Introduction
Cardiac hypertrophy occurs when the heart endures
overload or injury. Although hypertrophy is an adaptive process
that initially maintains cardiac output, sustained hypertrophy
ultimately leads to heart failure, which is the leading cause of
morbidity and mortality worldwide (1,2).
Various stimuli, including mechanical stress and neurohumoral
factors, such as angiotensin II (Ang II), endothelin-1,
catecholamine and growth factors, are involved in the progression
of cardiac hypertrophy (3). These
stimuli activate membrane receptors and intracellular signaling
pathways to mediate the transcription of hypertrophy-related genes
(4). Enlargement and apoptotic
loss of cardiomyocytes are the key pathological changes in cardiac
hypertrophy (5). To prevent
cardiomyocytes from enlargement and cell death, blocking the
transition between adaptive hypertrophy and heart failure is
necessary. However, the existing treatments that modify
hemodynamics and inhibit active neurohumoral factors are not
capable of successfully restoring the injured cardiomyocytes.
Disrupting the intracellular signaling pathways that mediate
cardiac hypertrophy has been increasingly studied and novel targets
have been identified that may be used to explore therapeutic
strategies for cardiac hypertrophy and heart failure.
Icariin (C33H40O15;
molecular weight, 676.66), a prenylated flavonol glycoside, is the
major active component isolated from plants of the Epimedium family
(6). Multiple pharmacological
properties of icariin have been revealed, including
immunoregulation, antioxidative stress, antiapoptosis and
stimulation of angiogenesis (6–9).
Song et al (8) identified
that icariin attenuated cardiac remodeling in rats with congestive
heart failure by inhibiting matrix metalloproteinase (MMP) activity
and protecting cardiomyocytes from apoptosis. This result
demonstrates the cardiac protective role of icariin. However, it is
not known whether icariin has a direct effect on cardiomyocytes and
the mechanism underlying its cardiac protective role remains
unclear.
Ang II functions as a significant hormonal mediator
in cardiac hypertrophy that can induce a direct injury on
cardiomyocytes. Reactive oxygen species (ROS)-dependent activation
of the c-Jun N-terminal kinase (JNK) and p38 pathways has been
shown to play a critical role in the effect Ang II exhibits on
cardiomyocytes (10). A previous
study demonstrated that icariin inhibits the production of ROS and
blocks the activity of the JNK and p38 pathways in
lipopolysaccaride (LPS)-treated microglial cells (11). However, the effect of icariin on
Ang II-induced cardiomyocyte injury and the underlying mechanisms
remain unknown. In the present study, a hypertrophic model was used
in Ang II-stimulated H9c2 cardiomyocytes. The aims were to
determine whether icariin treatment directly prevented
cardiomyocytes from hypertrophy and apoptosis and to determine
whether the cardioprotective effect of icariin was mediated via the
inhibition of the ROS-dependent JNK and p38 pathways.
Materials and methods
Reagents
Icariin (≥94% purity as determined by high
performance liquid chromatography analysis), Ang II and
2′,7′-dichlorofluorescein diacetate (DCFH-DA) were purchased from
Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s modified Eagle’s
medium: Nutrient mixture F-12 (DMEM/F12), fetal bovine serum (FBS),
trypsin, penicillin and streptomycin were purchased from Gibco-BRL
(Carlsbad, CA, USA). TRIzol, Alexa Fluor® 488 goat
anti-mouse IgG and SlowFade Gold antifade reagent with
4′,6-diamidino-2-phenylindole (DAPI) were purchased from Invitrogen
Life Technologies (Carlsbad, CA, USA). A Transcriptor First Strand
cDNA synthesis kit and Light Cycler 480 SYBR Green 1 Master Mix
were purchased from Roche Diagnostics (Basel, Switzerland).
Antibodies against α-actinin and an ApopTag® Plus
Fluorescein In Situ Apoptosis detection kit were purchased from
Millipore Corporation (Billerica, MA, USA). Primary antibodies were
purchased from Cell Signaling Technology, Inc. (Beverley, MA, USA)
and IRDye 800CW conjugated secondary antibodies were obtained from
LI-COR Biosciences (Lincoln, NE, USA).
H9c2 cardiomyocyte culture
The H9c2 embryonic rat heart-derived cell line was
obtained from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China). Icariin was dissolved in dimethyl sulfoxide at a
concentration of 10 mmol/l for storage. Cells were cultured in
DMEM/F12 1:1 medium, supplemented with 10% FBS, 100 U/ml penicillin
and 100 mg/ml streptomycin, in a humidified incubator with an
atmosphere of 5% CO2 at 37°C. Cells were seeded at a
density of 1×106 cells per well into six-well culture
plates for mRNA extraction, 5×105 cells per well into
six-well culture plates for cell surface area (CSA) and terminal
deoxynucleotidyl transferase-mediated dUTP nick end-labeling
(TUNEL) analysis, 5×103 cells per well in 96-well plates
for ROS detection and 1×107 cells per well into 100 mm
culture dishes for protein extraction. The cells were cultured in
serum-free DMEM/F12 1:1 medium for 24 h and pretreated with icariin
for 1 h prior to stimulation with Ang II.
Cell viability
Cell viability was analyzed using the Cell Counting
Kit-8 (CCK-8) assay. Following icariin treatment for 48 h, 10 μl
CCK-8 solution was added to each well of the 96-well plate and then
incubated for an additional 4 h. Absorbance was measured at 450 nm
using a microplate reader (Synergy HT; BioTek, Winooski, VT, USA).
The percentage of cell viability was calculated according to the
following formula: Cell viability (%) = optical density (OD) of the
treatment group/OD of the control group × 100%.
