Introduction
Osteogenesis imperfecta (OI) is a genetic disorder
that is characterized by recurrent fractures, low bone mass, blue
sclera and dentinogenesis imperfecta (DI). It is a rare disorder
with an overall incidence of ~1 in 10,000–20,000 births (1). The etiology remains unclear; however,
it is estimated that ~90% of cases are associated with mutations in
the collagen type I, α1 (COL1A1) or COL1A2 genes and the remaining
10% of cases are associated with other genes (2). OI is a heterogeneous disease and type
I is the most common and mild form (1). At present, there is no effective
therapy. The present report presents the case of a 15-year-old
Chinese male with OI type I. Informed consent was obtained from the
patient. The Ethics Committee of Shandong University (Jinan, China)
approved the study.
Case report
A 15-year-old male was admitted to Qilu Hospital
(Jinan, China) complaining of repeated fractures over the previous
three years, predominantly due to falling over or overexertion. The
fractures occurred approximately every 3–4 months, often in the
forearms and legs. The fractures healed with no significant delay
when treated with external fixations. The patient did not
experience bone pain, limb and joint deformities or muscle
weakness.
One year previously, the patient had been prescribed
a six-month treatment of Caltrate and calcitriol (one of each
tablet per day); during this period the patient experienced one
bone fracture. However, he suffered from two further fractures as a
result of minor impacts following discontinuation of the treatment
over the subsequent six months. The patient had not taken other
medications, such as steroids, which affect bone metabolism. The
patient received an appendectomy in 2006 due to acute appendicitis
and recovered well following the surgery.
The patient was born following a full-term pregnancy
by spontaneous vaginal delivery without fetal distress or asphyxia:
Birth height, 50 cm (50th percentile) and birth weight, 3,500 g
(50th percentile). The patient was breastfed with no delayed
eruption of milk or permanent teeth, and was able to sit at eight
months and walk independently at 16 months. At 8 and 12 months of
age, the patient presented with episodes of nondisplaced fractures
resulting from minor impacts. The patient grew more slowly than his
peers (with regard to height) until age eight years, but exhibited
normal intellectual development. The parents were
non-consanguineous and healthy and there was no family history of
