Introduction
Adrenocorticotrophic hormone (ACTH)-independent
macronodular adrenal hyperplasia (AIMAH) is a rare disorder
characterized by bilateral macronodular hyperplasia of the adrenal
glands and increased cortisol production with subclinical or overt
Cushing’s syndrome (CS) (1,2).
AIMAH represents <1% of cases of endogenous CS; however, since
10% of adrenal lesions are bilateral, AIMAH with subclinical
cortisol secretion is becoming increasingly recognized (3). Patients with AIMAH are diagnosed
following incidental radiological observations or the investigation
of an adrenal over-secretion syndrome (4,5).
AIMAH usually presents without symptoms, although in a number of
cases patients are diagnosed following clinical CS.
The most common cause (95% of patients) of AIMAH is
adrenocortical adenoma or carcinoma. The majority of the remaining
patients have primary pigmented nodular adrenal disease, a syndrome
that is characterized by multiple small bilateral pigmented
adrenocortical nodules, and is often associated with the Carney
complex (6). The diagnosis and
management of patients with ACTH-independent CS and bilateral
adrenal masses are problematic (7,8),
particularly for bilateral adrenal adenomas. In the current study,
a total of 23 AIMAH cases are presented; the patients were admitted
to the Tianjin Medical University General Hospital (Tianjin, China)
between July 1994 and 2010, and diagnosed by pathology.
Materials and methods
Patients
The present study was approved and registered by the
Ethics Committee of the Tianjin Medical University General Hospital
(Tianjin, China) in January 1993. The Ethics Committee approved all
associated screening, treatment, data collection and follow-up of
the patients; written informed consent was obtained from all
particpants. All experiments were undertaken in accordance with the
Declaration of Helsinki. A total of 23 patients (males, 14;
females, 9; mean age, 49 years) were admitted to the Tianjin
Medical University General Hospital between July 1994 and 2010. All
the patients presented with several symptoms characteristic of CS,
and hypertension was observed during examination. The duration of
the disease ranged between one and five years. Diabetes occurred in
10 cases (10/23), central obesity occurred in eight cases, a
sanguine appearance was observed in six cases and 14 patients
exhibited purple stripes on their body.
The levels of plasma cortisol, ACTH and urinary free
cortisol (UFC) were analyzed in all the patients using DPC Immulite
2000 (Siemens Healthcare, Los Angeles, CA, USA), and patients were
subjected to high and low dose dexamethasone suppression tests
(HDDST and LDDST). Plasma cortisol levels were monitored in these
patients, Magnetic resonance imaging (MRI) examinations (Discovery
MR750w 3.0T; GE Healthcare, Pittsburgh, PA, USA) and computed
tomography (CT) scans (Lightspeed VCT XT; GE Healthcare) were also
performed.
Operative procedures
Bilateral adrenalectomy was performed in eight
patients and unilateral adrenalectomy was performed in 15 patients.
The surgical procedures were performed as previously described by
Shinbo et al (9). Briefly,
under general anesthesia, patients undergoing a right adrenalectomy
were placed in a left lateral position. Next, 10/12-mm trocars were
placed at the mid-clavicular line below the costal margin and at
the median line 5 cm above the umbilicus, while 5-mm trocars were
placed at approximately three finger-widths below the xyphoid
process and at the anterior axillary line 5 cm below the costal
margin. Laparosonic coagulating shears with a suction and
irrigation device and with a cautery and L-hook tip were used when
necessary. The two trocars at the median line remained on the
abdominal wall while skin wounds at the other trocar sites were
closed.
For left laparoscopic adrenalectomy, patients were
repositioned in a right half lateral position, and two 5-mm trocars
were placed at the left region of the costal margin and at the
midclavicular line below the costal margin. The left adrenal gland
was isolated similarly to right side and enclosed in an endoscopic
pouch. Skin wounds were closed.