Quantitative polymerase chain reaction
(qPCR)
To detect the mRNA expression levels of hypertrophic
markers, including atrial natriuretic peptide (ANP) and B-type
natriuretic peptide (BNP), qPCR was performed as described
previously (12). Total RNA was
extracted from cultured H9c2 cells using TRIzol and 2-μg samples of
RNA were reverse-transcribed into cDNA using the Transcriptor First
Strand cDNA synthesis kit. PCR amplifications were quantified using
a LightCycler 480 SYBR Green 1 Master Mix and GAPDH was used as the
internal control.
CSA analysis
To assess CSA, cells were stained by
immunofluorescence for cardiac α-actinin (13). The cells were washed with
phosphate-buffered saline (PBS), fixed with RCL2 fixing liquid and
permeabilized in 0.1% Triton X-100 in PBS. The cells were stained
with anti-α-actinin at a dilution of 1:100 in 1% goat serum
overnight at 4°C, and then incubated with Alexa Fluor®
488 goat anti-mouse IgG for 1 h at 37°C. Cells on the coverslips
were mounted onto glass slides with SlowFade Gold antifade reagent
with DAPI and CSAs were measured using a quantitative digital image
analysis system (Image Pro-Plus version 6.0; Media Cybernetics,
Inc., Rockville, MD, USA).
TUNEL staining
Apoptotic nuclei were labeled using TUNEL staining
with a ApopTag® Plus Fluorescein In Situ Apoptosis
Detection kit, according to the manufacturer’s instructions
(14). Cells on the coverslips
were fixed with 1% paraformaldehyde in PBS, stained with TUNEL
reagents and the nuclei were stained with DAPI. The cell apoptotic
index was calculated as the percentage of apoptotic nuclei/total
number of nuclei.
ROS detection
Intracellular ROS generation was determined using
DCFH-DA, which becomes fluorescent on oxidation to DCF by
H2O2 produced within cells. Following Ang II
or/and Icariin treatments, H9c2 cells were washed twice and
incubated with 5 μM DCFH-DA solution in serum-free medium at 37°C
for 30 min in the dark. Data were then collected using a
fluorescent reader (Synergy HT; BioTek) at excitation/emission
wavelengths of 485/530 nm. A fluorescent microscope was also used
to evaluate the DCF fluorescence of the cells on the