bone disease.
The measurements of the patient on admittance to
hospital were: Height, 162 cm (25th percentile); body weight, 52 kg
(25th percentile) (3); body mass
index, 19.8 kg/m2; interphalangeal space, 158 cm; and
head circumference, 58 cm. On physical examination, the patient
displayed normal development, and was well-proportioned with a
normal gait and erect posture. The patient’s face was triangular in
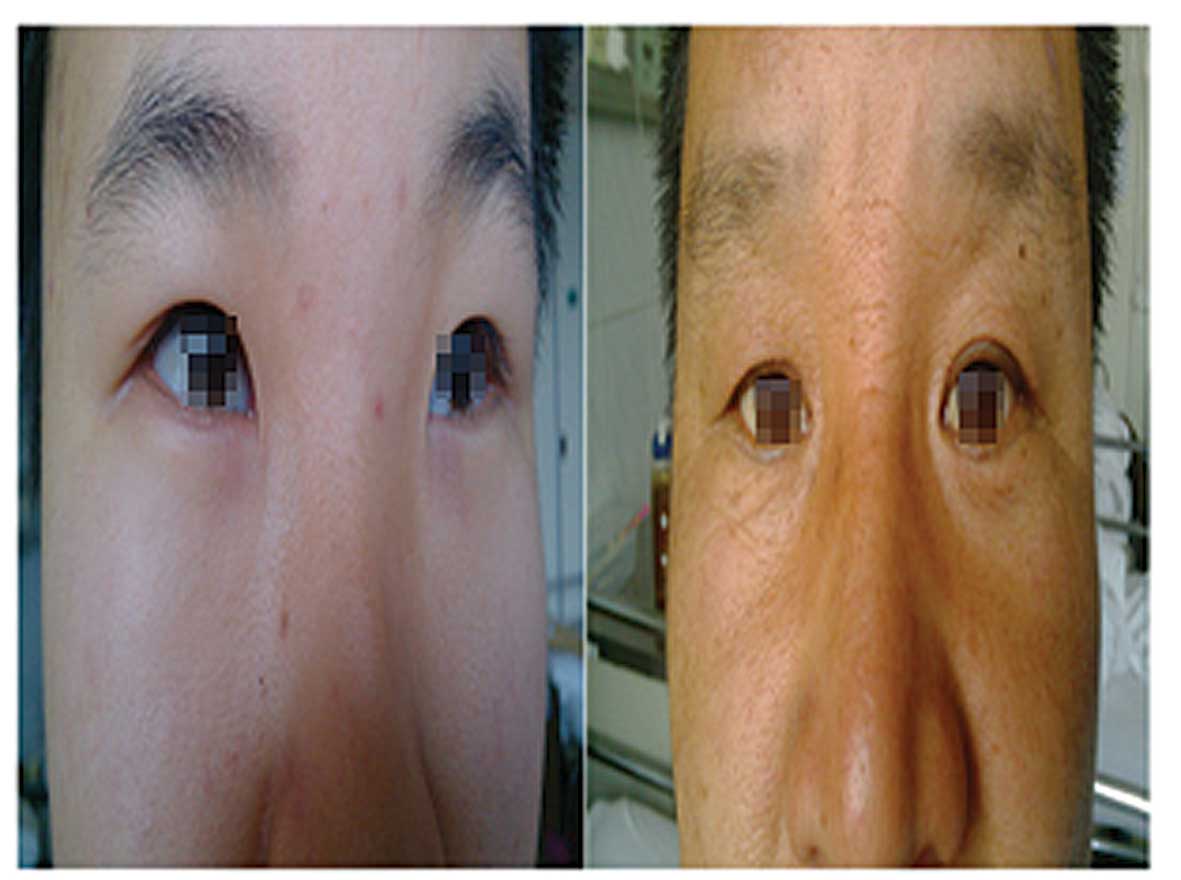
shape and blue sclera were apparent (Fig. 1). Intraoral examination revealed a
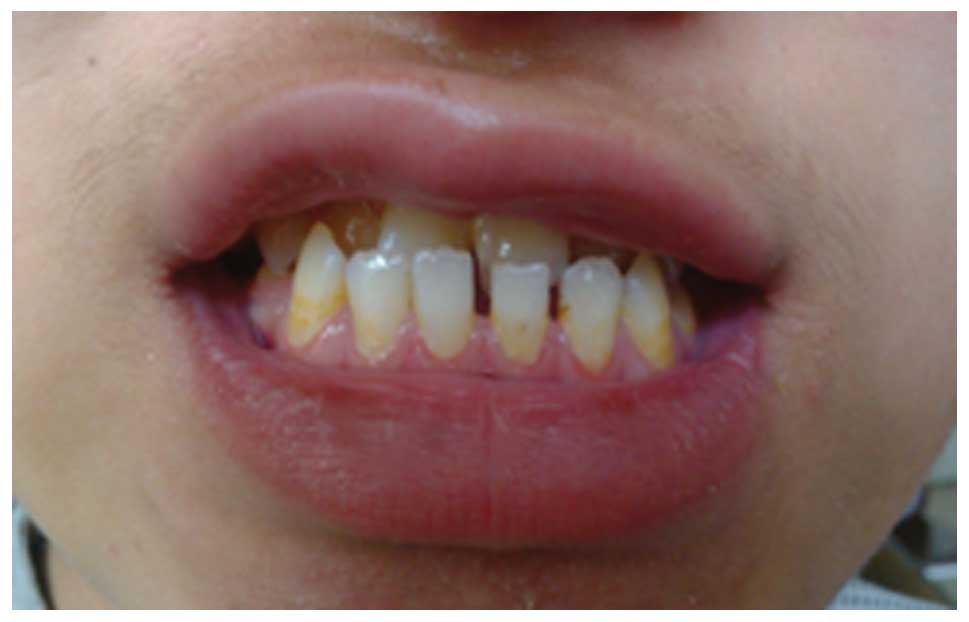
crossbite of the anterior teeth and no discolored or decayed teeth
(Fig. 2). In addition, altered
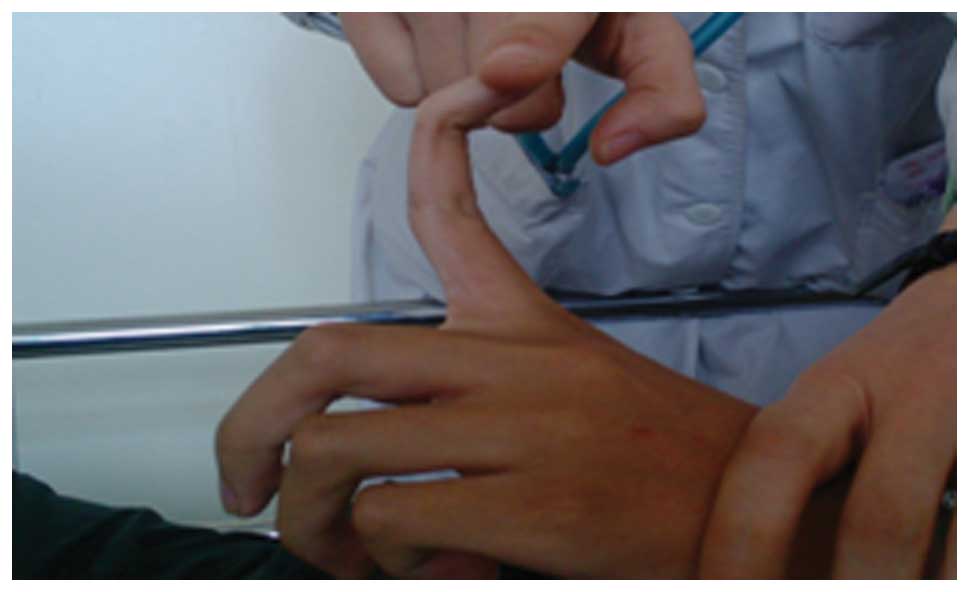
joint mobility and flexibility were observed (Fig. 3). The patient was estimated to be
at stage IV on the Tanner scale of genital and pubic hair
development (4). There was no
evidence of beading of the ribs, skeletal deformities, hearing
impairment, cardiac murmurs or respiratory difficulty and the
neurological examination was unremarkable. The laboratory
assessments, which included: erythrocyte sedimentation rate; levels
of C-reactive protein, serum creatinine, alkaline phosphatase,
calcium and phosphorus; and 24-hour urinary excretion of calcium,