Results
Laboratory testing results
Results of the HDDST and LDDST were negative. Four
patients received plasma cortisol rhythm determination, while the
plasma cortisol levels in the additional 19 cases were only
examined at 8:00 am. The results demonstrated that plasma cortisol
levels were elevated in 20 patients. In addition, the levels of UFC
were increased, while the levels of ACTH were decreased in the 23
patients (Table I).
 | Table ILevels of plasma cortisol, UFC and
ACTH in 23 patients with AIMAH. |
Table I
Levels of plasma cortisol, UFC and
ACTH in 23 patients with AIMAH.
| | | | Prior to surgery | Nodule volumea (ml) | Three years following
surgery |
|---|
| | | |
|
|
|
|---|
| Patient | Age (years) | Gender | Examination time | Cortisol
(nmol/l) | UFC (nmol/24 h) | ACTH (pmol/l) | Left | Right | Cortisol
(nmol/l) | UFC (nmol/24 h) | ACTH (pmol/l) |
|---|
| 1 | 36 | M | 8 am | 718 | 1684 | 0.2 | 14 | 18 | - | - | - |
| | | 0 am | 828 | - | - | - | - | - | - | - |
| | | 8 pm | 1132 | - | - | - | - | - | - | - |
| 2 | 42 | M | 8 am | 883 | 1601 | 0.1 | 30 | 32 | - | - | - |
| 3 | 47 | F | 8 am | 800 | 1341 | 0.6 | 27 | 18 | 497 | 400 | - |
| 4 | 39 | M | 8 am | 938 | 1311 | 0.9 | 14 | 14 | 580 | 428 | - |
| 5 | 57 | F | 8 am | 2070 | 1203 | 0.4 | 60 | 32 | - | - | - |
| 6 | 52 | F | 8 am | 828 | 828 | 0.1 | 32 | 14 | - | - | - |
| 7 | 55 | M | 8 am | 469 | 359 | 0.7 | 27 | 27 | 276 | 304 | - |
| 8 | 51 | M | 8 am | 552 | 2622 | 0.7 | 30 | 14 | 414 | 386 | - |
| | | 0 am | 552 | - | - | - | - | - | - | - |
| | | 8 pm | 331 | - | - | - | - | - | - | - |
| 9 | 61 | M | 8 am | 994 | 1573 | 0.6 | 32 | 50 | 524 | 320 | - |
| 10 | 59 | F | 8 am | 1104 | 1159 | 0.7 | 38 | 30 | - | - | - |
| 11 | 47 | M | 8 am | 1214 | 966 | 1.0 | 50 | 16 | 966 | 657 | 0.9 |
| 12 | 48 | M | 8 am | 1711 | 1242 | 0.9 | 55 | 18 | 1104 | 980 | 0.9 |
| 13 | 53 | F | 8 am | 1065 | 1423 | 1.2 | 50 | 26 | 450 | 380 | 1.8 |
| | | 0 am | 855 | - | - | - | - | - | - | - |
| | | 8 pm | 686 | - | - | - | - | - | - | - |
| 14 | 48 | M | 8 am | 960 | 630 | 0.7 | 45 | 45 | 500 | 350 | 0.8 |
| 15 | 46 | F | 8 am | 350 | 520 | 0.8 | 40 | 32 | 260 | 170 | 1.1 |
| | | 0 am | 440 | - | - | - | - | - | - | - |
| | | 8 pm | 460 | - | - | - | - | - | - | - |
| 16 | 47 | M | 8 am | 900 | 1420 | 0.9 | 33 | 42 | 430 | 290 | 2.1 |
| 17 | 39 | M | 8 am | 840 | 1125 | 1.2 | 46 | 28 | 410 | 260 | 2.0 |
| 18 | 48 | M | 8 am | 880 | 900 | 1.3 | 40 | 33 | - | - | - |
| 19 | 59 | F | 8 am | 1156 | 1236 | 0.5 | 45 | 48 | 650 | 780 | 0.9 |
| 20 | 42 | M | 8 am | 710 | 840 | 1.1 | 38 | 36 | 430 | 390 | 1.2 |
| 21 | 45 | F | 8 am | 850 | 960 | 0.8 | 40 | 38 | 600 | 620 | 1.0 |
| 22 | 57 | M | 8 am | 910 | 1022 | 0.7 | 45 | 40 | 710 | 510 | 0.8 |
| 23 | 49 | F | 8 am | 950 | 1150 | 0.9 | 50 | 29 | 580 | 420 | 1.2 |
Imaging examination
MRI examinations revealed that the pituitary gland
was normal in 17 patients; however, MRI of the pituitary gland was
not performed in the remaining six patients. Observations from the
CT scans revealed bilateral adrenal nodules of soft tissue density,
measuring ≤5 cm, and irregular nodular masses in the adrenal
glands. In addition, the CT scans demonstrated that the adrenal
lesions with macronodularity were significantly enlarged (Fig. 1), and the largest diameter of an
adrenal nodular was 6 cm.
