
mg/time, po, qd for 5 days and metoprolol 6.25 mg/time, po, bid for
2 days were used to improve myocardial remodeling. Spironolactone
20 mg/time, po, bid for 11 days, furosemide 20 mg/time, po, bid for
10 days and hydrochlorothiazide 25 mg/time, po, bid for 1 day,
diuresis. Aspirin 100 mg/time, po, qd for 3 days and clopidogrel 50
mg, po, qd for 3 days were used to prevent thrombosis formation.
After treatment for 11 days, the symptoms of the patient were
improved, she was discharged.
After returning home, her symptom has been improved.
The drugs were subscripted to her after discharge: Oral drug
spironolactone 10 mg/time qd for one year, hydrochlorothiazide 12.5
mg/times, qd for one year, metoprolol 6.25 mg/time, bid for one
year, hydrogen chloropidogrel 25 mg/time, qd for one year,
atorvastatin 20 mg/time, qN for one year. After 1 year of
treatment, the patient was retested for blood lipids (TC 10.03
mmol/l, HDL 0.48 mmol/l, LDL 852 mmol/l), function of liver and
kidney, electrolytes, blood glucose, blood coagulation, myocardial
damage markers were normal, ECG was sinus tachycardia, and ST
segment was low.
Ethical approval for the present study was obtained
from Children's Hospital of Chongqing Medical University and oral
informed consent was obtained from both the patient and the
patient's parent.
Discussion
FH is an autosomal dominant genetic disease. The
most common cause of FH is the mutations of the genes encoding
lipid handling proteins, such as LDL receptor (LDLR),
apolipoprotein B (ApoB) or proteins convertase
subtilisin/Kexin/kexin9 (PCSK 9), resulting in LDL metabolism
defects, and LDL cholesterol (LDL-C) levels are abnormally elevated
in plasma. The FH patients usually have xanthoma in peripheral
tissue, atherosclerosis and other clinical manifestations. Based on
a FH study in Jiangsu, China, the Chinese FH diagnostic standard
was established (2) (summarized in
Table I). Based on this standard,
the patient in current study met multiple criteria of FH, including
an early-onset coronary heart disease history and extremely high
level of LDL-C (13.44 mmol/l), and xanthoma formation at the tendon
sites. FH can further be divided into homozygote type (HoFH) and
heterozygote type (HeFH), where the HoFH is a serious rare disease
(3). The prevalence of HoFH was
1/(16–100)×104, and the patients' serum LDL-C >13
mmol/l. If without any treatment, the HoFH patients usually die
from coronary heart disease before the age of 30 (4). The incidence of coronary heart disease
in patients with HoFH is 100 times higher than that of the normal
population, and most of the patients with HoFH are able to develop
in childhood and adolescence (5).
The main clinical manifestations of HoFH are plasma LDL-C level is
extremely high (approximately 6–8 times higher than normal),
corneal arch, xanthoma at the tendon sites, premature coronary
heart disease and aortic valve disease (6). Aortic valve disease is mainly caused by
the involvement of the root of the aorta, which leads to the
stenosis and calcification of the aorta on the upper part of the
aortic valve and the aortic valve, and even the opening of the
coronary artery (7–9). Except absent of corneal arch, the
patients displayed all other symptoms.
 | Table I.Chinese FH diagnostic criteria. |
Table I.
Chinese FH diagnostic criteria.
| Standard | Score |
|---|
| First degree
relatives have a family history of early onset coronary heart
disease or vascular disease | 1 |
| Individual history of
early onset coronary heart disease | 2 |
| Personal history of
early cerebrovascular disease | 1 |
| LDL-C,
mmol/l−1 |
|
| ≥6.0 | 8 |
|
5.0–5.9 | 5 |
|
3.5–4.9 | 3 |
|
2.5–3.4 | 1 |
| FH diagnosis |
|
Diagnosis | >8 |
| Very
likely | 6–8 |
|
Probably | 3–5 |
|
Impossible | <3 |
The 2014 European Atherosclerotic Association
(European Atherosclerosis Society, EAS) HoFH management guide
proposed that the diagnosis of HoFH should be based on the standard
of genetic diagnosis or in conformity with the clinical diagnostic
criteria. The diagnostic genes include LDLR, ApoB, PCSK9, and
LDLRAP1. The clinical diagnostic criteria are: LDL-C>13 mmol/l
before treatment or LDL-C>8 mmol/l after treatment, and the
presence of xanthoma in the tendon at the age of 10 or the level of
parental LDL-C was consistent with heterozygote FH (HeFH) (3). The 2016 British HoFH management guide
also recommends the use of genetic diagnostic or clinical
diagnostic criteria, in which the genetic diagnostic criteria are
basically the same as the EAS HoFH management guidelines. Children
and adults diagnostic criteria are: Children LDL C>11 mmol/l and
skin tendon xanthoma before 10 years of age; adult LDL-C>13
mmol/l and there were obvious xanthoma of skin tendon, or LDL-C
level reached clinical diagnosis standard, while parents were
diagnosed as HeFH (8). Therefore, we
diagnosed the patient in this study as HoFH.
