Introduction
A number of studies have indicated that mushrooms
are rich sources of bioactive compounds. Among them, Ganoderma
lucidum is a polypore mushroom that grows on the lower trunks
of deciduous trees. This mushroom is a traditional Oriental
medicine that has been widely used as a tonic to promote longevity
and health for thousands of years in Asian countries, including
China, Japan and Korea (1–4). The pharmacological activities of
G. lucidum, particularly its intrinsic immunomodulating and
antitumor properties, have been well-documented (4,5).
Several studies have demonstrated that various G. lucidum
extracts interfere with cell cycle progression, induce apoptosis
and suppress angiogenesis in human cancer cells and thus act as
anticancer agents (6–11). Additionally, it has also been
suggested that G. lucidum extracts have a neuroprotective
effect and may be useful in future studies on the pathogenesis and
prevention of Alzheimer’s disease (AD) and Parkinson’s disease (PD)
(12–15). However, the precise biochemical
mechanisms underlying the G. lucidum extract-induced
anti-inflammatory effects have not yet been clarified in microglial
cells.
Microglia are the resident macrophage-like cells in
the brain that play a major role in host defense and tissue repair
in the central nervous system (CNS) (16,17).
However, under pathological conditions, activated microglia release
neurotoxic and pro-inflammatory mediators, including nitric oxide
(NO), prostaglandin E2 (PGE2), reactive
oxygen species and pro-inflammatory cytokines, including
interleukin (IL)-1β, IL-6 and tumor necrosis factor (TNF)-α
(18,19). Overproduction of these inflammatory
mediators and cytokines causes severe neurodegenerative diseases,
including AD, PD, cerebral ischemia, multiple sclerosis and trauma
(20–22). Activated microglia are a major
cellular source of pro-inflammatory and/or cytotoxic factors that
cause neuronal damage in the CNS. Previous studies have also
demonstrated that a decrease in the number of pro-inflammatory
mediators in microglia may attenuate the severity of these
disorders (23–25).
In the present study, we investigated the inhibitory
effects of an ethanol extract of G. lucidum (EGL) and the
mechanism of its anti-inflammatory action against the
lipopolysaccharide (LPS)-stimulated pro-inflammatory responses in
murine BV2 microglia. Our results indicate that EGL inhibits
inflammatory reactions by inhibiting nuclear factor κB (NF-κB) and
toll-like receptor (TLR) signaling pathways, suggesting that EGL
may be useful for the treatment of neuroinflammatory and
neurodegenerative diseases.
Materials and methods
Preparation of EGL
EGL was supplied by Dongeui University Oriental
Hospital (Busan, Korea). The freeze-dried and milled G.
lucidum fruiting bodies (200 g) were extracted with 25% ethanol
(4 liters) at room temperature for 10 h using a blender. The
extracts were filtered through a Whatman no. 2 filter (Maidstone,
UK), concentrated to 500 ml under vacuum conditions and then stored
at −20°C (11). The EGL solution
was directly diluted in medium prior to assay.
Cell culture
The murine BV2 cell line was maintained in
Dulbecco’s modified Eagle’s medium supplemented with 10% fetal
bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin
(Gibco-BRL, Grand Island, NY, USA) at 37°C in a humidified
incubator with 5% CO2. The cells were pre-treated with
the indicated EGL concentrations for 1 h before adding LPS (0.5
μg/ml, Sigma-Aldrich, St. Louis, MO, USA). Cell viability
was evaluated by the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma-Aldrich) reduction assay. In brief, cells (1×105
cells/ml) were seeded and treated with EGL and/or LPS for the
indicated time periods. Following treatment, the medium was removed
and the cells were incubated with 0.5 mg/ml MTT solution. After a 3
h incubation at 37°C in 5% CO2, the supernatant was
removed and formation of formazan was measured at 540 nm with a
microplate reader (Dynatech MR-7000; Dynatech Laboratories Inc., El
Paso, TX, USA).
NO production
NO concentrations in culture supernatants were
determined by measuring nitrite, which is a major stable product of
NO, using Griess reagent (Sigma-Aldrich). Cells (5×105
cells/ml) were stimulated in 24-well plates for 24 h, then 100
μl culture medium was mixed with an equal volume of Griess
reagent (1% sulfanilamide, 0.1% N-(1-naphthyl)-ethylenediamine
dihydrochloride and 2.5% H3PO4). Nitrite
levels were determined using an enzyme-linked immunosorbent assay
(ELISA) plate reader at 540 nm and nitrite concentrations were
calculated by reference to a standard curve generated using known
concentrations of sodium nitrite (26).
Measurement of PGE2
production
BV2 cells were sub-cultured in 6-well plates
(5×105 cells/ml) and incubated with the indicated
concentrations of EGL in the presence or absence of LPS (0.5
μ/ml) for 24 h. A 100 μl aliquot of the
culture-medium supernatant was collected for determination of the
PGE2 concentration by ELISA (Cayman Chemical, Ann Arbor,
MI, USA).
RNA isolation and reverse
transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from the cells using the
TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA).