phosphorus and parathyroid hormone (PTH) all appeared to be within
the normal ranges. The serum levels of osteocalcin and total type I
collagen telopeptide were above the normal values; however, the
level of 25-hydroxyvitamin D was marginally decreased. The levels
of thyroid hormone, growth hormone (GH), cortisol,
adrenocorticotropic hormone and sex hormones were observed to be
within the normal ranges. Protein electrophoresis, rheumatic
disease antibody and Bence-Jones protein tests were negative. An
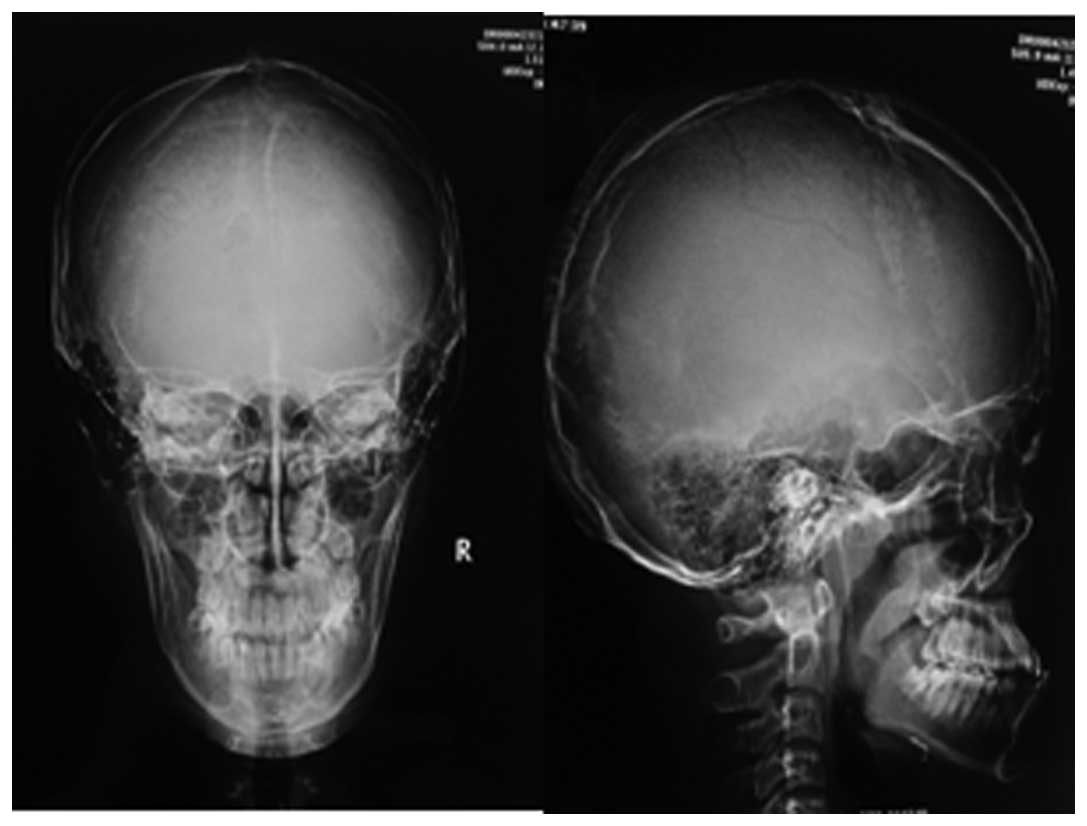
X-ray of the skull showed diffuse low bone density (Fig. 4), the absence of Wormian bones and
no indication of the ‘salt-and-pepper’ effect. Additional
observations obtained from the X-rays included biconcavity
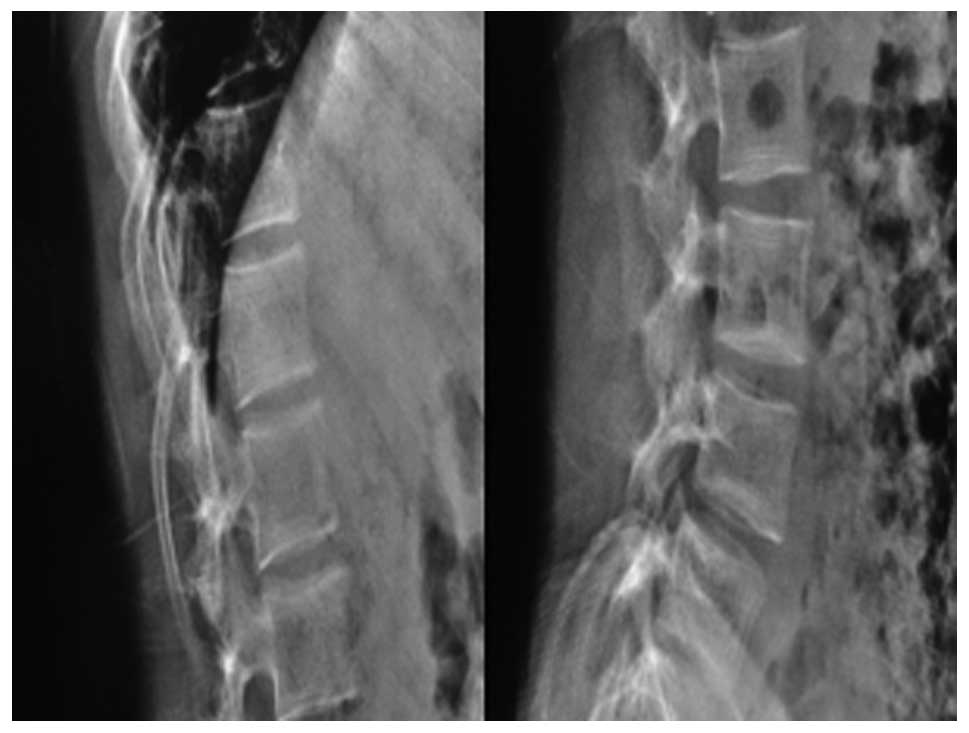
deformities in the lower thoracic and lumbar vertebrae (Fig. 5), a triradiate pelvis and
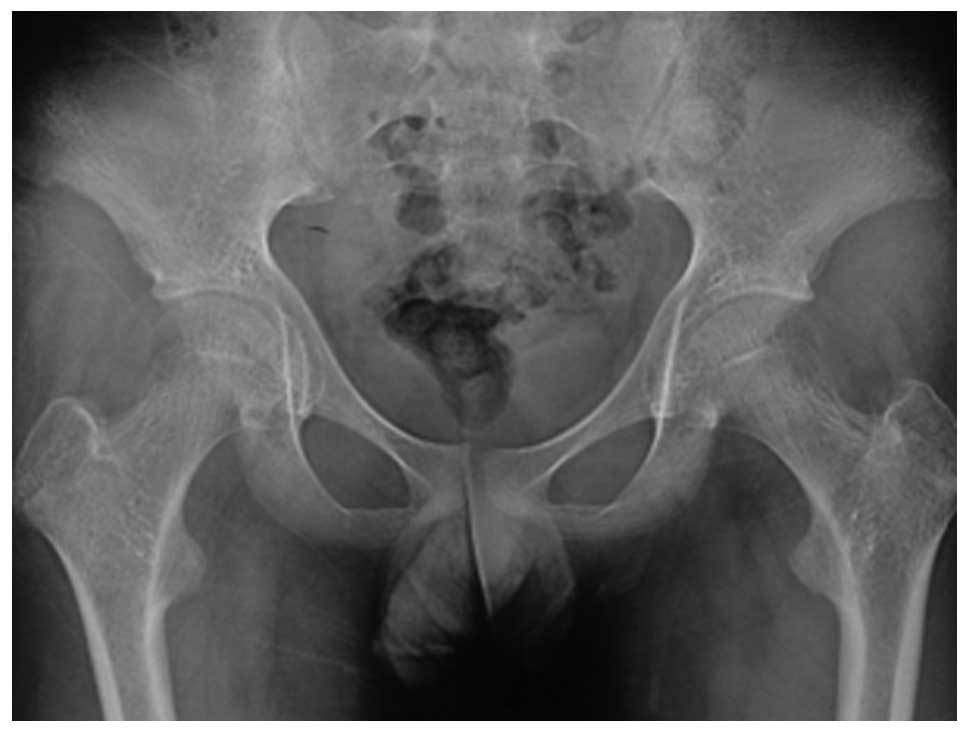
acetabular protrusion (Fig. 6),
growth arrest recovery lines in the two knee joints and former
fractures in the upper end of the fibula. Bone mineral density
(BMD) was measured via dual-energy X-ray absorptiometry and low
values (mean 0.633 g/cm2) for the lumbar vertebra L1–4
were observed. The radiographic features were characteristic of OI
and thus led to its diagnosis. A hearing threshold evaluation was
conducted and no issues were identified. An electrocardiogram
indicated sinus rhythm and an incomplete right bundle branch block.
Therefore, the clinical symptoms and radiologic features that were
observed resulted in the diagnosis of a mild form of OI type I.
The patient commenced treatment with a regular
intake of calcium (1,000 mg/day) in addition to an sufficient
intake of vitamin D (800 U/day), these were provided through an
ordinary protein-rich diet, extended exposure to sunlight as well
as prescriptions of Caltrate and Rocaltrol (calcitriol). The
patient was treated with pamidronate infusions intravenously, at a
maximum dosage (60 mg) for three days. Following the first
infusion, the patient developed an influenza-like reaction, which
was accompanied by a rash, fever and a maximum temperature of
38.8°C; the symptoms were controlled using indomethacin and fluid
therapy. Following four months of treatment for OI, the symptoms
and quality of life of the patient improved and according to the
follow-up, the patient continued to take Caltrate and Rocaltrol
(one tablet per day), enabling a return to normal schooling, with
no bone fractures occurring.