coverslips.
Western blotting
Western blotting was performed as described
previously (13). Cells were lysed
in radioimmunoprecipitation assay lysis buffer and 50-μg samples of
the cell lysates were electrophoresed on 10% SDS-PAGE gels. The
proteins were then transferred onto Immobilon-FL transfer membranes
(Millipore Corporation) and blocked with 5% non-fat milk for 2 h.
The membranes were incubated with antibodies specific for
phosphorylated (p)-JNK, p-extracellular signal-related kinase
(ERK), p-p38, total (T)-JNK, T-ERK, T-p38, Bcl-2, Bax,
cleaved-caspase 3 or GAPDH overnight at 4°C. The samples were then
incubated with IRDye 800CW conjugated secondary antibodies and the
blots were scanned by a two-color infrared imaging system (Odyssey,
LI-COR Biosciences).
Statistical analysis
Data are presented as the mean ± SEM and analyzed
using a statistical software (SPSS 16.0; SPSS Inc., Chicago, IL,
USA). Differences among the groups were determined by two-way
analysis of variance followed by Tukey’s post hoc test. A
comparison between the control and all the treatment groups was
performed using the unpaired Student’s t-test. P<0.05 was
considered to indicate a statistically significant difference.
Results
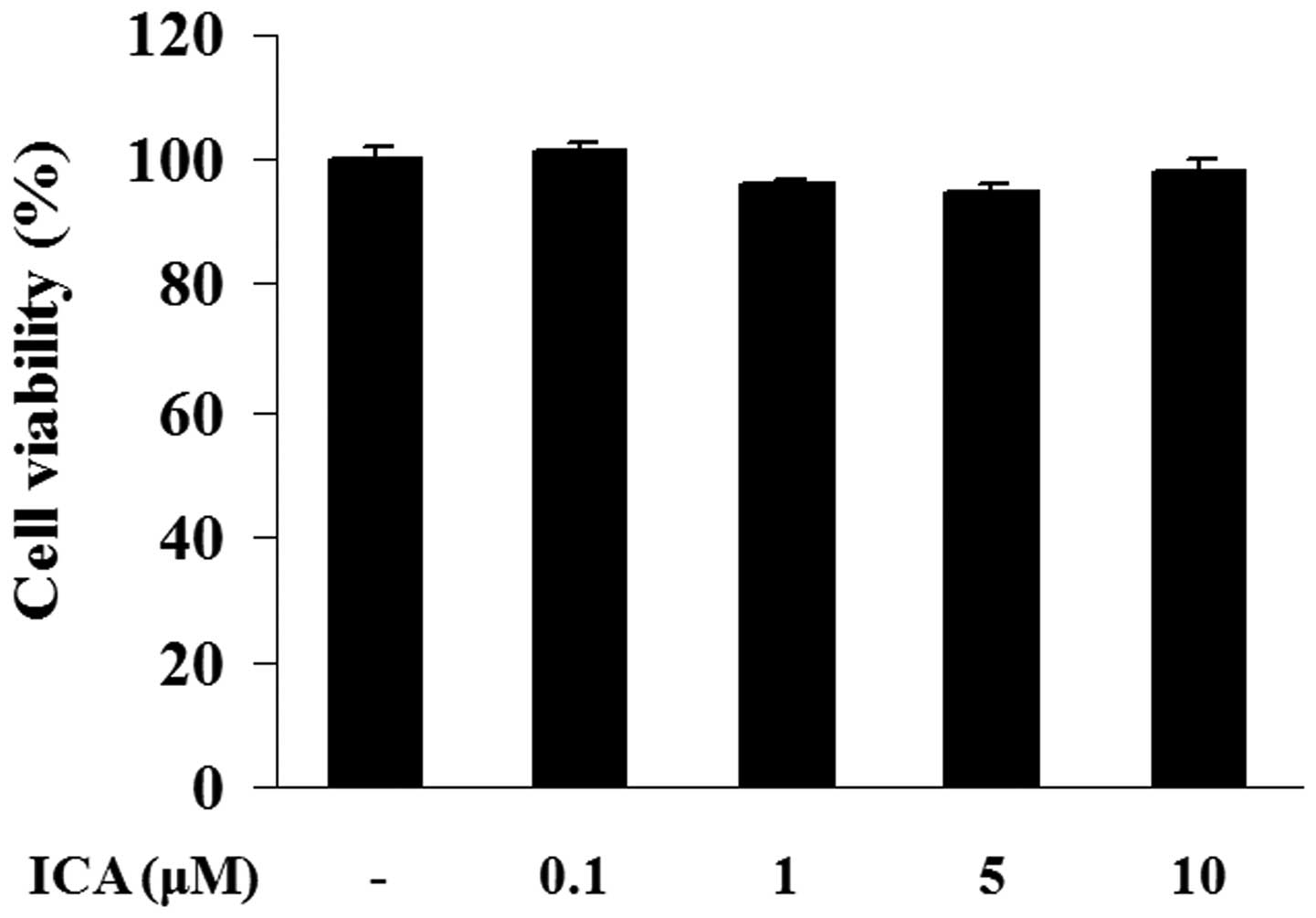
Effect of icariin on cell viability
The potential cytotoxicity of icariin was analyzed
using a CCK-8 assay. H9c2 cells were incubated with various
concentrations of icariin (0.1, 1, 5 or 10 μM) for 48 h. Cell
viability in icariin-treated cells exhibited no significant
differences when compared with the control cells, indicating that
icariin at a concentration of 0.1, 1, 5 or 10 μM did not possess
any cytotoxicity in H9c2 cells (Fig.
1).
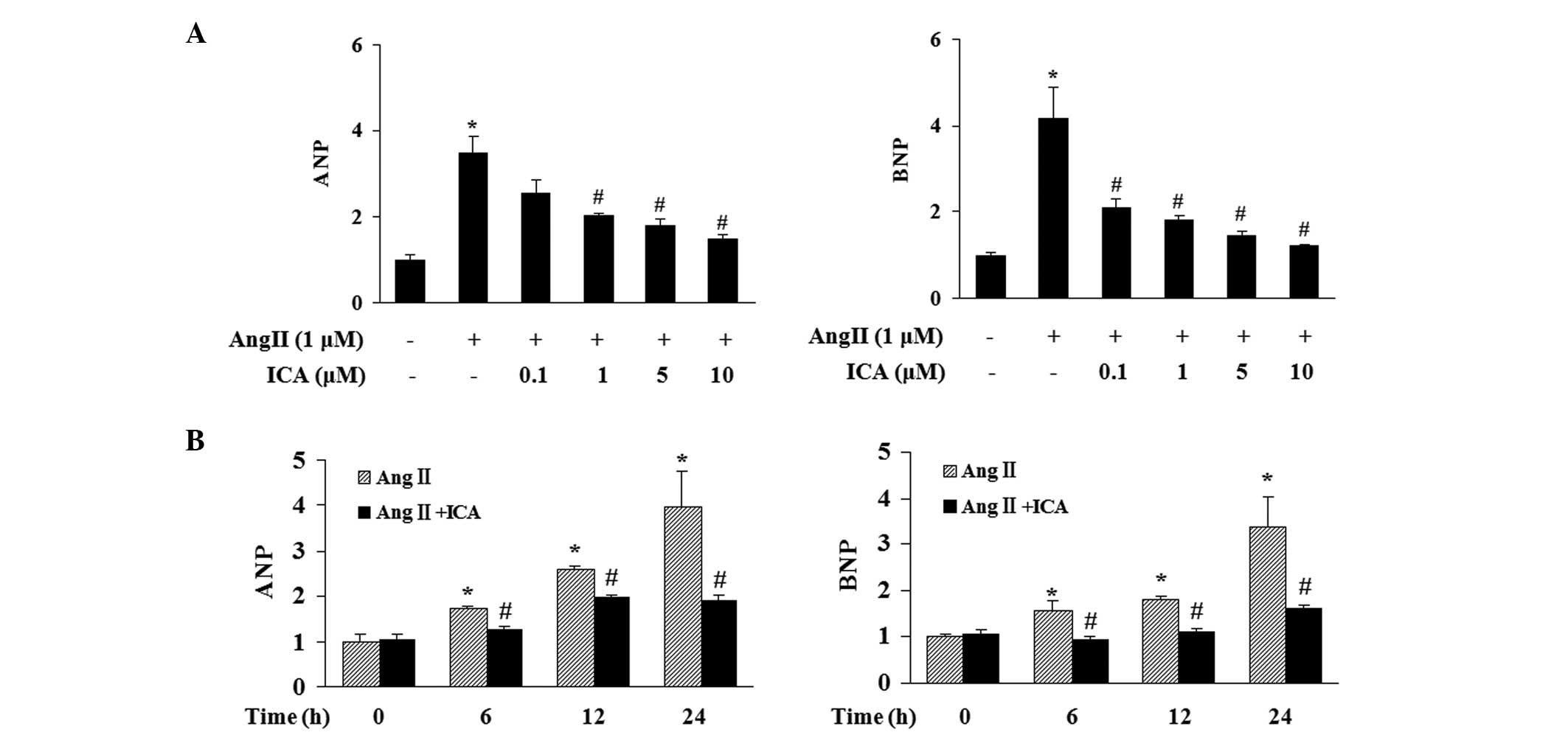
Effect of icariin on ANP and BNP
induction
The effect of icariin at various concentrations
(0.1, 1, 5 or 10 μM) on the induction of ANP and BNP in response to
Ang II was determined. Stimulation with Ang II for 24 h markedly
increased the mRNA expression levels of ANP and BNP in H9c2 cells
and icariin treatment markedly attenuated this increase in a
concentration-dependent manner (Fig.