Surgery and discharge
Following surgery, all the resected samples were
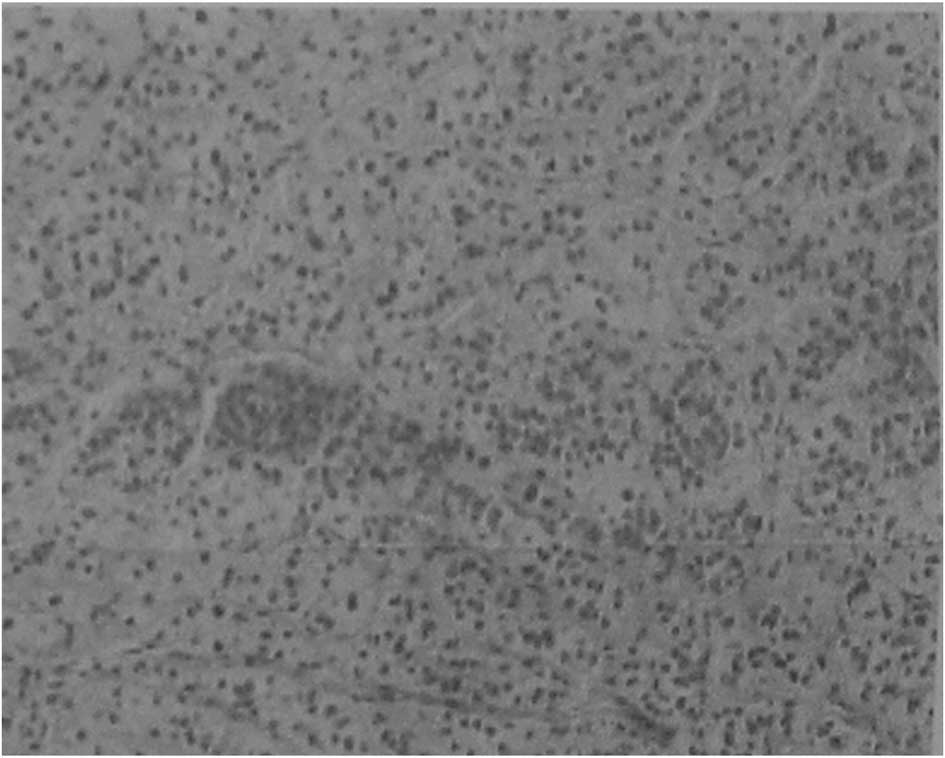
confirmed positive by histopathological analysis (Fig. 2). The nodules were observed as
bright cells under the light microscope and the normal cortical
structure had disappeared. The general clinical and biochemical
conditions of the patients are summarized in Table II. Lumbar open surgery was
performed in nine patients, retroperitoneal laparoscopic surgery
was performed in seven patients, single abdominal open surgery were
performed in three patients, multiple lumbar open surgery was
performed in two patients and laparoscopic surgery was performed in
two patients. Infection occurred in one patient following surgery,
which was controlled following treatment. The resected adrenal
nodular mass in all the patients was a diffused grayish yellow or
golden yellow color (Fig. 3). The
largest nodular was 6×4×4 cm, weighing 40 g and with no clear coat
covering the resected adrenal tissue. The surface of the resected
slide was golden brown and had a diameter of 0.6–1.7 cm. Patients
with unilateral resection were not treated with hormone drug
therapy at discharge; however, bilateral resection patients
received 5 or 10 mg prednisone treatment.
| Table IIGeneral clinical and biochemical
conditions of the patients following surgery. |
Table II
General clinical and biochemical
conditions of the patients following surgery.
