Introduction
Rosai-Dorfman disease (RDD), originally known as
sinus histiocytosis with massive lymphadenopathy, is a
non-Langerhans cell histiocytosis that was first described in 1965
by Destombes (1) and subsequently
recognized as a distinct entity by Rosai and Dorfman in 1969
(2). There are two main forms of
RDD: One form that affects the lymph nodes and in certain cases the
extranodal organs (3), and the
other form is purely cutaneous RDD (CRDD) (4). CRDD is extremely rare, with only a
few reported cases. The etiology of CRDD remains unknown with a
number of viral and immune causes hypothesized. CRDD presents as
solitary or numerous papules, nodules, plaques or as a combination
of these. Based on previous case reports, certain treatment
options, including dapsone, thalidomide and isotretinoin, have been
suggested for patients with varying efficacy (5–7). The
present study reports a case of purely CRDD and discusses the
clinical and histological features, etiology and course in
conjunction with the literature review.
Case report
A 25-year-old Chinese female patient presented with
a two-month history of red plaques on her face. The lesion grew
progressively in size, with occasional pain and pruritus. The
patient had no particular previous medical history, and was
otherwise healthy, with no history of fever, malaise or weight
loss. There was no mucosal involvement. The results of a general
physical examination were normal, with no signs of lymph node
enlargement. However, a dermatological examination showed indurated
erythematous plaques with a number of reddish-yellow nodules on the
left side of the patient’s face (Fig.
1).
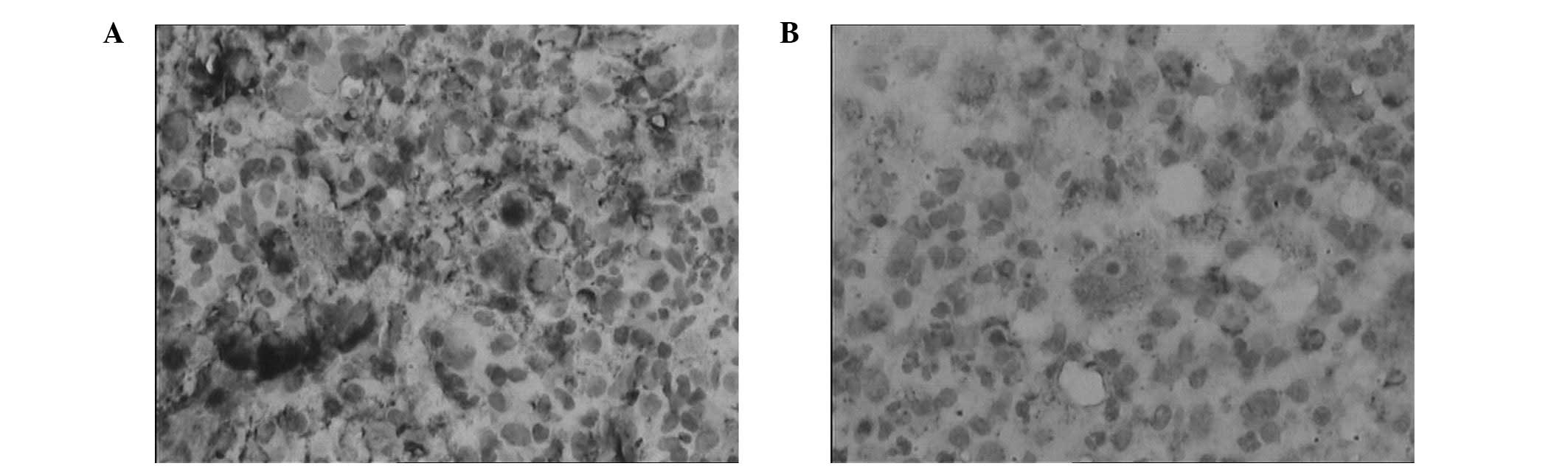
The biopsy specimen obtained from the afflicted area
showed that the epidermis was normal, but revealed dense
inflammatory infiltrates, composed of neutrophils, plasma cells,
lymphocytes and histiocytes, in the dermis. No significant
histiocytic atypia was identified. The histiocytes exhibited
abundant, focally foamy cytoplasm and large vesicular nuclei;
lymphocytes and plasma cells were observed within the cytoplasm of
these histiocytes (emperipolesis; Fig.