2A). Icariin at a concentration of 10 μM significantly blocked
the induction of ANP and BNP in response to Ang II at various time
points, thus, this concentration was selected for further
investigations (Fig. 2B).
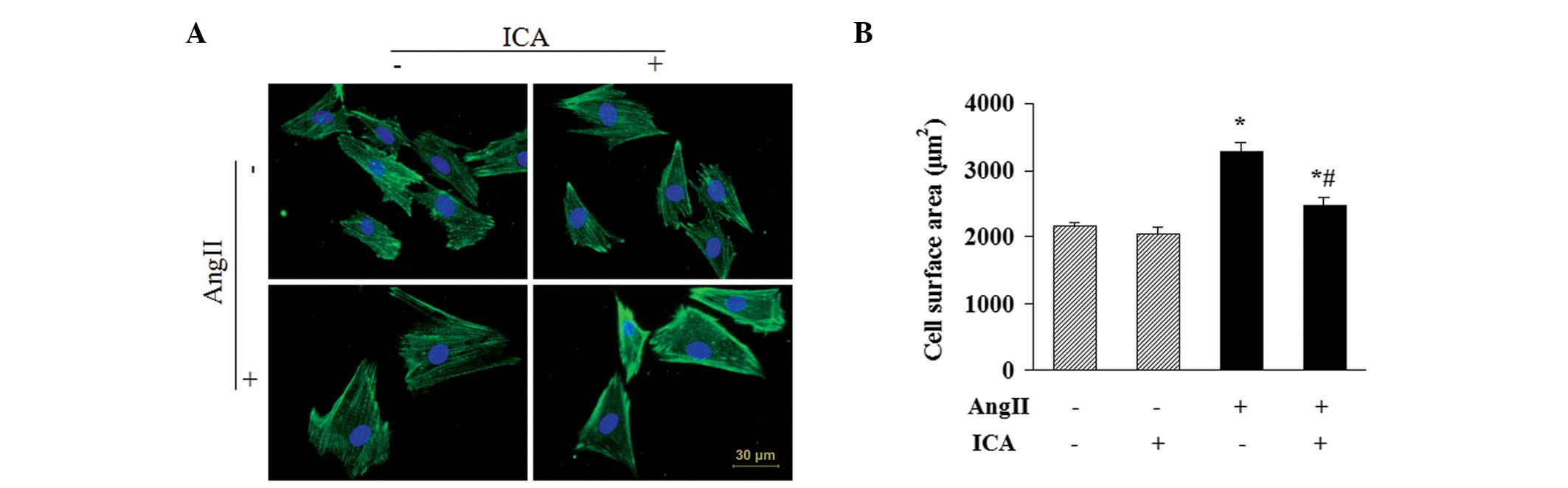
Icariin attenuates the Ang II-induced
increase in CSA
CSAs of H9c2 cells were determined by α-actinin
staining to further evaluate the antihypertrophic effect of
icariin. Ang II stimulation for 48 h resulted in a significant
increase in the CSAs of H9c2 cells. However, icariin treatment
markedly attenuated the increase, indicating that Ang II-induced
enlargement of H9c2 cells was suppressed by icariin (Fig. 3).