The total RNA (1.0 μg) was reverse-transcribed using M-MLV
reverse transcriptase (Promega Corporation, Madison, WI, USA) to
produce cDNAs. The inducible nitric oxide synthase (iNOS),
cyclooxygenase (COX)-2, IL-1β and TNF-α genes were amplified from
the cDNA using PCR. The PCR primers were as follows: mouse iNOS,
5′-ATG TCC GAA GCA AAC ATC AC-3′ and 5′-TAA TGT CCA GGA AGT AGG
TG-3′; COX-2, 5′-CAG CAA ATC CTT GCT GTT CC-3′ and 5′-TGG GCA AAG
AAT GCA AAC ATC-3′; IL-1β, 5′-ATG GCA ACT GTT CCT GAA CTC AAC T-3′
and 5′-TTT CCT TTC TTA GAT ATG GAC AGG AC-3′; and TNF-α, 5′-ATG AGC
ACA GAA AGC ATG ATC-3′ and 5′-TAC AGG CTT GTC ACT CGA ATT-3′.
Following amplification, the PCR products were electrophoresed on
1% agarose gels and visualized by ethidium bromide (Sigma-Aldrich)
staining. Glyceraldehyde-3-phosphate dehydrogenase was used as an
internal control.
Protein extraction and western blot
analysis
Cells were washed three times with
phosphate-buffered saline (PBS) and lysed in lysis buffer (1%
Triton X-100, 1% deoxycholate and 0.1% NaN3) containing protease
inhibitors (Roche Diagnostics, Mannheim, Germany). In a parallel
experiment, nuclear and cytosol proteins were prepared using
NE-PER® nuclear and cytoplasmic extraction reagents
(Pierce Biotechnology Inc., Rockford, IL, USA) according to the
manufacturer’s instructions. For the western blot analysis, equal
amounts of protein were subjected to electrophoresis on sodium
dodecyl sulfate-polyacrylamide gels and transferred to
nitrocellulose membranes (Schleicher & Schuell Inc., Keene, NH,
USA) by electroblotting. Blots were probed with the desired
antibodies for 1 h, incubated with the diluted enzyme-linked
secondary antibodies and visualized by enhanced chemiluminescence
(Amersham Co., Arlington Heights, IL, USA) according to the
manufacturer’s instructions. Actin and nucleolin were used as
internal controls for the cytosolic and nuclear fractions,
respectively. Antibodies against iNOS, COX-2, IκB-α, NF-κB p65,
TLR4 and myeloid differentiation factor 88 (MyD88) were purchased
from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies
against actin and nucleolin were obtained from Sigma-Aldrich.
Peroxidase-labeled goat anti-rabbit immunoglobulin and fluorescein
isothiocyanate (FITC)-conjugated donkey anti-rabbit IgG were
purchased from Amersham Co. and Sigma-Aldrich, respectively.
Cytokine assays
The levels of IL-1β and TNF-α were measured using
ELISA kits (R&D Systems, Minneapolis, MN, USA) according to the
manufacturer’s instructions. Briefly, BV2 cells (5×105
cells/ml) were plated in 24-well plates and pre-treated with the
indicated EGL concentrations for 1 h prior to treatment with 0.5
μg/ml LPS for 24 h. A 100 μl aliquot of each
culture-medium supernatant was collected for determination of the
IL-1β and TNF-α concentrations by ELISA.
NF-κB luciferase assay
A total of 1×106 BV2 cells were
transfected with 2 μg NF-κB-luciferase reporter plasmids (BD
Biosciences, San Jose, CA, USA) using Lipofectamine according to
the manufacturer’s instructions (Gibco-BRL). Then, the cells were
pre-incubated in the presence or absence of EGL before being
stimulated with LPS for 6 h. Cells were washed twice with PBS and
lysed with reporter lysis buffer (Promega Corporation). Following
vortexing and centrifuging at 12,000 × g for 1 min at 4°C, the
supernatant was stored at −70°C. For the luciferase assay, 20
μl cell extract was mixed with 100 μl luciferase
assay reagent at room temperature, then, the mixture was analyzed
using a LB96V microplate luminometer (Perkin-Elmer, Waltham, MA,
USA) (27).
Statistical analysis
Data are presented as mean ± standard deviation.
Statistical significance was determined using an analysis of
variance followed by the Student’s t-test. P<0.05 was considered
to indicate a statistically significant difference.
Results
Effects of EGL on NO and PGE2
production in LPS-stimulated BV2 microglia
The potential anti-inflammatory properties of EGL
were evaluated against the production of two major inflammatory
mediators, NO and PGE2, in LPS-stimulated BV2 microglia.
To determine the levels of NO and PGE2 production, the
amounts of nitrite and PGE2 released into the culture
medium were measured using Griess reagent and ELISA, respectively.
According to the NO detection assay result, LPS alone markedly
induced NO production from cells compared with the control
(Fig. 1A). However, pre-treatment
with EGL significantly repressed the levels of NO production in the
LPS-stimulated BV2 microglia in a concentration-dependent manner,
up to 1 μg/ml. Under the same conditions, stimulating the
cells with LPS also resulted in a significant increase in
PGE2 production; however, this was markedly repressed by
pre-treatment with EGL (Fig.
1B).
Effects of EGL on cell viability in
LPS-stimulated BV2 microglia
The cells were exposed to EGL for 24 h in the
presence or absence of LPS to exclude a cytotoxic effect of EGL on
BV2 cell growth. Cell viability was then measured by the MTT assay.
