Introduction
Lymphangioleiomyomatosis (LAM) is a rare,
genetically determined, progressive disease that occurs frequently
in females of a childbearing age. Sporadic LAM affects one in
400,000 adult females, while the condition occurs in ∼40% of women
with tuberous sclerosis complex (TSC) (1). LAM is a slowly progressive lung disease
that is associated with mutations in TSC genes. In the disease,
neoplastic smooth muscle cells infiltrate the pulmonary parenchyma
and lymphatic system, resulting in extensive tissue remodeling and
architectural distortion of the lung. In addition, LAM is
associated with tumors of the chest and abdomen, including
lymphangiomyomas and angiomyolipomas (2). A diagnosis of LAM can be confirmed with
the presence of pulmonary cysts in computed tomography (CT) scans,
or the proliferation of abnormal smooth muscle cells, as detected
by a lung biopsy (3). Patients with
LAM usually develop progressive dyspnoea and recurrent
pneumothorax, chylous collections and occasional hemoptysis
(4). In the majority of cases, a
biopsy is obtained using video-assisted thoracoscopy. However,
misdiagnosis is common and may result in inappropriate therapeutic
procedures that can further complicate treatment. The present study
reports the case of a patient with sporadic LAM who presented with
bloody sputum as the initial symptom, which subsequently resulted
in a delay in diagnosis. The current study discusses the diagnosis
and treatment protocols of the patient with sporadic LAM, and
compares the etiology, diagnosis and treatment of the case with
previously reported cases of LAM in the literature.
Case report
A 43-year-old female had presented with a small
amount of bloody sputum over the previous five years. The bloody
sputum was considered to be the result of inflammation and was
improved following anti-infection and hemostasis treatment. A chest
X-ray examination performed in 2009 revealed no evident
abnormalities. However, a chest computed tomography (CT) scan
performed in the Traditional Chinese Medical Hospital of Taizhou
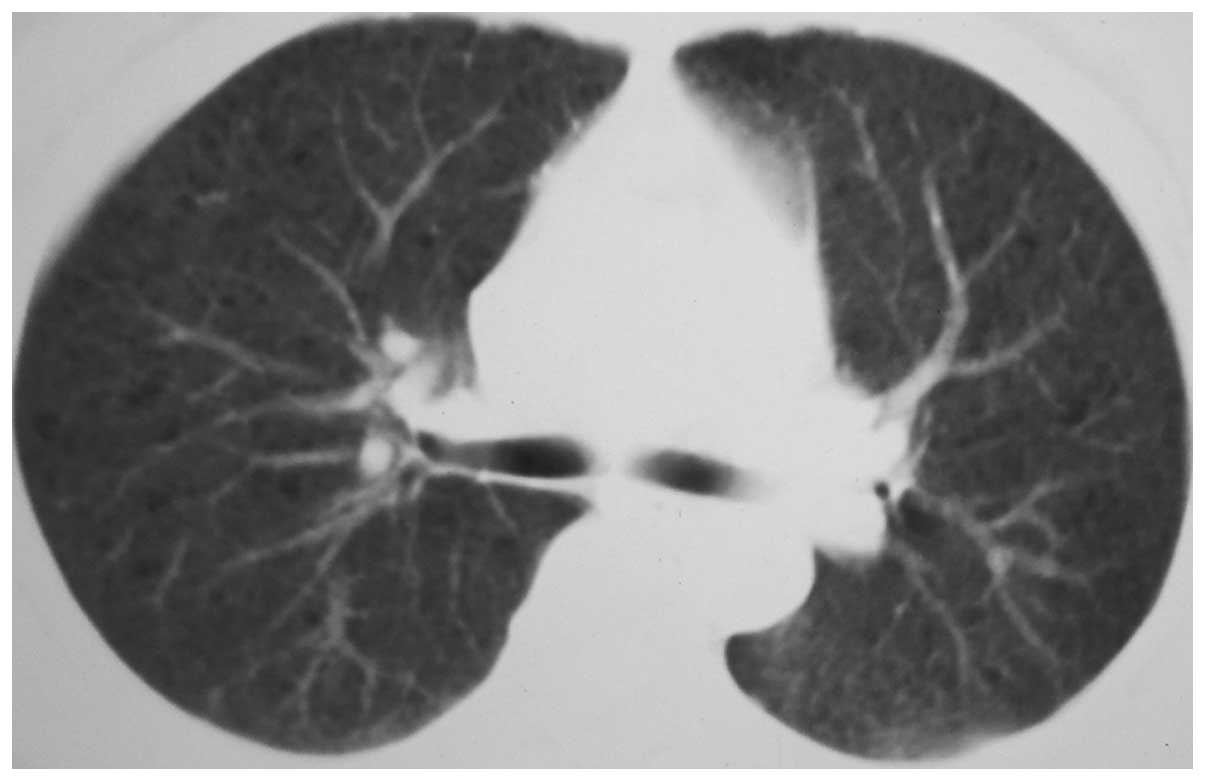
(Taizhou, China) in February 2011 revealed a small area of floc
density shadow with a blurred edge in the tip section of the right
lung, frosting-like changes in the remaining lung fields and a
scattered bullous emphysema shadow in both lung fields (Fig. 1).