Discussion
OI, also termed brittle bone disease, is a genetic
disorder comprising a heterogeneous group of diseases. It is
characterized by a susceptibility to bone fractures with a severity
ranging from slight fracture to prenatal fracture. Additional
typical clinical features include blue sclera, DI, hyperlaxity of
ligaments and skin, hearing impairment, short stature and bone
deformities (1). OI presents a
complex clinical challenge; it results in abnormal blood
coagulation and airway obstruction, cardiovascular anomalies and
delayed wound healing (5,6). It is a rare disorder, with an
incidence rate of ~1 in 10,000–20,000 births (1).
As OI is a heterogeneous disease and the clinical
manifestations are variable, Sillence et al divided OI into
four types based on their clinical severity and genetic features
(7). The classification was as
follows: Type I, mild non-deforming; type II, perinatal lethal;
type III, severely deforming; and type IV, moderately deforming.
However, an increased number of OI cases have subsequently been
identified and investigated; therefore, based on the novel
clinical, radiological and molecular features, OI types V to VIII
have been added to the original Sillence classification (8). These are: Type V, moderate deforming
with normal teeth and sclera; type VI, moderate disease with
fishscale pattern of bone lamellation, normal sclera and teeth;
type VII, which is clinically similar to type II, with the
exception that the patients have a smaller head and normal sclera;
and type VIII, patients exhibit defects in growth and
mineralization (9). OI has been
defined as an autosomal dominant disorder resulting from a
quantitative or qualitative defect in the synthesis of type I
collagen, a predominant and principal extracellular matrix protein
within bone tissues (10). The
mutation may be in one of the two genes, COL1A1 and COL1A2, which
encode for the pro-α1(I) or pro-α2(I) chains of type I collagen
(11). However, the resulting
phenotypes vary widely, depending on which chains are affected, the
position in the collagen structure at which the mutation occurs,
and the nature of the amino acid substituent (1). To date, certain forms of OI
exhibiting autosomal recessive inheritance have been identified.
These genetic causes comprise defects in collagen chaperones and
proteins that are involved in type I procollagen assembly,
processing and maturation, as well as in proteins that are involved
in the formation and homeostasis of bone tissue (12). In the present case, the OI type I
was considered to be an autosomal dominant type resulting from a
quantitative defect of collagen within normal structures. The
clinical diagnosis of OI is predominantly based on the signs and
symptoms that are mentioned above, specifically the presence of
blue sclera and DI (8). With
regards to an etiological diagnosis, collagen analysis and genetic
testing is advantageous, but not always necessary. Biopsies from
cultured skin fibroblasts may be used to determine the quantity and
structure of the type I procollagen molecules, whereas genomic DNA
obtained from white blood cells can be screened for mutations
(13). However, the association
between genotype and phenotype is currently not fully defined.
Therefore, a positive result may confirm the diagnosis of OI
whereas a negative result does not completely dismiss it (1,8).
Thus, the definition of OI is dependent on signs and symptoms.
In the present case, the patient had experienced
repeated fractures resulting from minor impacts for three years and
routine calcium treatment showed little effect. The physical
examination demonstrated blue sclera, hypermobility of the joints,
a triangular-shaped face and no hearing impairment or DI. The
radiological images revealed osteoporosis and although the clinical
features indicated a diagnosis of OI, juvenile idiopathic
osteoporosis was also considered to be an alternative diagnosis.
The latter disorder is a transient form of childhood osteoporosis
without the extraskeletal features that distinguish it from OI. It
usually develops in prepubertal, previously healthy infants and
persists for 3–5 years without heredity (14).
Once the diagnosis of OI has been established, an
evaluation of the patient by a multidisciplinary team is necessary.
Physiotherapy, rehabilitation and orthopedic surgery are the
mainstays of OI management. The goal of multimodality therapy is to
maximize the mobility and functional capabilities of patients
(15), and in the present case of
mild type I OI, the aim was also to attain a normal quality of life
for the patient. Oral and intravenous bisphosphonates (BPs) and
potent antiresorptive agents are widely administered for the
treatment of all types of OI. Clinical trials have indicated the
effectiveness of BPs in the improvement of BMD and the remission of
clinical symptoms (10). Although
BPs are not a cure for OI, they provide an effective adjunct to
comprehensive care. However, optimal usage of BPs, the
appropriateness of use by patients with milder symptoms and the
side-effects remain unknown. A large scale, randomized double-blind
placebo-controlled trial is required to refine their clinical
applications. Additional medical therapies are available, such as
GHs and PTHs; however, the outcome and adverse effects associated
with their use require further assessment and analysis (10).