As indicated in Fig. 2, the
concentrations of EGL (0.1–1 mg/ml) used to inhibit NO and
PGE2 production did not affect cell viability. The
results clearly indicate that the inhibition of NO and
PGE2 production in the LPS-stimulated BV2 cells was not
due to a cytotoxic effect of EGL.
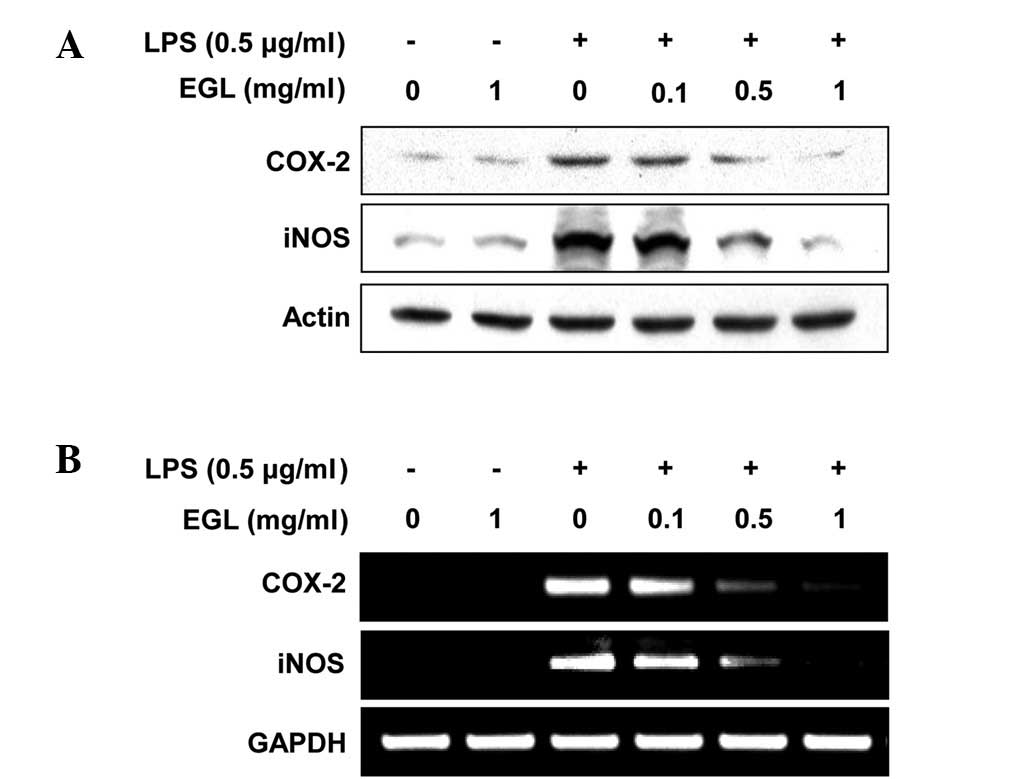
Effects of EGL on LPS-stimulated iNOS and
COX-2 expression
RT-PCR and western blot analyses were carried out to
determine whether the inhibition of NO and PGE2
production by EGL in LPS-stimulated BV2 cells was associated with
reduced levels of iNOS and COX-2 expression. As shown in Fig. 3, iNOS and COX-2 protein levels were
markedly upregulated following 24 h of treatment with LPS (0.5
μg/ml) alone; however, EGL significantly inhibited the iNOS
and COX-2 protein expression in the LPS-stimulated BV2 microglia in
a concentration-dependent manner (Fig.
3A). The effects of EGL on iNOS and COX-2 mRNA expression were
evaluated to investigate whether EGL suppressed the LPS-mediated
induction of iNOS and COX-2 at the pre-translational level. RT-PCR
data revealed that the reduction in iNOS and COX-2 mRNAs correlated
with the reduction in the corresponding protein levels (Fig. 3B). These results suggest that
EGL-induced reductions in iNOS and COX-2 expression were the cause
of the reduced NO and PGE2 production levels.
Effects of EGL on LPS-induced IL-1β and
TNF-α production and mRNA expression
The effects of EGL on the production of
pro-inflammatory cytokines, including IL-1β and TNF-α, were
analyzed using ELISA. BV2 cells were incubated with various
concentrations of EGL in the presence or absence of LPS (0.5
μg/ml) for 24 h and cytokine levels in the culture media
were measured. As shown in Fig. 4,
the IL-1β and TNF-α levels were markedly increased in the culture
media of the LPS-stimulated BV2 microglia. However, pre-treatment
with EGL significantly reduced the release of these
pro-inflammatory cytokines in a concentration-dependent manner. In
a parallel experiment, the effects of EGL on LPS-induced IL-1β and
TNF-α mRNA expression were studied using RT-PCR. As shown in
Fig. 5, IL-1β and TNF-α mRNA
transcription levels also decreased following EGL treatment. These
results suggest that EGL was effective in suppressing
pro-inflammatory cytokine production by altering IL-1β and TNF-α
transcription levels in activated microglia.