The patient was admitted to the Department of
Respiratory Medicine at Taizhou People's Hospital (Taizhou, China)
in January 2012, reporting symptoms of a cough, shortness of breath
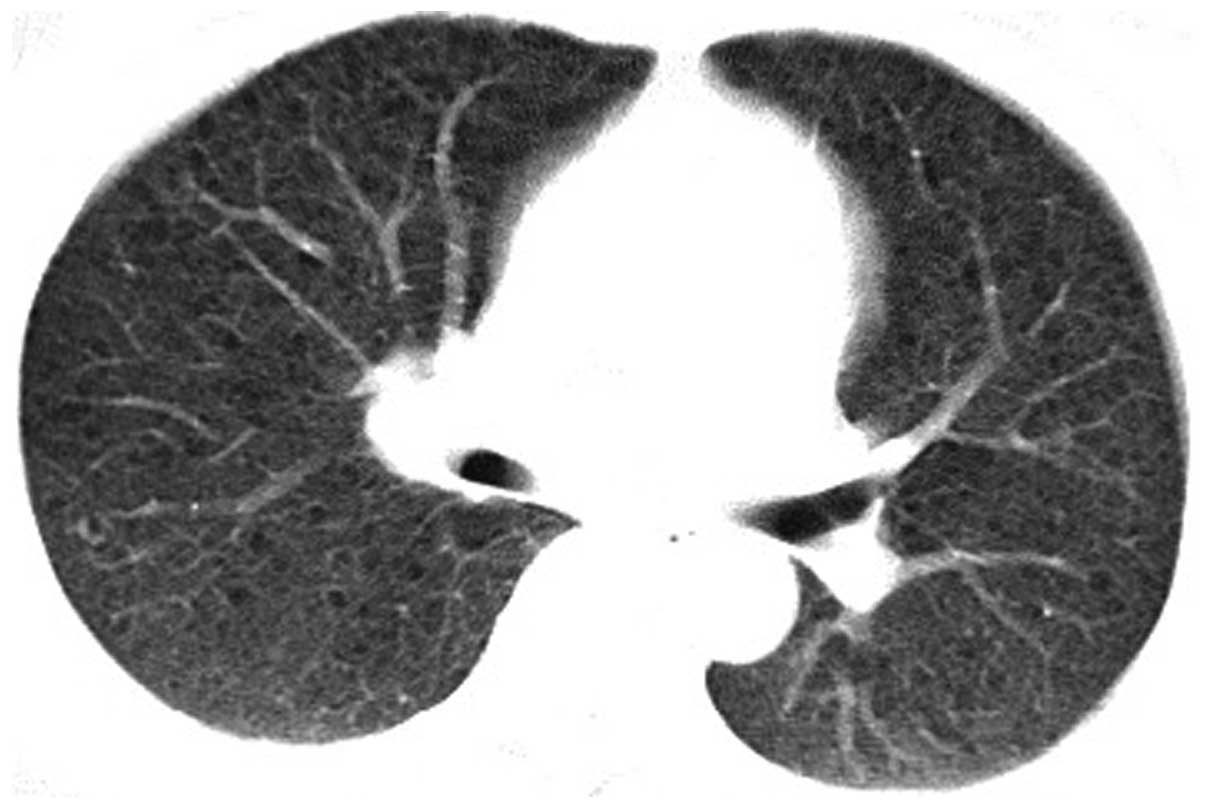
and bloody sputum over the previous 10 days. A high-resolution
computed tomography (HRCT) scan and an enhanced chest CT scan
revealed increasing, thickening and blurred lung markings, ground
glass-like changes, multiple small circular thin-walled translucent
areas in both lung fields and mild mediastinal lymphadenopathy
(Fig. 2). A complete blood test and
biochemical tests produced normal results. In addition, tests for
exfoliated cells and acid-fast bacilli in the sputum were negative.