2). Immunohistochemical staining revealed that the histiocytes
were strongly positive for S100 protein, weakly positive for CD68
and negative for CD1a (Fig. 3).
Thus, the diagnosis of CRDD was confirmed. Laboratory examination
revealed no abnormality in routine tests, including the blood
count, urinalysis, erythrocyte sedimentation rate, C-reactive
protein levels, liver and kidney function tests and muscle enzymes.
In addition, the serum protein level and collagen vascular
screening tests (antinuclear antibodies, C3, C4) were normal.
The patient was treated with a twice-daily topical
application of mometasone furoate 0.1% cream for one month. In
addition, local injections of 1 mg compound betamethasone were
administered once a month for two months. The plaque treated with
local injections of the steroid showed improvement. However, the
patient declined to continue with the injection therapy due to
side-effects, including increased blood pressure and weight gain,
from the systematic use of corticosteroids. The patient showed
slight improvement in the three months of outpatient follow-up
which involved a telephone call every month. Written informed
consent was obtained from the patient prior to participation in the
present study.
Discussion
The term CRDD is used to describe the forms of RDD
that only involve the skin. CRDD differs from RDD in that RDD
exhibits systemic involvement (4,8).
Extranodal forms occur in 43% of RDD cases, with the skin being the
most common site. Approximately 10% of RDD patients exhibit skin
lesions, and in 3% of cases, the disease presents exclusively in
the skin (5). Purely CRDD was
reported for the first time by Thawerani et al (4) in 1978 in a 48-year-old male patient
who presented with a solitary nodule on the shoulder. Since then, a
number of cases of CRDD have been reported (8,9). It
has been suggested that the CRDD and RDD variants of the disease
are distinct clinical entities (10). The purely cutaneous form of RDD, as
observed in the patient of the present study, is very rare. CRDD
has a later age of onset (median age, 43.5 years) compared with
RDD. In addition, CRDD shows a marked female predominance (2:1) and
most commonly affects Asian and Caucasian individuals. By contrast,
RDD has a median onset age of 20.6 years and is slightly more
common in males (1.4:1). The majority of RDD patients are of
African descent and the disease is rarely reported in Asian
patients (10).
The etiology of CRDD remains unknown with viral and
immune causes hypothesized. The polyclonal nature of the cell
infiltrate and the clinical progression of RDD suggest a reactive
process rather than a neoplastic disorder (11). The cell of origin in RDD is
uncertain; however, Middel et al found that stimulation of
monocytes and macrophages via macrophage colony-stimulating factor
generated immunosuppressive macrophages, which may represent the
primary mechanism for the pathogenesis of RDD (12). An additional study by Mannan and
Karak proposed a dendritic cell origin (13). In certain cases, human herpesvirus
(HHV)-6 and Epstein-Barr virus infections were found to be
associated with the pathogenesis of RDD (14). Luppi et al identified HHV-6
viral antigen expression in two cases of RDD (15). Furthermore, Levine et al
used in situ hybridization to detect HHV-6 in the tissues of
RDD patients (14). Parvovirus B19
has also been identified in four RDD patients in an
immunohistochemical study (16).
Histological findings in CRDD are usually similar to
those in RDD. Typically, the epidermis is normal. In the dermis, a
diffuse infiltrate of histiocytes is accompanied by a background
infiltrate of lymphocytes and plasma cells. Lymphoid follicles with
reactive germinal centers may also be present. The phenomenon of
emperipolesis, which represents the presence of intact lymphocytes
within histiocytes, is common in CRDD. Less often, the cytoplasm
may contain plasma cells, neutrophils and red blood cells. Mitoses
and nuclear atypia are rare, while necrosis is absent. CRDD
histiocytes stain positively for S100 protein and CD68, but
negatively for CD1a, which can be used to confirm the diagnosis of
CRDD. Electronic microscopy of CRDD tissue reveals no signs of
Birbeck granules, which eliminates the possibility of Langerhans
cell histiocytosis (17).