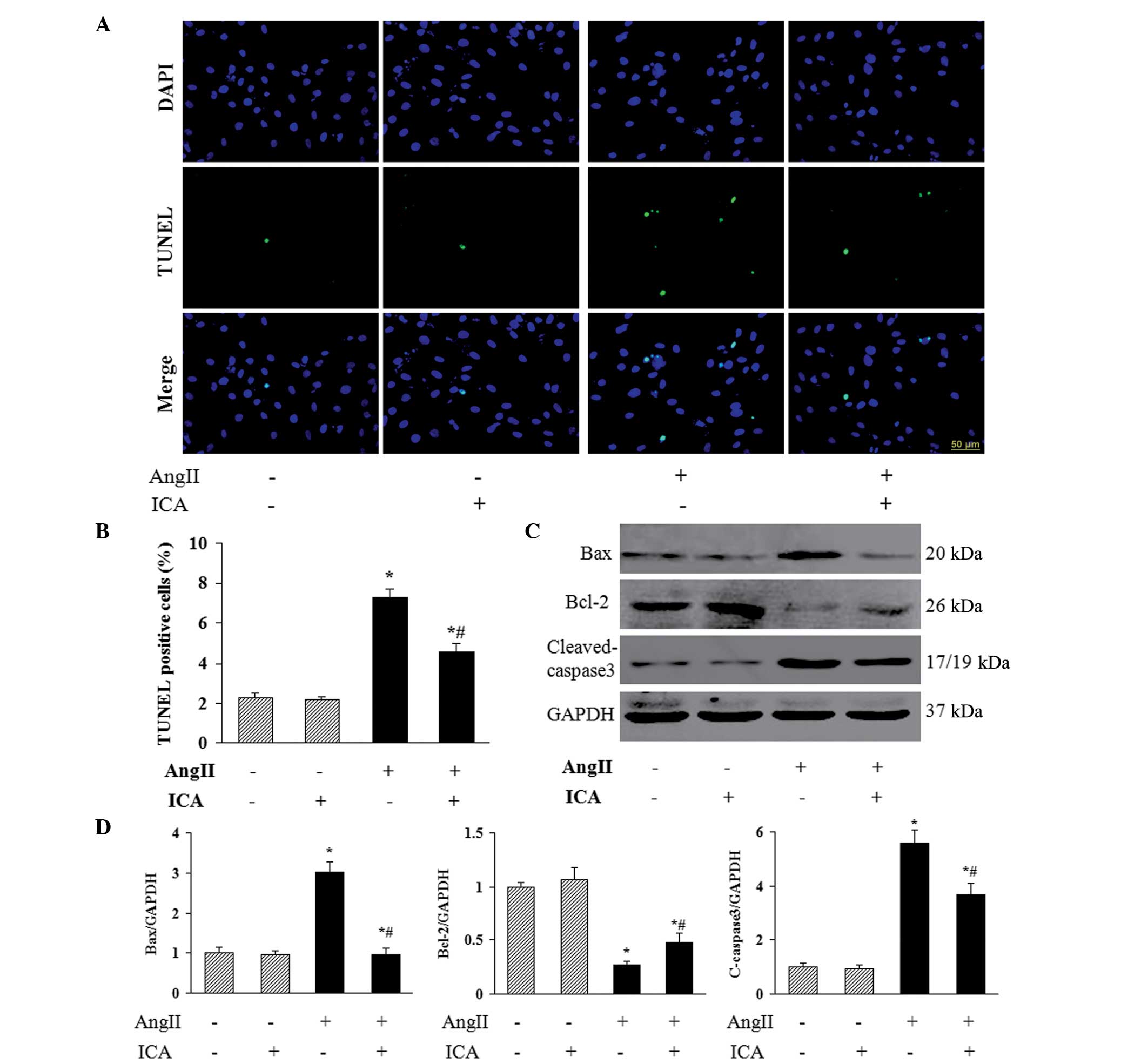
Icariin inhibits Ang II-induced
apoptosis
To investigate the role of icariin in Ang II-induced
apoptosis of H9c2 cells, TUNEL staining was used to identify the
apoptotic nuclei. A marked increase in the number of TUNEL-positive
nuclei was observed in cells that had been incubated with Ang II,
and icariin treatment markedly reduced Ang II-induced cell
apoptosis (Fig. 4A and B). In
addition, icariin decreased the protein expression levels of Bax
and cleaved-caspase 3 in H9c2 cells in response to Ang II (Fig. 4C and D), which may mediate the
antiapoptotic effect of icariin. Furthermore, the decreased level
of antiapoptotic protein Bcl-2 in Ang II-treated H9c2 cells was
restored with icariin treatment (Fig.
4C and D).
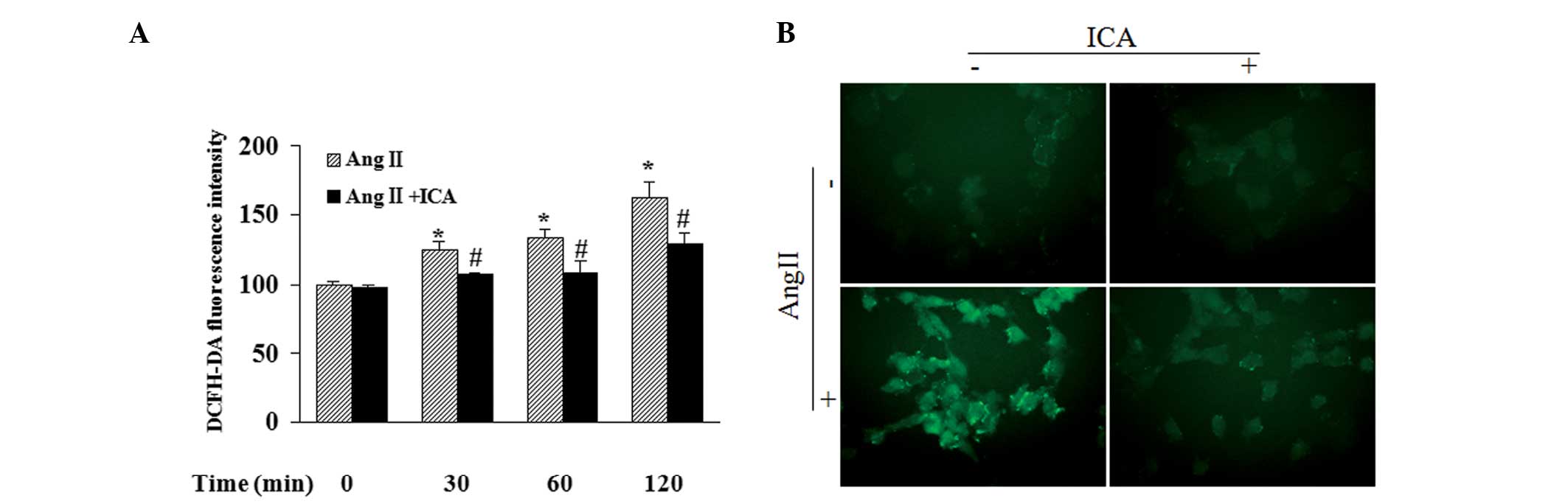
Icariin decreases the production of
ROS
Fluorescence intensity in cells following incubation
with DCFH-DA, exhibited by a fluorescent reader, revealed that Ang
II increased the ROS content in a time-dependent manner. In
addition, icariin treatment markedly blocked Ang II-induced ROS
production at the indicated time points (Fig. 5A). DCF-derived fluorescence
observed with a microscope also demonstrated that icariin inhibited
the accumulation of intracellular ROS in Ang II-treated cells,
which was in accordance with the results of the fluorescent reader
(Fig. 5B).