Color ultrasonography of the spleen, pancreas and gallbladder were
normal. However, a uterine, ovary and fallopian tube color
ultrasonography examination revealed the presence of fibroids in
the uterus. Pulmonary function tests revealed a mild obstructive
ventilatory dysfunction. After hospitalization, the symptoms were
shown to improve following the administration of anti-infection
(levofloxacin 0.4 qd and aztreonam 2.0 bid) and hemostasis
(aminomethylbenzoic acid 0.5 qd) treatment in 10 days. The initial
diagnosis of the patient was LAM. However, the patient refused to
undergo a further biopsy to confirm the diagnosis.
A positron emission tomography-CT examination
performed on February 14 2012 revealed extensive ground glass-like
changes in both lung fields, multiple and scattering diffuse cystic
lesions of low density, ground glass nodules with slightly higher
intake of 18F-fluorodeoxyglucose (18F-FDG) in the right upper lobe,
mediastinal lymph node with no increasing intake of 18F-FDG and
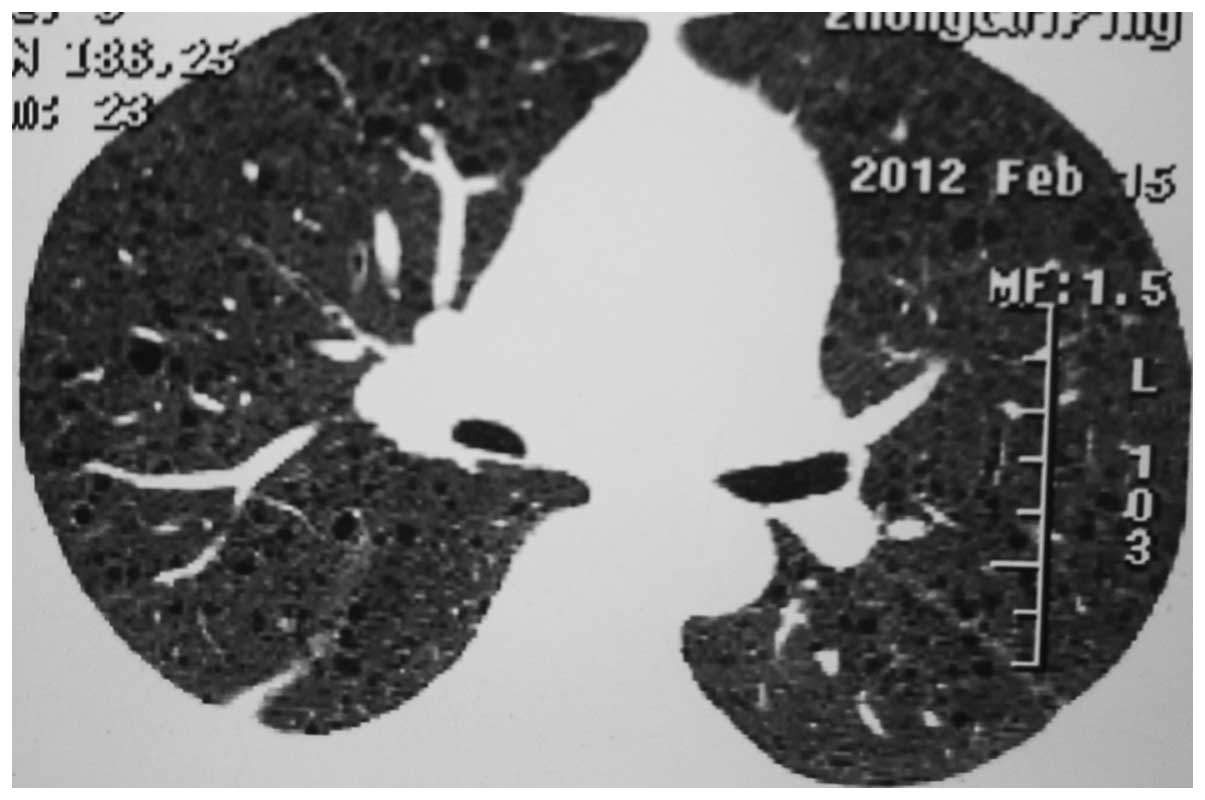
uterus fibroids with an increased 18F-FDG intake. In addition, a
HRCT scan conducted on February 15 2012 revealed ground glass-like
changes and multiple small, circular, thin-walled, translucent
areas in both lung fields, small patchy in the right upper
pulmonary, mediastinal lymph node (Fig.