| Case | Resected
position | Blood loss
(ml) | Resected adrenal
gland weight (L/R/g) | Discharge time
following surgery (days) | Discharge
medication | Complication | Surgical
method |
|---|
| 1 | R | 250 | −/36 | 9 | - | - | Lumbar open
surgery |
| 3 | L | 200 | 54/− | 11 | - | - | Lumbar open
surgery |
| 4 | L | 150 | 28/− | 11 | - | - | Lumbar open
surgery |
| 6 | L | 250 | 64/− | 9 | - | - | Lumbar open
surgery |
| 7 | R | 300 | −/54 | 11 | - | - | Lumbar open
surgery |
| 8 | L | 250 | 60/− | 14 | - | - | Lumbar open
surgery |
| 11 | L | 250 | 100/− | 11 | - | - | Lumbar open
surgery |
| 12 | L | 200 | 110/− | 10 | - | - | Lumbar open
surgery |
| 13 | L | 150 | 100/− | 10 | - | - | Lumbar open
surgery |
| 15 | L | 350 | 80/− | 9 | - | - | Retroperitoneal
laparoscopic surgery |
| 16 | R | 150 | −/84 | 12 | - | - | Retroperitoneal
laparoscopic surgery |
| 17 | L | 100 | 92/− | 13 | - | - | Retroperitoneal
laparoscopic surgery |
| 18 | L | 90 | 80/− | 11 | - | - | Retroperitoneal
laparoscopic surgery |
| 20 | L | 90 | 76/− | 11 | - | - | Retroperitoneal
laparoscopic surgery |
| 23 | L | 100 | 100/− | 12 | - | - | Retroperitoneal
laparoscopic surgery |
| 2 | Bi | 350 | 60/64 | 21 | Prednisone (5 mg,
bid) | - | Retroperitoneal
laparoscopic surgery |
| 5 | Bi | 400 | 120/64 | 20 | Prednisone (5 mg,
bid) | - | Single abdominal
open surgery |
| 10 | Bi | 400 | 76/60 | 19 | Prednisone (10 mg,
bid) | - | Single abdominal
open surgery |
| 9 | Bi | 450 | 64/100 | 21 | Prednisone (5 mg,
bid) | - | Single abdominal
open surgery |
| 14 | Bi | 510 | 90/90 | 19 | Prednisone (10 mg,
bid) | - | Multiple lumbar
open surgery |
| 19 | Bi | 250 | 90/96 | 20 | Prednisone (10 mg,
bid) | - | Multiple lumbar
open surgery |
| 21 | Bi | 150 | 80/76 | 21 | Prednisone (5 mg,
bid) | - | Laparoscopic
surgery |
| 22 | Bi | 200 | 90/80 | 30 | Prednisone (5 mg,
bid) | Infection | Laparoscopic
surgery |
Postoperative follow-up
Following surgery, hypertension of the patients was
significantly alleviated. The eight patients who underwent
bilateral adrenalectomy were followed-up for 2–8 years and were
treated with glucocorticoid replacement therapy. The patients had
normal blood pressure, breathing rate returned to the normal level,
and the obesity and sanguine appearance was alleviated.
Decreased blood pressure following surgery was
observed in 15 patients. However, after three years, 12 of the 15
patients had hypertension of ~150–170/90–100 mmHg, which was
maintained at 130–150/70–85 mmHg following oral administration of
antihypertensive drugs. The additional three patients (cases 11, 12
and 23) had no response to oral antihypertensive drugs and their
blood pressure reached 170–180/90–110 mmHg. Plasma cortisol, UFC,
ACTH and CT analyses were performed, and the contralateral adrenal
gland was found to have increased to 35, 32 and 38 ml,
respectively. The blood pressure of these patients returned to
normal following the removal of the contralateral adrenal gland and
hormone replacement therapy. Nelson’s syndrome was not observed
following therapy.
Discussion
CS is caused by excessive cortisol secretion and is
associated with increased mortality and severe morbidity. The
condition is not fully reversible, despite biochemical control. CS
is characterized by the loss of normal feedback regulation and
circadian rhythm of the hypothalamic-pituitary axis due to
inappropriate secretion of ACTH from a pituitary tumor (Cushing’s
disease) or an ectopic source (ectopic ACTH secretion). The
remaining causes (20%) are ACTH independent. Once a diagnosis is
established, the therapeutic goal is the removal of the tumor.
Whenever surgery is not curative, the management of patients with
CS requires a major effort to control hypercortisolemia and the
associated symptoms (10).
The diagnosis of CS is based on the clinical
features of hypercortisolism, the absence of serum cortisol diurnal
rhythm, elevated midnight sleeping cortisol levels and incomplete
cortisol suppression test (11).
CS due to AIMAH was first reported in an isolated case (12).
AIMAH is characterized by bilateral macronodular
hyperplasia of the adrenal glands and is one of the causes of CS
(13). Clinical manifestations
include hypertension, weight gain, impaired glucose tolerance or
diabetes mellitus, osteoporosis and an increased susceptibility to
bruising. Hypogonadism and gynecomastia have been reported in males
and hirsutism in females (14).
The precise etiology of AIMAH is unknown; however, previous studies
have demonstrated that aberrant adrenal expression and aberrant
function of a number of peptide hormone receptors, including
receptors for glucose-dependent insulinotropic hormone,
vasopressin, luteinizing hormone/human chorionic gonadotropin,
β-adrenergic agonists and serotonin, may lead to adrenal cell
proliferation and abnormal regulation of steroidogenesis in AIMAH
(15,16).