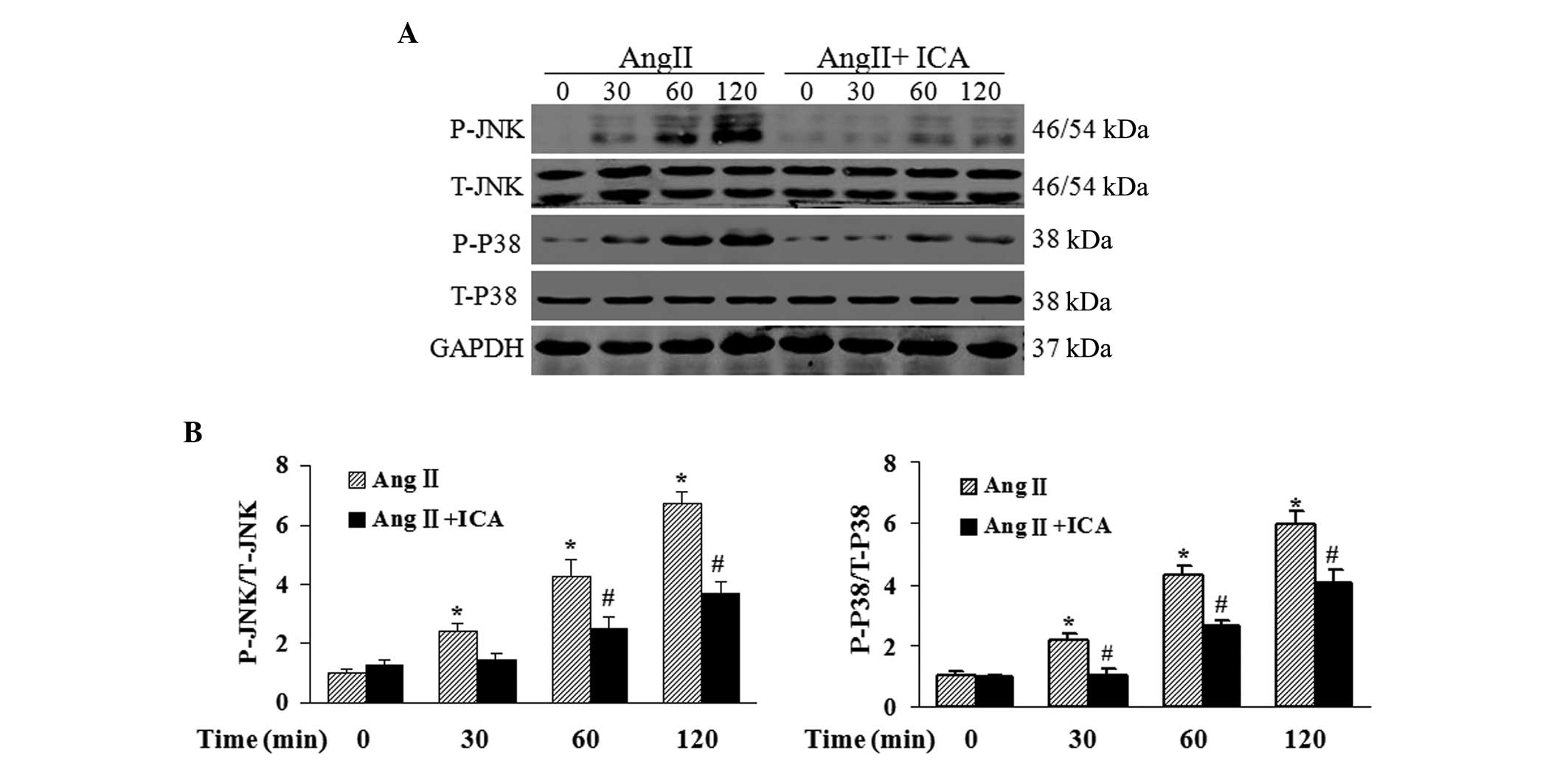
Icariin blocks the activation of JNK and
p38 pathways in response to Ang II
To further explore the mechanisms underlying the
antihypertrophic and antiapoptotic effects of icariin in Ang
II-treated H9c2 cells, western blotting was used to detect the
phosphorylation levels of JNK and p38, which are key mediators of
cardiac hypertrophy and apoptosis. Phosphorylated levels of JNK and
p38 were shown to be markedly elevated by Ang II. However, icariin
treatment inhibited the phosphorylation of JNK and p38 in response
to Ang II at the indicated time points (Fig. 6).
Discussion
Icariin, the major active component isolated from
plants of the Epimedium family, has been reported to exhibit
potential protective effects on the cardiovascular system (8,9).
However, it is not known whether icariin has a direct effect on Ang
II-induced cardiomyocyte injury. In the present study, icariin was
found to protect H9c2 cells from hypertrophy and apoptosis in
response to Ang II. The beneficial effect of icariin may be
mediated by inhibiting the ROS-dependent JNK and p38 pathways.