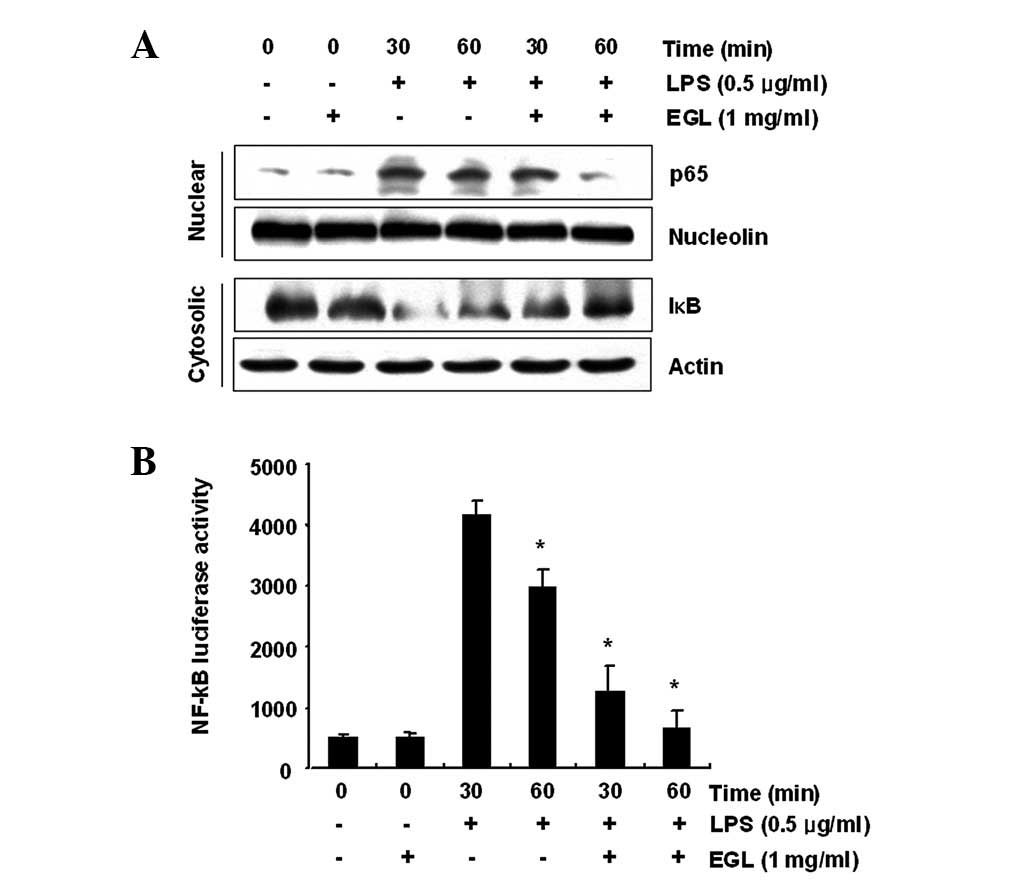
Effects of EGL on LPS-induced NF-κB
activity
Activation of NF-κB is crucial for the induction of
iNOS, COX-2, TNF-α and IL-1β genes in activated BV2 microglia
(28,29). To further characterize the
mechanism through which EGL inhibits pro-inflammatory and/or
cytotoxic factor expression, the ability of EGL to prevent
translocation of the NF-κB p65 subunit to the nucleus was first
examined. Western blot analysis revealed that the amount of NF-κB
p65 in the nucleus markedly increased following exposure to LPS
alone; however, the LPS-induced p65 level in the nuclear fraction
was reduced following EGL pre-treatment (Fig. 6A). In addition, the ability of EGL
to block the LPS-stimulated degradation of IκB-α was investigated
by western blotting. As shown in Fig.
6A, IκB-α was markedly degraded at 30 min after LPS treatment;
however, this LPS-induced IκB-α degradation was significantly
reversed by EGL. Furthermore, the inhibition of LPS-induced NF-κB
activation by EGL was confirmed using a luciferase assay. BV2 cells
transfected with NF-κB-luciferase reporter plasmids were
pre-treated with EGL for 1 h then, following stimulation with LPS
for 6 h, luciferase activity was measured. As shown in Fig. 6B, LPS significantly enhanced the
NF-κB activity up to ∼8-fold over the basal level, whereas EGL
significantly inhibited the LPS-induced NF-κB activity. These
findings show that the anti-inflammatory effect of EGL in
LPS-stimulated BV2 cells involves the NF-κB pathway.