Cross-sectional imaging is commonly used to identify
adrenal disease in patients with CS. CT and MRI scans are used to
document the lesion size and shape, presence or absence of
calcification, hemorrhage and necrosis. With regard to MRI,
T1-weighted images are hypointense relative to the liver and
isointense relative to muscle, while T2-weighted images tend to be
hyperintense relative to the liver (17,18).
By contrast, the nodules of patients with chronic ACTH stimulation
appear isointense relative to the liver on T2-weighted MRI scans
(19).
Histological analysis revealed a marked increase in
the number of small clear cells, which are predominantly derived
from the upper fascicular zone. The amount of cortisol produced by
each cell is small; thus, significant enlargement of the adrenal
gland is necessary before excessive cortisol production causes CS
(20). The definitive treatment of
CS consists of surgical resection of the tumor secreting ACTH. When
the source of excessive cortisol secretion is the pituitary gland,
the standard approach is to perform an endoscopic endonasal
trans-sphenoidal exploration, with excision of the tumor if
identified. This surgical procedure is demanding and should only be
performed in centers with extensive experience to minimize the
surgical risks, reduce the possibility of remission and maintain
other pituitary gland functions.
Bilateral adrenalectomy via an overt or laparoscopic
approach is the most useful treatment for patients with AIMAH and
hormonal hypersecretion (21,22).
However, in patients exhibiting moderately increased hormonal
production, unilateral adrenalectomy is proposed as a safe and
effective alternative; it is expected that as the cell mass
increases in the contralateral adrenal gland, a second
adrenalectomy may be required (23,24).
In conclusion, a total of 23 cases diagnosed with
AIMAH were presented in the current study. High and low dose
dexamethasone failed to suppress cortisone secretion in the
suppression tests and ACTH levels were low in all the cases.
Bilateral enlarged adrenal glands were observed on CT scans, and
bilateral adrenal macronodular hyperplasia was confirmed in all the
cases by pathological examination. In accordance with previous
studies (25,26), the results of the present study
demonstrated that AIMAH had unique endocrinological, radiological
and pathological features. Diagnosis of AIMAH is predominantly
derived from pathological examination and long term remission may
be achieved by unilateral adrenalectomy. Contralateral
adrenalectomy should be performed in cases of recurrence, when
followed with periodical examination of the symptoms and serum
concentration of cortisol.
References
|
1
|
Bertagna X, Guignat L, Groussin L and
Bertherat J: Cushing’s disease. Best Pract Res Clin Endocrinol
Metab. 23:607–623. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Goñi Iriarte MJ: Cushing’s syndrome:
special issues. Endocrinol Nutr. 56:251–261. 2009.(In Spanish).
View Article : Google Scholar
|
|
3
|
Lacroix A: ACTH-independent macronodular
adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab.
23:245–259. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bourdeau I, D’Amour P, Hamet P, Boutin JM
and Lacroix A: Aberrant membrane hormone receptors in incidentally
discovered bilateral macronodular adrenal hyperplasia with
subclinical Cushing’s syndrome. J Clin Endocrinol Metab.
86:5534–5540. 2001.PubMed/NCBI
|
|
5
|
Yamada Y, Sakaguchi K, Inoue T, et al:
Preclinical Cushing’s syndrome due to
adrenocorticotropin-independent bilateral adrenocortical
macronodular hyperplasia with concurrent excess of gluco- and
mineralocorticoids. Intern Med. 36:628–632. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stratakis CA, Carney JA, Lin JP, et al:
Carney complex, a familial multiple neoplasia and lentiginosis
syndrome. Analysis of 11 kindreds and linkage to the short arm of
chromosome 2. J Clin Invest. 97:699–705. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Karapanou O, Vlassopoulou B, Tzanela M,
Stratigou T, Tsatlidis V and Tsirona S: Adrenocorticotropic hormone
independent macronodular adrenal hyperplasia due to aberrant
receptor expression: is medical treatment always an option? Endocr
Pract. 19:e77–e82. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yoshida M, Umeda H, Iwama S, et al:
Assessment of long-term efficacy and safety of metyrapone
monotherapy in a patient with ACTH-independent macronodular adrenal
hyperplasia. Endocrine. 41:160–161. 2012. View Article : Google Scholar
|
|
9
|
Shinbo H, Suzuki K, Sato T, Kageyama S,
Ushiyama T and Fujita K: Simultaneous bilateral laparoscopic
adrenalectomy in ACTH-independent macronodular adrenal hyperplasia.