Despite the distinctive histological features, the
clinical diagnosis of CRDD is hard to confirm, as the clinical
presentation is variable in the absence of lymphadenopathy. The
lesions in CRDD, which may be solitary or numerous, usually present
as papules, nodules, plaques or as a combination of these (8,9). In
certain cases, the lesions may present as pustules, acneiform
lesions and lesions mimicking vasculitis and panniculitis, or even
a breast mass (18). The patient
in the present study exhibited a facial profusion of indurated
erythematous plaques with a number of reddish-yellow nodules. The
most common site of lesions in CRDD is the face, followed by the
back, chest, thigh, flank and shoulder. The prognosis in CRDD is
reasonably good; however, the condition may be associated with the
involvement of other disorders, including bilateral uveitis,
antinuclear antibody positive lupus erythematosus, rheumatoid
arthritis, hypothyroidism, lymphoma and HIV infection (19,20).
The treatment of CRDD is challenging. Numerous
treatment methods have been attempted; however, an ideal option has
not been identified in all cases and the response is often poor. As
RDD is characterized as a benign and self-limiting disease, it has
been suggested that less aggressive therapeutic approaches should
be used if possible. Surgical excision of the lesions has been
helpful in certain cases. Cryotherapy and local radiation have also
been found to improve the condition. In addition, dapsone and
thalidomide have been effective in cases refractory to other
treatments (5,6). Mixed results have been observed with
isotretinoin and imatinib (7,21); a
number of patients improved, while the condition in other patients
remained poor. Utikal et al described a patient with
complete remission of CRDD after receiving imatinib therapy
(21); however, a different study
reported a patient with CRDD who was completely resistant to this
treatment (22). The patient in
the present study was treated with topical steroids and showed
improvement. However, the patient decided to discontinue treatment
due to side-effects.
In conclusion, CRDD is an unusual clinical entity
comprising a wide-spectrum of lesions, which vary clinically and
histologically. The clinical morphology is variable; however, often
histological features may be characterized to aid the confirmation
of a diagnosis. Generally, CRDD follows a benign clinical course,
with a possibility of spontaneous remission. However, further
molecular and genetic studies are required to explain the
predominant involvement of skin and the higher incidence of the
disease in Asian patients.
References
|
1
|
Destombes P: Adenitis with lipid excess,
in children or young adults, seen in the Antilles and in Mali (4
cases). Bull Soc Pathol Exot Filiales. 58:1169–1175. 1965.(In
French). PubMed/NCBI
|
|
2
|
Rosai J and Dorfman RF: Sinus
histiocytosis with massive lymphadenopathy. A newly recognized
benign clinicopathological entity. Arch Pathol. 87:63–70.
1969.PubMed/NCBI
|
|
3
|
Rosai J and Dorfman RF: Sinus
histiocytosis with massive lymphadenopathy: a pseudolymphomatous
benign disorder. Analysis of 34 cases. Cancer. 30:1174–1188. 1972.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thawerani H, Sanchez RL, Rosai J and
Dorfman RF: The cutaneous manifestations of sinus histiocytosis
with massive lymphadenopathy. Arch Dermatol. 114:191–197. 1978.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chan CC and Chu CY: Dapsone as a potential
treatment for cutaneous Rosai-Dorfman disease with neutrophilic
predominance. Arch Dermatol. 142:428–430. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tjiu JW, Hsiao CH and Tsai TF: Cutaneous
Rosai-Dorfman disease: remission with thalidomide treatment. Br J
Dermatol. 148:1060–1061. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chang LY, Kuo TT and Chan HL: Extranodal
Rosai-Dorfman disease with cutaneous, ophthalmic and laryngeal
involvement: report of a case treated with isotretinoin. Int J
Dermatol. 41:888–891. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Frater JL, Maddox JS, Obadiah JM and
Hurley MY: Cutaneous Rosai-Dorfman disease: comprehensive review of
cases reported in the medical literature since 1990 and
presentation of an illustrative case. J Cutan Med Surg. 10:281–290.