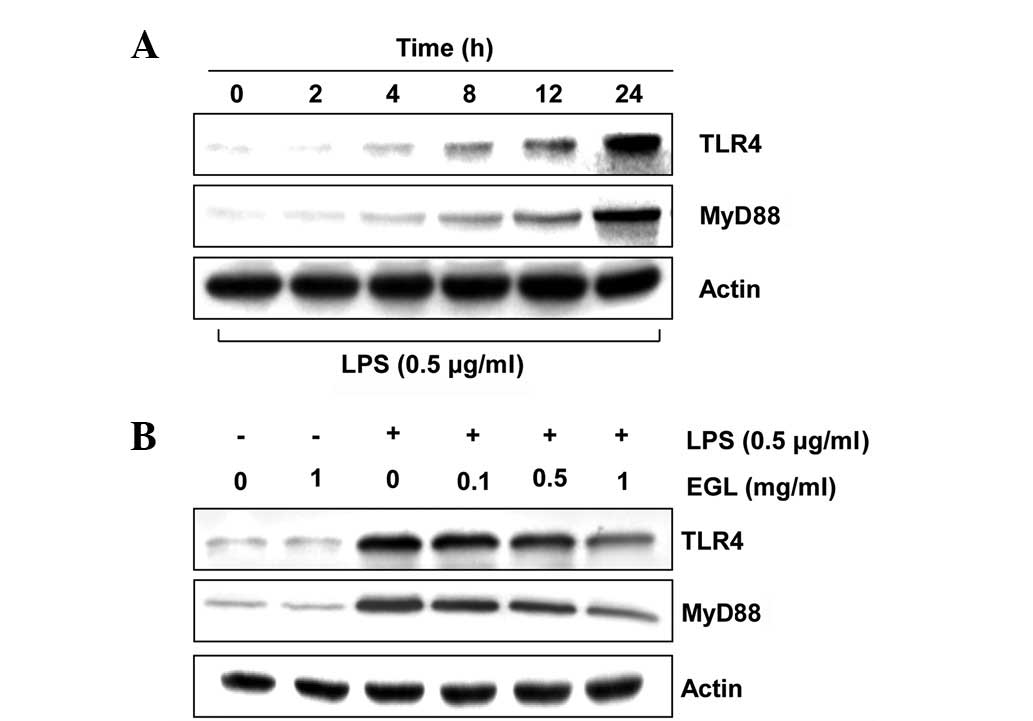
Effects of EGL on the levels of
LPS-induced TLR4 and MyD88
Previous studies established that the inflammatory
response to LPS completely relies on the presence of TLR4, which
triggers the intracellular association of MyD88 with its cytosolic
domain (30–32). To investigate whether the
anti-inflammatory activity of EGL is associated with the modulation
of these proteins, the effects of EGL on LPS-induced upregulation
of TLR4 and MyD88 in BV2 cells were examined. As shown in Fig. 7, EGL concentration-dependently
attenuated the LPS-stimulated increase in TLR4 and MyD88
expression. This finding indicates that EGL is capable of
disrupting key signal transduction pathways, including TLR
signaling pathways activated by LPS in BV2 microglia, which
subsequently prevents the production of pro-inflammatory mediators
and cytokines.
Discussion
G. lucidum is an edible basidiomycete
white-rot macrofungus mushroom that has long been prescribed to
prevent and treat various human diseases, particularly in Asian
countries (1–4). Extensive study has been carried out
on the therapeutic potential of G. lucidum. The exact
ingredients in G. lucidum have not yet been identified;
however, the major active ingredients include polysaccharides,
triterpenoids, unsaturated fatty acids and ergosterol (2,33–35).
Although previous reports have indicated that G. lucidum
extracts exhibit inhibitory actions on neurotoxicity (12–15),
the anti-inflammatory and related immune responses remain poorly
understood. Therefore, we investigated the inhibitory effects of
EGL on the production of LPS-stimulated pro-inflammatory mediators
and cytokines in BV2 microglia to evaluate the cellular and
molecular mechanisms of the anti-inflammatory effects of this
traditional medicine.
COXs are enzymes that catalyze the conversion of
arachidonic acid to PGH2, which is the precursor of a
variety of biologically active mediators, including
PGE2, prostacyclin and thromboxane A2. COXs
exist as two major isozymes: COX-1, a constitutive COX, and COX-2,
an isoform that is induced during the responses to a number of
stimulants and is activated at the inflammation site (36,37).
Several studies have reported that COX-2 is associated with
cytotoxicity in brain diseases, since the inhibition of COX-2
induction and/or activity reduces brain injury following ischemia
and slows the progression of AD and PD (38). Additionally, NO is an important
regulatory molecule in diverse physiological functions, including
vasodilation, neural communication and host defense (39,40).
In mammalian cells, NO is synthesized from three different isoforms
of NOS, including endothelial NOS, neuronal NOS and iNOS. Activated
microglial cells are a major cellular source of iNOS in the brain
and the excessive release of NO by activated microglia is
correlated with the progression of neurodegenerative disorders. The
major producers of TNF-α in the brain are microglia and they may
play a role in a number of pathological conditions of the brain
(17,41,42).
Therefore, TNF-α overexpression has been implicated in the
pathogenesis of several human CNS disorders (18,43,44).
IL-1β is also a potent pro-inflammatory cytokine that acts through
IL-1 receptors on numerous cell types, including neurons and
microglia. Moreover, IL-1β is an important mediator of neuroimmune
interactions that participate directly in neurodegeneration
(45). Thus, inhibiting
inflammatory mediator and cytokine production or function serves as
a key mechanism for identifying a treatment for inflammatory
diseases, including brain injury. In the present study, EGL
significantly suppressed LPS-stimulated PGE2 and NO
production in BV2 microglia in a concentration-dependent manner
without affecting cell viability (Figs. 1 and 2), which appeared to be due to the
transcriptional suppression of COX-2 and iNOS (Fig. 3). Our data also indicate that
treatment with EGL prior to LPS significantly attenuates the
production of the inflammatory cytokines TNF-α and IL-1β, by
inhibiting their expression at the transcriptional level (Figs. 4 and 5). These results suggest that the
anti-inflammatory activity of EGL may contribute to the suppressive
effects of COX-2, iNOS, TNF-α and IL-1β in LPS-activated BV2
microglia.