Enlargement and apoptotic loss of cardiomyocytes
play critical roles in the transition from cardiac hypertrophy to
heart failure (5). H9c2 cells, an
embryonic rat-heart-derived cell line, maintain similar
characteristics to primary cardiomyocytes, including morphology,
protein expression, electrophysiological properties and
hypertrophic responses (15,16).
Ang II functions as a significant hormonal mediator in cardiac
hypertrophy, which can induce pathological growth and apoptosis in
cardiomyocytes (17). In the
current study, an Ang II-induced injury model in H9c2 cells was
used to evaluate the direct protection of icariin on
cardiomyocytes. A previous study demonstrated that icariin
attenuated cardiac remodeling in rats with congestive heart failure
by inhibiting MMP activity and cardiomyocyte apoptosis (8), indicating the protective role of
icariin in the cardiovascular system. However, whether icariin can
provide a direct benefit on cardiomyocytes and the underlying
mechanisms remain unclear. The results of the present study
revealed that icariin directly repressed Ang II-induced H9c2 cell
enlargement and the expression of ANP and BNP, which are considered
to be molecular markers of cardiomyocyte hypertrophy. Furthermore,
icariin blocked apoptosis in Ang II-treated H9c2 cells by
regulating the protein expression of the proapoptotic proteins, Bax
and cleaved-caspase 3, as well as the antiapoptotic protein,
Bcl-2.
Clinical and experimental studies have provided
substantial evidence that ROS production, reflecting the status of
oxidative stress, is enhanced in hypertrophic and failing hearts
(18,19). Ang II, norepinephrine and
mechanical stretch can induce the production of ROS associated with
the hypertrophic response in cardiomyocytes (20–22).
Increased intracellular ROS levels in a hypertrophic heart
contribute to the progression of cardiac remodeling and heart
failure. H2O2, a major source of ROS,
directly induces cardiomyocyte hypertrophy and apoptosis in
vitro (23,24). Therefore, antioxidants that block
ROS production exhibit therapeutic potential in treating cardiac
hypertrophy. Previous studies have shown that icariin attenuated
H2O2-induced neurotoxicity (25) and inhibited ROS production in
LPS-treated microglia (11),
demonstrating the antioxidative effect of icariin. In the present
study, icariin was shown to block the production of ROS in H9c2
cells, which may mediate the cardioprotective effect of
icariin.
Increased ROS levels not only lead to the oxidation
and damage of macromolecules, membranes and DNA, but also function
as a secondary messengers in cellular signaling (26). Activation of JNK and p38, members
of the mitogen-activated protein kinase family, has been reported
to be induced by ROS (27).
Following activation, JNK and p38 phosphorylate a wide array of
intracellular targets, including numerous transcription factors,
resulting in the reprogramming of cardiac gene expression, the
hypertrophic phenotype and apoptosis of cardiomyocytes (28). Inhibition of the JNK or p38
pathways in cardiomyocytes results in attenuated hypertrophic
growth induced by agonist stimulation (29). Previous evidence revealed that Ang
II-induced activation of JNK and p38 in cardiomyocytes is
ROS-dependent (10), indicating
that treatment targeting the production of ROS may suppress the JNK
and p38 pathways and subsequently result in a protective effect on
Ang II-induced injury in cardiomyocytes. The results of the present
study demonstrated that icariin downregulated ROS levels and the
phosphorylation of JNK and p38 in Ang II-treated H9c2 cells. These
observations indicate that icariin protects H9c2 cells from Ang
II-induced hypertrophy and apoptosis via the inhibition of the
ROS-dependent JNK and p38 pathways.
In conclusion, the current study has demonstrated a
previously unknown effect of icariin on Ang II-induced
cardiomyocyte hypertrophy and apoptosis through inhibiting the
ROS-dependent JNK and p38 pathways. The results of the present
study provide experimental evidence for the application of icariin
in the treatment of cardiac hypertrophy.
Acknowledgements
The study was supported by grants from the National
Natural Science Foundation of China (nos. 81300070 and 81270303)
and the Fundamental Research Funds for the Central Universities of
China (no. 2012302020212).
References
|
1
|
Bui AL, Horwich TB and Fonarow GC:
Epidemiology and risk profile of heart failure. Nat Rev Cardiol.
8:30–41. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shah AM and Mann DL: In search of new
therapeutic targets and strategies for heart failure: recent
advances in basic science. Lancet. 378:704–712. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rohini A, Agrawal N, Koyani CN and Singh
R: Molecular targets and regulators of cardiac hypertrophy.
Pharmacol Res. 61:269–280. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
van Berlo JH, Maillet M and Molkentin JD:
Signaling effectors underlying pathologic growth and remodeling of
the heart. J Clin Invest. 123:37–45. 2013.PubMed/NCBI
|
|
5
|
Mann DL, Barger PM and Burkhoff D:
Myocardial recovery and the failing heart: myth, magic, or
molecular target? J Am Coll Cardiol. 60:2465–2472. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu CQ, Liu BJ, Wu JF, et al: Icariin
attenuates LPS-induced acute inflammatory responses: involvement of
PI3K/Akt and NF-kappaB signaling pathway. Eur J Pharmacol.
642:146–153. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li WW, Gao XM, Wang XM, Guo H and Zhang
BL: Icariin inhibits hydrogen peroxide-induced toxicity through
inhibition of phosphorylation of JNK/p38 MAPK and p53 activity.