Finally, after literature search, we found one
previous study that reported a 17 years old female patient. She had
hypercholesterolemia on 2 year old, xanthoma at age of 9, and the
clinical manifestations of coronary heart disease at age of 12. In
addition, her TC and LDL-C was significantly increased, the
electrocardiogram has ST segment depression, cardiac color Doppler
showed stenosis and mitral regurgitation. There was sign of heart
failure. The aorta stenosis and coronary artery stenosis were
detected by angiography. The genetic examination revealed that she
has exon 541T>C homozygous mutation of LDLR gene (9). Based on the clinical manifestation, we
believe our results are in agreement with those reported by Zhao
et al (9).
Thus, we showed the feasibility of a clinical
diagnosis of HoFH without a genetic diagnosis. We believe this is
extremely importantly in developing countries, where genetic
diagnosis might be challenging and not available. In this case, we
had successfully used lipid-lowering therapy, anti-thrombosis
therapy and cardiac remodeling therapy to improve cardiac function
of the patient.
According to the clinical diagnostic criteria, only
about 1/4 of FH cases can be predicted. Most patients were
diagnosed until middle age, thus missing the chance of early
treatments. It has been estimated that there are about 26 million
potential FH patients in China (10), but the rate of diagnosis and
treatment in clinics is still very low (11). According to the international FH
foundation, population screening should be based on age, sex and
specific LDL-C level. Systematic screening strategy is very
important for identifying FH proband, which is the key to discover
new cases for family members (12).
Therefore, we should perform blood lipids, electrocardiogram,
echocardiography and carotid ultrasound examination on parents,
siblings and other family members, and complete family genetic
testing, in order to identify FH patients and perform early
treatment.
The current treatment options for FH include
lifestyle changes, drug therapy and liver transplantation.
Lifestyle changes are the most important aspect of lipid-lowering
therapy, including reducing uptake of saturated fatty acids and
cholesterol, eating cellulose rich foods, stopping smoking and
controlling salt consumption, proper physical activity, and weight
loss. Conventional lipid-lowering drugs include statins and
ezetimibe. The National Lipid Association of the United States
suggested that the level of LDL-C in patients with FH decreased to
below 2.5 mmol/l or decreased by >50% compared with that before
treatment (12). But HoFH is a
serious hereditary disease. Even when multiple drugs combined with
lifestyle intervention, it is still difficult to achieve LDL-C
compliance (13). The EAS HoFH
management guide recommends that HoFH patients take high-intensity
tolerable dose statins as initial treatment, gradually combine with
other types of lipid-lowering drugs such as ezembo, and PCSK9
inhibitor, or plasma lipoprotein replacement, in addition, some
patients may choose to undergo liver transplantation (14). In recent years, with the in-depth
study of the mechanism of cholesterol metabolism, new target drugs
are emerging. The most representative new drugs are mainly two
oligonucleotide inhibitors (mipomersen and lomitapide) (15) and two PCSK9 monoclonal antibody
inhibitors (alirocumab and evolocumab) (16). Mipomersen and lomitapide can
significantly reduce blood lipids in patients with HoFH, but the
cost is high and the incidence of adverse reactions is high. In
contrast, PCSK9 inhibitors are better choices at present because of
their therapeutic effects and high potency ratio.
Clearly, our case report has limitations. One of
them was there was no genetic test for the patient to finally
confirm the case was a HoFH at the gene level. Luckily, some
clinical evidences strongly suggested it was a HoFH case. First,
the immediate family members are all normal without known skin
xanthoma, indicating they might be heterozygous carriers. Second,
the symptoms of the patients are consistent to the diagnostic
criteria from both the 2014 European Atherosclerotic Association
and the 2016 British HoFH management guidance. Most importantly,
after following treatment of HoFH, the patient has recovered and
was finally discharged. Thus, we feel our case is deserved to be
achieved and valuable for the physicians from developing countries,
where the genetic tests might not be feasible. The other
limitations include that we do not have information for the family
history of coronary heart disease neither did we check the
patient's second-degree family members for the symptom. We failed
to obtain this important information due to quick improvement of
the patient's symptom and discharge of the patient.
Acknowledgements
Not applicable.
Funding
The present study was supported by internal funding
from Chongqing Medical University.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YL, TT, HZ and JH participated in the manuscript
writing; YL, TT, MZ, YZ, SH and LZ participated in data collection;
XL participated in manuscript editing and data analysis; HZ and JH
participated in the data analysis and manuscript revision. All
authors have read and approved the manuscript.