NF-κB, as a result of its key role in several
pathological conditions, is a major drug target for a variety of
diseases. NF-κB is also a primary regulator of genes that are
involved in the production of pro-inflammatory cytokines and
enzymes involved in the process of inflammation (28,29).
NF-κB is normally located in the cytoplasm where it is complexed
with an inhibitory IκB protein. In response to pro-inflammatory
stimuli, IκB is phosphorylated and subsequently degraded. NF-κB is
then released and translocated to the nucleus where it promotes the
expression of inflammation-related genes. Moreover, blocking the
transcriptional activity of NF-κB in microglial nuclei also
suppresses the expression of iNOS, COX-2 and pro-inflammatory
cytokines, including IL-1β and TNF-α (46,47).
Our results indicate that EGL inhibits LPS-induced IκB-α
degradation, nuclear translocation of the NF-κB p65 subunit and
NF-κB transcriptional activity in BV2 microglia (Fig. 6). Therefore, inhibition of the
NF-κB signaling pathway in microglia by EGL may result in the
downregulation of pro-inflammatory mediators, resulting in an
anti-inflammatory effect.
TLR family members are receptors of the innate
immune system that recognize pathogen-associated molecular
patterns. Among them, TLR4 acts as a major LPS signaling receptor,
leading to the activation of key transcription factors, including
NF-κB and activator protein (AP)-1, which, in turn, enhance the
synthesis of effector inflammatory genes, including cytokines and
chemokines (30–32,48).
Stimulation of the TLR4 extracellular domain by LPS or endotoxins
sequentially triggers the intracellular association of MyD88 with
its cytosolic domain (30,49). Therefore, MyD88 serves as a key
TLR4 adaptor protein, linking the receptors to downstream kinases,
suggesting that TLR4 and MyD88 act as specific targets for
inflammatory responses (30,48,50).
Our data clearly demonstrate that EGL markedly inhibits LPS-induced
TLR4 and MyD88 expression in BV2 microglia (Fig. 7). These results suggest that TLR4
and MyD88 are involved in the inhibitory effects of EGL on the
LPS-induced expression of NO, PGE2 and pro-inflammatory
cytokines.
In summary, we identified that EGL significantly
attenuates the LPS-induced release of inflammatory mediators and
cytokines, including NO, PGE2, TNF-α and IL-1β.
Moreover, it acts at the transcriptional level in BV2 microglia.
The anti-inflammatory action of EGL is mediated by the prevention
of the activation of NF-κB and inhibition of IκB-degradation and
possibly by inhibition of the TLR signaling pathway. Thus, EGL may
provide an effective treatment for a number of neurodegenerative
diseases; however, the pharmacology and mode of action of its
active components require further investigation.
Acknowledgements
This study was supported by a research
grant from Dong-A University, Republic of Korea.
References
|
1.
|
Mahajna J, Dotan N, Zaidman BZ, Petrova RD
and Wasser SP: Pharmacological values of medicinal mushrooms for
prostate cancer therapy: the case of Ganoderma lucidum. Nutr
Cancer. 61:16–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Boh B, Berovic M, Zhang J and Zhi-Bin L:
Ganoderma lucidum and its pharmaceutically active compounds.
Biotechnol Annu Rev. 13:265–301. 2007. View Article : Google Scholar
|
|
3.
|
Paterson RR: Ganoderma - a
therapeutic fungal biofactory. Phytochemistry. 67:1985–2001. 2006.
View Article : Google Scholar
|
|
4.
|
Yuen JW and Gohel MD: Anticancer effects
of Ganoderma lucidum: a review of scientific evidence. Nutr
Cancer. 53:11–17. 2005.
|
|
5.
|
Chien CM, Cheng JL, Chang WT, Tien MH,
Tsao CM, Chang YH, Chang HY, Hsieh JF, Wong CH and Chen ST:
Polysaccharides of Ganoderma lucidum alter cell
immunophenotypic expression and enhance CD56+ NK-cell
cytotoxicity in cord blood. Bioorg Med Chem. 12:5603–5609.
2004.PubMed/NCBI
|
|
6.
|
Hu H, Ahn NS, Yang X, Lee YS and Kang KS:
Ganoderma lucidum extract induces cell cycle arrest and
apoptosis in MCF-7 human breast cancer cell. Int J Cancer.
102:250–253. 2002. View Article : Google Scholar
|
|
7.
|
Lin SB, Li CH, Lee SS and Kan LS:
Triterpene-enriched extracts from Ganoderma lucidum inhibit
growth of hepatoma cells via suppressing protein kinase C,
activating mitogen-activated protein kinases and G2-phase cell
cycle arrest. Life Sci. 72:2381–2390. 2003.PubMed/NCBI
|
|
8.
|
Hong KJ, Dunn DM, Shen CL and Pence BC:
Effects of Ganoderma lucidum on apoptotic and
anti-inflammatory function in HT-29 human colonic carcinoma cells.
Phytother Res. 18:768–770. 2004.
|
|
9.
|
Stanley G, Harvey K, Slivova V, Jiang J
and Sliva D: Ganoderma lucidum suppresses angiogenesis
through the inhibition of secretion of VEGF and TGF-β1 from
prostate cancer cells. Biochem Biophys Res Commun. 330:46–52. 2005.