Surgical intervention is an alternative method of
treatment when medical therapies fail. In the present case, no
deformities were observed and the reaction to pamidronate was
positive; thus, no surgical intervention was required.
Additionally, after the patient was discharged from hospital, it
was necessary for monitored, moderate physical activity programs to
be conducted in order to prevent contractures and
immobility-induced bone loss (16). The authors propose psychosocial
therapy as a provision for children to address the issues that may
arise as a result of repeated bone fractures. In addition, if the
patient has children in the future, his partner would require
routine prenatal screening, by ultrasound, in addition to genetic
counseling since individuals exhibiting a dominant OI trait present
a 50% risk of transmission with each pregnancy.
In conclusion, experimental approaches, such as bone
marrow and stem cell transplantation, in addition to gene-based
therapy, provide potential cures for OI. However, these approaches
are currently not ready for clinical trials (10).
References
|
1
|
Raunch F and Glorieux FH: Osteogenesis
imperfecta. Lancet. 363:1377–1385. 2004. View Article : Google Scholar
|
|
2
|
van Dijk FS, Cobben JM, Kariminejad A, et
al: Osteogenesis Imperfecta: A review with clinical examples. Mol
Syndromol. 2:1–20. 2011.PubMed/NCBI
|
|
3
|
Li H, Ji CY, Zong XN and Zhang YQ: Height
and weight growth curves of Chinese children and adolescents 0–18
years. Zhonghua Er Ke Za Zhi. 47:487–492. 2009.(In Chinese).
|
|
4
|
Carel JC and Léger J: Precocious puberty.
N Engl J Med. 22:2366–2377. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Edge G, Okafor B, Fennelly ME and Ransford
AO: An unusual manifestation of bleeding diathesis in a patient
with osteogenesis imperfecta. Eur J Anaesthesiol. 14:215–219. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oakley I and Reece LP: Anesthetic
implications for the patient with osteogenesis imperfecta. AANA J.
78:47–53. 2010.PubMed/NCBI
|
|
7
|
Sillence DO, Senn A and Danks DM: Genetic
heterogeneity in osteogenesis imperfecta. J Med Genet. 16:101–116.
1979. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Glourieux FH: Osteogenesis imperfecta.
Best Pract Res Clin Rheumatol. 22:85–100. 2008. View Article : Google Scholar
|
|
9
|
Starr SR, Roberts TT and Fischer PR:
Osteogenesis imperfecta: primary care. Pediatr Rev. 31:e54–e64.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Monti E, Mottes M, Fraschini P, et al:
Current and emerging treatments for the management of osteogenesis
imperfecta. Ther Clin Risk Manag. 6:367–381. 2010.PubMed/NCBI
|
|
11
|
Byers PH, Krakow D, Nunes ME and Pepin M;
American college of medical genetics. Genetic evaluation of
suspected osteogenesis imperfecta (OI). Genet Med. 8:383–388. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rohrbach M and Giunta C: Recessive
osteogenesis imperfecta: clinical, radiological, and molecular
findings. Am J Med Genet C Semin Med Genet. 160C:175–189. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Körkkö J, Ala-Kokko L, De Paepe A, et al:
Analysis of the COL1A1 and COL1A2 genes by PCR amplification and
scanning by conformation-sensitive gel electrophoresis identifies
only COL1A1 mutations in 15 patients with osteogenesis imperfecta
type I: identification of common sequences of null-allele
mutations. Am J Hum Genet. 62:98–110. 1998.
|
|
14
|
Smith R: Idiopathic juvenile osteoporosis:
experience of twenty-one patients. Br J Rheumatol. 34:68–77. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Engelbert RH, Pruijs HE, Beemer FA and
Helders PJ: Osteogenesis imperfecta in childhood: treatment
strategies. Arch Phys Med Rehabil. 79:1590–1594. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zeitlin L, Fassier F and Glorieux FH:
Modern approach to children with osteogenesis imperfecta. J Pediatr
Orthop B. 12:77–87. 2003.PubMed/NCBI
|