2006.
|
|
9
|
Lu CI, Kuo TT, Wong WR and Hong HS:
Clinical and histopathologic spectrum of cutaneous Rosai-Dorfman
disease in Taiwan. J Am Acad Dermatol. 51:931–939. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brenn T, Calonje E, Granter SR, Leonard N,
Grayson W, Fletcher CD and McKee PH: Cutaneous Rosai-Dorfman
disease is a distinct clinical entity. Am J Dermatopathol.
24:385–391. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Paulli M, Bergamaschi G, Tonon L, Viglio
A, Viglio A, Rosso R, Facchetti F, Geerts ML, Magrini U and Cazzola
M: Evidence for a polyclonal nature of the cell infiltrate in sinus
histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease).
Br J Haematol. 91:415–418. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Middel P, Hemmerlein B, Fayyazi A, Kaboth
U and Radzun HJ: Sinus histiocytosis with massive lymphadenopathy:
evidence for its relationship to macrophages and for a
cytokine-related disorder. Histopathology. 35:525–533. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mannan AA and Karak AK: Sinus
histiocytosis with massive lymphadenopathy (Rosai Dorfman disease):
evidence for a dendritic cell derivation? Indian J Pathol
Microbiol. 48:300–304. 2005.
|
|
14
|
Levine PH, Jahan N, Murari P, Manak M and
Jaffe ES: Detection of human herpesvirus 6 in tissues involved by
sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman
disease). J Infect Dis. 166:291–295. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Luppi M, Barozzi P, Garber R, Maiorana A,
Bonacorsi G, Artusi T, Trovato R, Marasca R and Torelli G:
Expression of human herpesvirus-6 antigens in benign and malignant
lymphoproliferative diseases. Am J Pathol. 153:815–823. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mehraein Y, Wagner M, Remberger K, Füzesi
L, Middel P, Kaptur S, Schmitt K and Meese E: Parvovirus B19
detected in Rosai-Dorfman disease in nodal and extranodal
manifestations. J Clin Pathol. 59:1320–1326. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weitzman S and Jaffe R: Uncommon
histiocytic disorders: the non-Langerhans cell histiocytoses.
Pediatr Blood Cancer. 45:256–264. 2005. View Article : Google Scholar
|
|
18
|
Puppin D Jr, Chavaz P and Harms M:
Histiocytic lymphophagocytic panniculitis (Rosai-Dorfman disease):
a case report. Dermatology. 184:317–320. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Silvestre JF and Aliaga A: Cutaneous sinus
histiocytosis and chronic uveitis. Pediatr Dermatol. 17:377–380.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Perry BP, Gregg CM, Myers S, Lilly S, Mann
KP and Prieto V: Rosai-Dorfman disease (extranodal sinus
histiocytosis) in a patient with HIV. Ear Nose Throat J.
77:855–858. 1998.PubMed/NCBI
|
|
21
|
Utikal J, Ugurel S, Kurzen H, Erben P,
Reiter A, Hochhaus A, Nebe T, Hildenbrand R, Habenkorn U, Goerdt S
and Schadendorf D: Imatinib as a treatment option for systemic
non-Langerhans cell histiocytoses. Arch Dermatol. 143:736–740.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gebhardt C, Averbeck M, Paasch U, Ugurel
S, Kurzen S, Stumpp P, Simon JC and Treudler R: A case of cutaneous
Rosai-Dorfman disease refractory to imatinib therapy. Arch
Dermatol. 145:571–574. 2009. View Article : Google Scholar : PubMed/NCBI
|