View Article : Google Scholar
|
|
10.
|
Müller CI, Kumagai T, O’Kelly J, Seeram
NP, Heber D and Koeffler HP: Ganoderma lucidum causes
apoptosis in leukemia, lymphoma and multiple myeloma cells. Leuk
Res. 30:841–848. 2006.
|
|
11.
|
Jang KJ, Han MH, Lee BH, Kim BW, Kim CH,
Yoon HM and Choi YH: Induction of apoptosis by ethanol extracts of
Ganoderma lucidum in human gastric carcinoma cells. J
Acupunct Meridian Stud. 3:24–31. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Cheung WM, Hui WS, Chu PW, Chiu SW and Ip
NY: Ganoderma extract activates MAP kinases and induces the
neuronal differentiation of rat pheochromocytoma PC12 cells. FEBS
Lett. 486:291–296. 2000. View Article : Google Scholar
|
|
13.
|
Zhao HB, Lin SQ, Liu JH and Lin ZB:
Polysaccharide extract isolated from Ganoderma lucidum
protects rat cerebral cortical neurons from hypoxia/reoxygenation
injury. J Pharmacol Sci. 95:294–298. 2004.PubMed/NCBI
|
|
14.
|
Lai CS, Yu MS, Yuen WH, So KF, Zee SY and
Chang RC: Antagonizing beta-amyloid peptide neurotoxicity of the
anti-aging fungus Ganoderma lucidum. Brain Res.
1190:215–224. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Zhou ZY, Tang YP, Xiang J, Wua P, Jin HM,
Wang Z, Mori M and Cai DF: Neuroprotective effects of water-soluble
Ganoderma lucidum polysaccharides on cerebral ischemic
injury in rats. J Ethnopharmacol. 131:154–164. 2010.PubMed/NCBI
|
|
16.
|
Perry VH and Gordon S: Macrophages and
microglia in the nervous system. Trends Neurosci. 92:273–274.
1998.
|
|
17.
|
Sawada M, Kondo N, Suzumura A and
Marunouchi T: Production of tumor necrosis factor-alpha by
microglia and astrocytes in culture. Brain Res. 491:394–397. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Meda L, Cassatella MA, Szendrei GI, Otvos
L Jr, Baron P, Villalba M, Ferrari D and Rossi F: Activation of
microglial cells by β-amyloid protein and interferon-γ. Nature.
373:647–650. 1995.
|
|
19.
|
Dandona P, Ghanim H, Chaudhuri A, Dhindsa
S and Kim SS: Macronutrient intake induces oxidative and
inflammatory stress: potential relevance to atherosclerosis and
insulin resistance. Exp Mol Med. 42:245–253. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Gonzalez-Scarano F and Baltuch G:
Microglia as mediators of inflammatory and degenerative diseases.
Annu Rev Neurosci. 22:219–240. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Amor S, Puentes F, Baker D and van der
Valk P: Inflammation in neurodegenerative diseases. Immunology.
129:154–169. 2010. View Article : Google Scholar
|
|
22.
|
Dheen ST, Kaur C and Ling EA: Microglial
activation and its implications in the brain diseases. Curr Med
Chem. 14:1189–1197. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Eikelenboom P and van Gool WA:
Neuroinflammatory perspectives on the two faces of Alzheimer’s
disease. J Neural Transm. 111:281–294. 2004.PubMed/NCBI
|
|
24.
|
Gao HM, Liu B, Zhang W and Hong JS: Novel
anti-inflammatory therapy for Parkinson’s disease. Trends Pharmacol
Sci. 24:395–401. 2003.
|
|
25.
|
Liu B and Hong JS: Role of microglia in
inflammation-mediated neurodegenerative diseases: mechanisms and
strategies for therapeutic intervention. J Pharmacol Exp Ther.
304:1–7. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Bae DS, Kim YH, Pan CH, Nho CW, Samdan J,
Yansan J and Lee JK: Protopine reduces the inflammatory activity of
lipopolysaccharide-stimulated murine macrophages. BMB Rep.
45:108–113. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Jin CY, Moon DO, Lee KJ, Kim MO, Lee JD,
Choi YH, Park YM and Kim GY: Piceatannol attenuates
lipopolysaccharide-induced NF-kappaB activation and
NF-kappaB-related proinflammatory mediators in BV2 microglia.
Pharmacol Res. 54:461–467. 2006. View Article : Google Scholar
|
|
28.
|
Lee JW, Lee MS, Kim TH, Lee HJ, Hong SS,
Noh YH, Hwang BY, Ro JS and Hong JT: Inhibitory effect of
inflexinol on nitric oxide generation and iNOS expression via
inhibition of NF-κB activation. Mediators Inflamm.