Ethics approval and consent to
participate
The study has been approved by IRB of Children's
Hospital of Chongqing Medical University.
Patient consent for publication
The patient provided oral consent for publication of
this study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Selvan JP, Uthaman B, Abushaban L and
Jebaraj R: Homozygous familial hypercholesterolemia with
generalized arterial disease. Medical principles and practice:
international journal of the Kuwait University. Health Sci Centre.
16:75–78. 2007.
|
|
2
|
Shi Z, Yuan B, Zhao D, Taylor AW, Lin J
and Watts GF: Familial hypercholesterolemia in China: Prevalence
and evidence of underdetection and undertreatment in a community
population. Int J Cardiol. 174:834–836. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cuchel M, Bruckert E, Ginsberg HN, Raal
FJ, Santos RD, Hegele RA, Kuivenhoven JA, Nordestgaard BG, Descamps
OS, Steinhagen-Thiessen E, et al: Homozygous familial
hypercholesterolaemia: New insights and guidance for clinicians to
improve detection and clinical management. A position paper from
the Consensus Panel on Familial Hypercholesterolaemia of the
European Atherosclerosis Society. Turk Kardiyol Dern Ars. 43:1–4.
2015.(In Turkish). PubMed/NCBI
|
|
4
|
Page MM, Bell DA, Hooper AJ, Watts GF and
Burnett JR: Lipoprotein apheresis and new therapies for severe
familial hypercholesterolemia in adults and children. Best Pract
Res Clin Endocrinol Metab. 28:387–403. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vuorio A, Tikkanen MJ and Kovanen PT:
Inhibition of hepatic microsomal triglyceride transfer protein-a
novel therapeutic option for treatment of homozygous familial
hypercholesterolemia. Vasc Health Risk Manag. 10:263–270. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gidding SS: The complexities of homozygous
familial hypercholesterolemia management. Pediatr Transplant.
20:1020–1021. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
France M: Homozygous familial
hypercholesterolaemia: Update on management. Paediatr Int Child
Health. 36:243–247. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
France M, Rees A, Datta D, Thompson G,
Capps N, Ferns G, Ramaswami U, Seed M, Neely D, Cramb R, et al:
HEART UK statement on the management of homozygous familial
hypercholesterolaemia in the United Kingdom. Atherosclerosis.
255:128–139. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao X, Bu L, Qin S, Kong D, Fan B and Ge
J: Early development of xanthoma and coronary disease in a young
female with homozygous familial hypercholesterolemia. Int J
Cardiol. 176:e15–e19. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jiang L, Gao F, Hu LB, Sun LY, Pan XD, Lin
J, Zhang HB, Yong Q, Wang Q, Yang Y, et al: Seven-year clinical
follow-up of a Chinese homozygous familial hypercholesterolemia
child with premature xanthomas and coronary artery disease-a need
for early diagnosis and aggressive treatment. Int J Cardiol.
177:188–191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vallejo-Vaz AJ, Kondapally Seshasai SR,
Cole D, Hovingh GK, Kastelein JJ, Mata P, Raal FJ, Santos RD, Soran
H, Watts GF, et al: Familial hypercholesterolaemia: A global call
to arms. Atherosclerosis. 243:257–259. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Watts GF, Gidding S, Wierzbicki AS, Toth
PP, Alonso R, Brown WV, Bruckert E, Defesche J, Lin KK, Livingston
M, et al: Integrated guidance on the care of familial
hypercholesterolaemia from the International FH Foundation. Eur J
Prev Cardiol. 22:849–854. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ito MK and Watts GF: Challenges in the
diagnosis and treatment of homozygous familial
hypercholesterolemia. Drugs. 75:1715–1724. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Besseling J, Sjouke B and Kastelein JJ:
Screening and treatment of familial hypercholesterolemia-Lessons
from the past and opportunities for the future (based on the
Anitschkow Lecture 2014). Atherosclerosis. 241:597–606. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cuchel M, Meagher EA, du Toit Theron H,
Blom DJ, Marais AD, Hegele RA, Averna MR, Sirtori CR, Shah PK,
Gaudet D, et al: Efficacy and safety of a microsomal triglyceride
transfer protein inhibitor in patients with homozygous familial
hypercholesterolaemia: A single-arm, open-label, phase 3 study.
Lancet. 381:40–46. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Raal FJ, Honarpour N, Blom DJ, Hovingh GK,
Xu F, Scott R, Wasserman SM and Stein EA; TESLA Investigators, :
Inhibition of PCSK9 with evolocumab in homozygous familial
hypercholesterolaemia (TESLA Part B): A randomised, double-blind,
placebo-controlled trial. Lancet. 385:341–350. 2015. View Article : Google Scholar : PubMed/NCBI
|