3). Tests for the hepatitis B surface antibody and hepatitis B
core antibody were positive, whereas the other hepatitis markers
were negative. The level of anti-rheumatoid factor was 22 IU/ml,
while the level of anti-streptolysin O was 47 IU/ml. Tests for
perinuclear and cytoplasmic anti-neutrophil cytoplasmic antibodies
were negative. In addition, tests for anti-nuclear antibodies, such
as anti-ribonucleoprotein/Sm, anti-Sm, anti-SSA, anti-SSB,
anti-SCl-70 and anti-JO-1 antibodies, were all negative. Estradiol,
progesterone and human chorionic gonadotropin levels were within
the normal range. High-risk types of human papilloma virus DNA were
negative. A transbronchial lung biopsy examination failed to
confirm the diagnosis in the Department of Respiratory Medicine.
Thus, the patient underwent video-assisted thoracoscopic surgery to
perform a resection of the right upper lobe and the right lower
lobe tissue on February 29 2012. Pathological analysis confirmed
the diagnosis of LAM (Figs. 4 and
5). In addition,
immunohistochemistry analysis revealed positive staining for human
melanoma black (HMB)-45, melanin A, α-smooth muscle actin (SMA;
Fig. 6), desmin, CD34 and D2-40, and
negative expression for the estrogen receptor (ER) and progesterone
receptor (PR). An additional chest X-ray revealed changes following