2007:93148–93157. 2007.PubMed/NCBI
|
|
29.
|
Baima ET, Guzova JA, Mathialagan S, Nagiec
EE, Hardy MM, Song LR, Bonar SL, Weinberg RA, Selness SR, Woodard
SS, Chrencik J, Hood WF, Schindler JF, Kishore N and Mbalaviele G:
Novel insights into the cellular mechanisms of the
anti-inflammatory effects of NF-κB essential modulator binding
domain peptides. J Biol Chem. 285:13498–13506. 2010.
|
|
30.
|
Miller SI, Ernst RK and Bader MW: LPS,
TLR4 and infectious disease diversity. Nat Rev Microbiol. 3:36–46.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Lee YH, Jeon SH, Kim SH, Kim C, Lee SJ,
Koh D, Lim Y, Ha K and Shin SY: A new synthetic chalcone
derivative, 2-hydroxy-3′,5,5′-trimethoxychalcone (DK-139),
suppresses the Toll-like receptor 4-mediated inflammatory response
through inhibition of the Akt/NF-κB pathway in BV2 microglial
cells. Exp Mol Med. 44:369–377. 2012.PubMed/NCBI
|
|
32.
|
Romanovsky AA, Steiner AA and Matsumura K:
Cells that trigger fever. Cell Cycle. 5:2195–2197. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Fukuzawa M, Yamaguchi R, Hide I, Chen Z,
Hirai Y, Sugimoto A, Yasuhara T and Nakata Y: Possible involvement
of long chain fatty acids in the spores of Ganoderma lucidum
(Reishi Houshi) to its anti-tumor activity. Biol Pharm Bull.
31:1933–1937. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Sanodiya BS, Thakur GS, Baghel RK, Prasad
GB and Bisen PS: Ganoderma lucidum: a potent pharmacological
macrofungus. Curr Pharm Biotechnol. 10:717–742. 2009. View Article : Google Scholar
|
|
35.
|
Xu Z, Chen X, Zhong Z, Chen L and Wang Y:
Ganoderma lucidum polysaccharides: immunomodulation and
potential anti-tumor activities. Am J Chin Med. 39:15–27. 2011.
View Article : Google Scholar
|
|
36.
|
Vane JR, Mitchell JA, Appleton I,
Tomlinson A, Bishop-Bailey D, Croxtall J and Willoughby DA:
Inducible isoforms of cyclooxygenase and nitric-oxide synthase in
inflammation. Proc Natl Acad Sci USA. 91:2046–2050. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Hawkey CJ: Cox-2 inhibitors. Lancet.
353:307–314. 1999. View Article : Google Scholar
|
|
38.
|
Giovannini MG, Scali C, Prosperi C,
Bellucci A, Pepeu G and Casamenti G: Experimental brain
inflammation and neurode-generation as model of Alzheimer’s
disease: protective effects of selective COX-2 inhibitors. Int J
Immunopathol Pharmacol. 16:31–40. 2003.
|
|
39.
|
MacMicking J, Xie QW and Nathan C: Nitric
oxide and macrophage function. Ann Rev Immunol. 15:323–330. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Mitchell JA, Larkin S and Williams TJ:
Cyclooxygenase-2: regulation and relevance in inflammation. Biochem
Pharmacol. 50:1535–1542. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Boka G, Anglade P, Wallach D, Javoy-Agid
F, Agid Y and Hirsch EC: Immunocytochemical analysis of tumor
necrosis factor and its receptors in Parkinson’s disease. Neurosci
Lett. 172:151–154. 1994.PubMed/NCBI
|
|
42.
|
Murphy S: Production of nitric oxide by
glial cells: regulation and potential roles in the CNS. Glia.
29:1–13. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Akassoglou K, Bauer J, Kassiotis G,
Pasparakis M, Lassmann H, Kollias G and Probert L: Oligodendrocyte
apoptosis and primary demyelination induced by local TNF/p55TNF
receptor signaling in the central nervous system of transgenic
mice: models for multiple sclerosis with primary
oligodendrogliopathy. Am J Pathol. 153:801–813. 1998. View Article : Google Scholar
|
|
44.
|
Sriram K, Matheson JM, Benkovic SA, Miller
DB, Luster MI and O’Callaghan JP: Mice deficient in TNF receptors
are protected against dopaminergic neurotoxicity: implications for
Parkinson’s disease. FASEB J. 16:1474–1476. 2002.PubMed/NCBI
|
|
45.
|
Rothwell N, Allan S and Toulmond S: The
role of interleukin 1 in acute neurodegeneration and stroke:
pathophysiological and therapeutic implications. J Clin Invest.
100:2648–2652. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Baldwin AS Jr: The NF-kappaB and I-kappaB
proteins: new discoveries and insights. Ann Rev Immunol.
14:649–683. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Moon DO, Choi YH, Kim ND, Park YM and Kim
GY: Anti-inflammatory effects of β-lapachone in
lipopolysaccharide-stimulated BV2 microglia. Int Immunopharmacol.
7:506–514. 2007.
|
|
48.
|
Trinchieri G and Sher A: Cooperation of
Toll-like receptor signals in innate immune defence. Nat Rev
Immunol. 7:179–190. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Medzhitov R: Toll-like receptors and
innate immunity. Nat Rev Immunol. 1:135–145. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Seki E, Tsutsui H, Iimuro Y, Naka T, Son
G, Akira S, Kishimoto T, Nakanishi K and Fujimoto J: Contribution
of Toll-like receptor/myeloid differentiation factor 88 signaling
to murine liver regeneration. Hepatology. 41:443–450. 2005.
View Article : Google Scholar : PubMed/NCBI
|