Mutat Res. 708:1–10. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Song YH, Cai H, Gu N, Qian CF, Cao SP and
Zhao ZM: Icariin attenuates cardiac remodelling through
down-regulating myocardial apoptosis and matrix metalloproteinase
activity in rats with congestive heart failure. J Pharm Pharmacol.
63:541–549. 2011. View Article : Google Scholar
|
|
9
|
Chung BH, Kim JD, Kim CK, et al: Icariin
stimulates angiogenesis by activating the MEK/ERK- and
PI3K/Akt/eNOS-dependent signal pathways in human endothelial cells.
Biochem Biophys Res Commun. 376:404–408. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nishida M, Tanabe S, Maruyama Y, et al: G
alpha 12/13- and reactive oxygen species-dependent activation of
c-Jun NH2-terminal kinase and p38 mitogen-activated
protein kinase by angiotensin receptor stimulation in rat neonatal
cardiomyocytes. J Biol Chem. 280:18434–18441. 2005.PubMed/NCBI
|
|
11
|
Zeng KW, Fu H, Liu GX and Wang XM: Icariin
attenuates lipopolysaccharide-induced microglial activation and
resultant death of neurons by inhibiting TAK1/IKK/NF-kappaB and
JNK/p38 MAPK pathways. Int Immunopharmacol. 10:668–678. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou H, Bian ZY, Zong J, et al: Stem cell
antigen 1 protects against cardiac hypertrophy and fibrosis after
pressure overload. Hypertension. 60:802–809. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhou H, Shen DF, Bian ZY, et al:
Activating transcription factor 3 deficiency promotes cardiac
hypertrophy, dysfunction, and fibrosis induced by pressure
overload. PLoS One. 6:e267442011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou H, Yang HX, Yuan Y, et al:
Paeoniflorin attenuates pressure overload-induced cardiac
remodeling via inhibition of TGFβ/Smads and NF-κB pathways. J Mol
Histol. 44:357–367. 2013.PubMed/NCBI
|
|
15
|
Hescheler J, Meyer R, Plant S, Krautwurst
D, Rosenthal W and Schultz G: Morphological, biochemical, and
electrophysiological characterization of a clonal cell (H9c2) line
from rat heart. Circ Res. 69:1476–1486. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Watkins SJ, Borthwick GM and Arthur HM:
The H9c2 cell line and primary neonatal cardiomyocyte cells show
similar hypertrophic responses in vitro. In Vitro Cell Dev Biol
Anim. 47:125–131. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fortuño MA, Ravassa S, Fortuño A, Zalba G
and Díez J: Cardiomyocyte apoptotic cell death in arterial
hypertension: mechanisms and potential management. Hypertension.
38:1406–1412. 2001.PubMed/NCBI
|
|
18
|
Tsutsui H, Kinugawa S and Matsushima S:
Oxidative stress and heart failure. Am J Physiol Heart Circ
Physiol. 301:H2181–H2190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Anilkumar N, Sirker A and Shah AM: Redox
sensitive signaling pathways in cardiac remodeling, hypertrophy and
failure. Front Biosci (Landmark Ed). 14:3168–3187. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brasier AR, Jamaluddin M, Han Y, Patterson
C and Runge MS: Angiotensin II induces gene transcription through
cell-type-dependent effects on the nuclear factor-kappaB
(NF-kappaB) transcription factor. Mol Cell Biochem. 212:155–169.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Amin JK, Xiao L, Pimental DR, et al:
Reactive oxygen species mediate alpha-adrenergic
receptor-stimulated hypertrophy in adult rat ventricular myocytes.
J Mol Cell Cardiol. 33:131–139. 2001. View Article : Google Scholar
|
|
22
|
Pimentel DR, Amin JK, Xiao L, et al:
Reactive oxygen species mediate amplitude-dependent hypertrophic
and apoptotic responses to mechanical stretch in cardiac myocytes.
Circ Res. 89:453–460. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Essick EE, Ouchi N, Wilson RM, et al:
Adiponectin mediates cardioprotection in oxidative stress-induced
cardiac myocyte remodeling. Am J Physiol Heart Circ Physiol.
301:H984–H993. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Long X, Goldenthal MJ, Wu GM and
Marín-García J: Mitochondrial Ca2+ flux and respiratory
enzyme activity decline are early events in cardiomyocyte response
to H2O2. J Mol Cell Cardiol. 37:63–70.
2004.
|
|
25
|
Zhang L, Huang S, Chen Y, Wang Z, Li E and
Xu Y: Icariin inhibits hydrogen peroxide-mediated cytotoxicity by
up-regulating sirtuin type 1-dependent catalase and peroxiredoxin.
Basic Clin Pharmacol Toxicol. 107:899–905. 2010.PubMed/NCBI
|
|
26
|
Maulik SK and Kumar S: Oxidative stress
and cardiac hypertrophy: a review. Toxicol Mech Methods.
22:359–366. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Han MJ, Kim BY, Yoon SO and Chung AS: Cell
proliferation induced by reactive oxygen species is mediated via
mitogen-activated protein kinase in Chinese hamster lung fibroblast
(V79) cells. Mol Cells. 15:94–101. 2003.PubMed/NCBI
|
|
28
|
Balakumar P and Jagadeesh G: Multifarious
molecular signaling cascades of cardiac hypertrophy: can the muddy
waters be cleared? Pharmacol Res. 62:365–383. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liang Q and Molkentin JD: Redefining the
roles of p38 and JNK signaling in cardiac hypertrophy: dichotomy
between cultured myocytes and animal models. J Mol Cell Cardiol.
35:1385–1394. 2003. View Article : Google Scholar : PubMed/NCBI
|