the right lung surgery, which included an increased number of fuzzy
lung markings, multiple small, circular, thin-walled, translucent
areas in both lung fields, a small amount of pneumothorax and
pleural effusion in the right lung on March 19 2012. Blood gas
analysis revealed a pH of 7.44, a PaO2 of 67 mmHg and a
PaCO2 of 36 mmHg. A small amount of blood remained
visible in the sputum, and dyspnea on exertion was observed after
discharge. Following discharge the patient was recommended to go to
a post-disease clinic of LAM in Zhongshan Hospital (Shanghai,
China) but since the patient did not attend, anti-infection and
hemostasis treatment was performed in the outpatient clinic of
Taizhou People's Hospital (Taizhou, China).
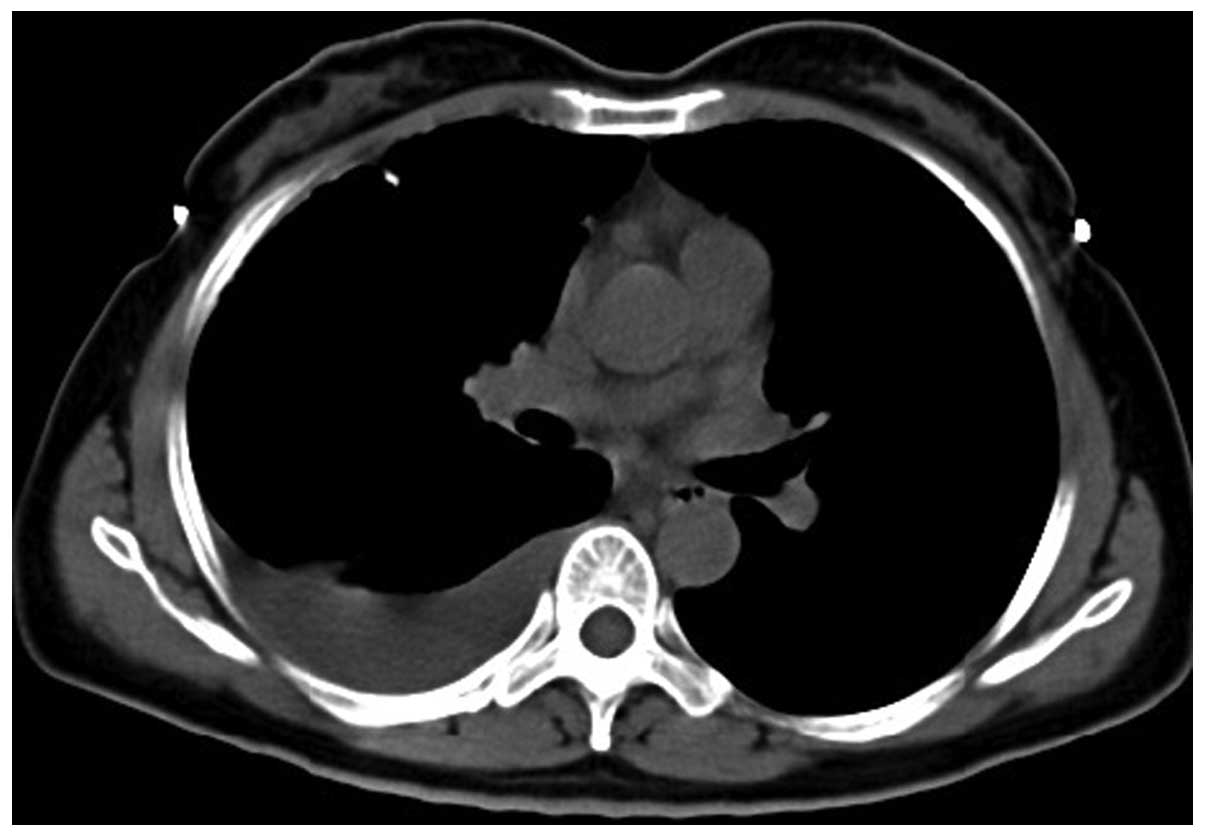
The patient was readmitted to Taizhou People's
Hospital on December 3 2012 due to the presence of blood in the
sputum and pain in the right back. Physical examinations revealed a
bulging right thorax, percussive flatness in the right lower lung,
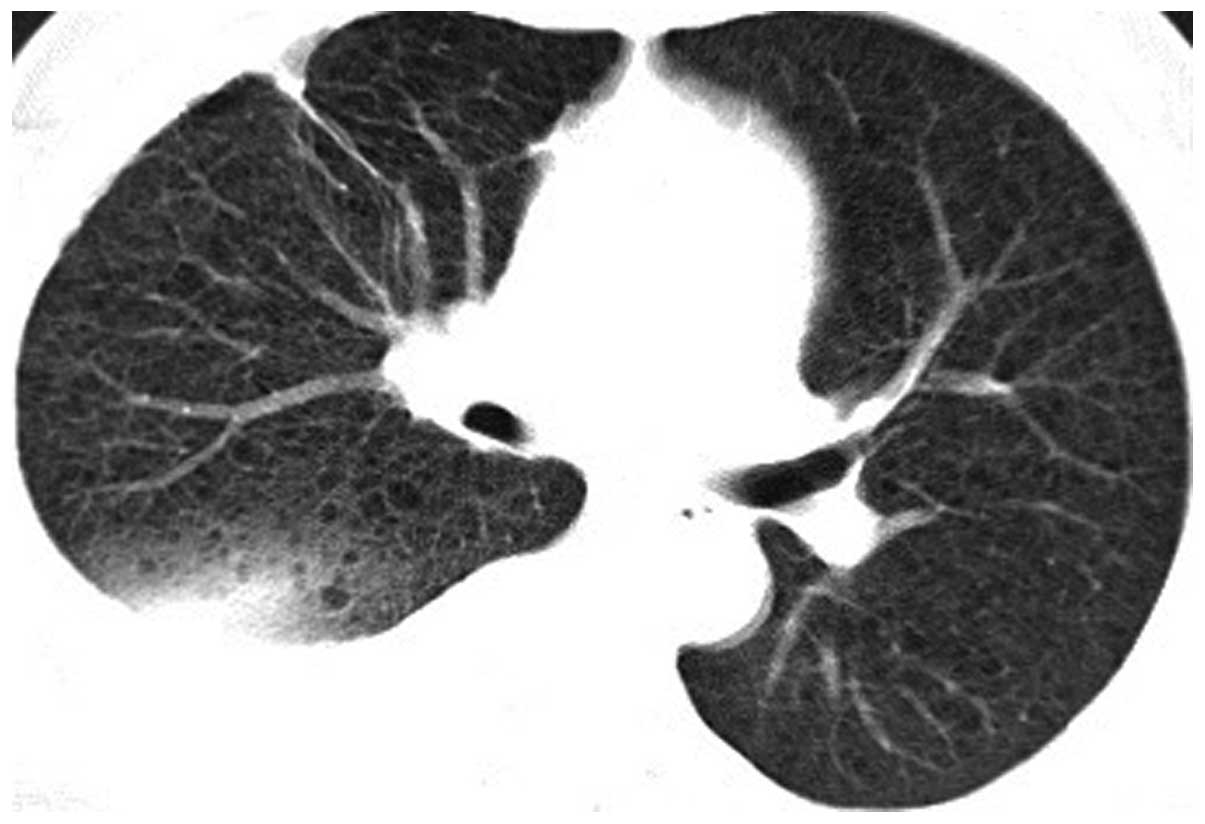
and significantly lower auscultation of the right side. A chest CT
scan of the patient revealed bilateral, diffuse, thin cysts within
the lung parenchyma, right pleural effusion and right lower lung
atelectasis (Figs. 7 and 8). A tube thoracostomy was performed with
an ARROW® catheter (Arrow International, Inc., Reading, PA, USA).