Int J Urol. 8:315–318. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pozza C, Graziadio C, Giannetta E, Lenzi A
and Isidori AM: Management strategies for aggressive cushing’s
syndrome: from macroadenomas to ectopics. J Oncol. 2012:6852132012.
View Article : Google Scholar
|
|
11
|
Newell-Price J, Trainer P, Besser M and
Grossman A: The diagnosis and differential diagnosis of Cushing’s
syndrome and pseudo-Cushing’s states. Endocr Rev. 19:647–672.
1998.PubMed/NCBI
|
|
12
|
Kirschner MA, Powell RD Jr and Lipsett MB:
Cushing’s syndrome: nodular cortical hyperplasia of adrenal glands
with clinical and pathological features suggesting adrenocortical
tumor. J Clin Endocrinol Metab. 24:947–955. 1964. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lacroix A, Ndiaye N, Tremblay J and Hamet
P: Ectopic and abnormal hormone receptors in adrenal Cushing’s
syndrome. Endocr Rev. 22:75–110. 2001.PubMed/NCBI
|
|
14
|
Doppman JL, Chrousos GP, Papanicolaou DA,
Stratakis CA, Alexander HR and Nieman LK:
Adrenocorticotropin-independent macronodular adrenal hyperplasia:
an uncommon cause of primary adrenal hypercortisolism. Radiology.
216:797–802. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mircescu H, Jilwan J, N’Diaye N, et al:
Are ectopic or abnormal membrane hormone receptors frequently
present in adrenal Cushing’s syndrome? J Clin Endocrinol Metab.
85:3531–3536. 2000.PubMed/NCBI
|
|
16
|
Bertagna X, Groussin L, Luton JP and
Bertherat J: Aberrant receptor-mediated Cushing’s syndrome. Horm
Res. 59(Suppl 1): 99–103. 2003. View Article : Google Scholar
|
|
17
|
Doppman JL, Nieman LK, Travis WD, et al:
CT and MR imaging of massive macronodular adrenocortical disease: a
rare cause of autonomous primary adrenal hypercortisolism. J Comput
Assist Tomogr. 15:773–779. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rockall AG, Babar SA, Sohaib SA, et al: CT
and MR imaging of the adrenal glands in ACTH-independent cushing
syndrome. Radiographics. 24:435–452. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Verma A, Mohan S and Gupta A:
ACTH-independent macronodular adrenal hyperplasia: imaging findings
of a rare condition: A case report. Abdom Imaging. 33:225–229.
2008. View Article : Google Scholar
|
|
20
|
Sasano H, Suzuki T and Nagura H:
ACTH-independent macronodular adrenocortical hyperplasia:
immunohistochemical and in situ hybridization studies of
steroidogenic enzymes. Mod Pathol. 7:215–219. 1994.PubMed/NCBI
|
|
21
|
Swain JM, Grant CS, Schlinkert RT, et al:
Corticotropin-independent macronodular adrenal hyperplasia: a
clinicopathologic correlation. Arch Surg. 133:541–546. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Stratakis CA and Kirschner LS: Clinical
and genetic analysis of primary bilateral adrenal diseases (micro-
and macronodular disease) leading to Cushing’s syndrome. Horm Metab
Res. 30:456–463. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Boronat M, Lucas T, Barcelo B, Alameda C,
Hotait H and Estrada J: Cushing’s syndrome due to autonomous
macronodular adrenal hyperplasia: long-term follow-up after
unilateral adrenalectomy. Postgrad Med J. 72:614–616. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Imöhl M, Köditz R, Stachon A, et al:
Catecholamine-dependent hereditary Cushing’s syndrome - follow-up
after unilateral adrenalectomy. Med Klin (Munich). 97:747–753.
2002.(In German).
|
|
25
|
Zhang Q, Dou J, Gu W, Yang G, Mu Y and Lu
J: In silico analyses reveal nuclear asymmetry of spongiocytes and
compact cells of adrenocorticotrophic hormone-independent
macronodular adrenocortical hyperplasia. Am J Med Sci. 347:400–405.
2014. View Article : Google Scholar
|
|
26
|
Imai T, Kikumori T, Shibata A, Fujiwara M,
Hibi Y and Nakao A: Laparoscopic adrenalectomy for incidentaloma
and bilateral adrenal disease. Asian J Surg. 26:64–70. 2003.
View Article : Google Scholar : PubMed/NCBI
|