In total, ∼6,000 ml chylous pleural fluid was drained. Rivalta
test: positive (+); nucleated cell count, 3.1×109/l;
monocyte percentage, 80%; polynuclear cell percentage, 20%.
Biochemistry analysis of the pleural fluid revealed 41.5 g/l total
protein, 5.4 mmol/l glucose, 2.55 mmol/l total cholesterol, 23.6
mmol/l triglycerides, 101 U/l lactate dehydrogenase catalase and 5
U/l adenosine deaminase. Furthermore, blood gas analysis revealed a
pH of 7.39, a PaO2 of 75 mmHg and a PaCO2 of
39 mmHg. These examinations were performed on December 7 2012.
The pleural fluid exudation was significantly
reduced following treatment with a fat-free diet, chest tube
drainage, anti-infection (mezlocillin sulbactam 5.0 bid) and
hemostasis (aminomethylbenzoic acid 0.5 qd), nutritional supportive
treatment in 14 days. Lung function following discharge revealed a
mild obstructive ventilatory dysfunction and decreased lung
diffusion capacity, as assessed on December 20 2012. A chest X-ray
was performed on December 28 2012, which revealed a number of
changes following the right lung surgery, including an increased
number of fuzzy lung markings, multiple small, circular,
thin-walled, translucent areas in both lung fields and sharp
costophrenic angles on both sides of the lungs (Fig. 9). Base on the monitoring of an HRCT
scan and pulmonary function test at 3 month intervals, the doctor
suggested to use mTOR inhibitors, such as sirolimus, if the
patient's condition progressed.
The study was approved by the Ethics Committee of
Taizhou People's Hospital (Jiangsu, China) and according to the
Declaration of Helsinki. Written informed consent was obtained from
the patient.
Discussion
Lymphangioleiomyomatosis (LAM) is a rare lung
disease of unknown etiology. The condition is characterized by
cystic remodeling of the lung parenchyma, caused by the
proliferation of abnormal smooth muscle-like LAM cells and the
presence of extrapulmonary manifestations, including
lymphadenopathy, angiomyolipoma and abdominal lymphangioleiomyoma
(5). The pulmonary disease is
characterized by various symptoms, including dyspnea, pleural
effusion, hemoptysis and spontaneous pneumothorax. Chylothorax is
one of the most frequent complications in the course of LAM,
appearing in up to 30% of cases. Pulmonary LAM can be diagnosed
through characteristic features in high-resolution computed
tomography (HRCT) images, or pathological examination via biopsy.
The condition can occur sporadically or in association with
tuberous sclerosis complex (TSC). Currently, there is no curative
treatment for the disease (6). LAM
is a rare, slowly progressive lung disease that almost exclusively
affects young women of a reproductive age (7).
Progressive dyspnea on exertion, pneumothorax and
chylous pleural effusions are the common pulmonary symptoms of LAM,
while hemoptysis may be present occasionally (8). Slow and progressive dyspnea is the
result of airway obstruction and cystic degradation of the
parenchyma (9). Pneumothorax may be
associated with the abnormal proliferation of small airway smooth
muscle, which leads to the obstruction of distal airways.
Spontaneous pneumothorax is the primary symptom of LAM in ∼40% of
cases, and ∼80% of patients have experienced pneumothorax in their
medical history (10). In addition,
chylothorax may be the first symptom of LAM, occurring as a primary
manifestation in ∼25% of patients (11,12). The
obstruction of the lymphatic system is the most important mechanism
underlying the establishment of chylothorax, while the obstruction
of blood vessels leads to the formation of focal areas of
hemorrhage and hemoptysis (13). The
present study reported a sporadic case of LAM, in which the onset
symptom was bloody sputum, and the diagnosis of LAM was confirmed 5
years after the initial presentation. A variety of factors, such as
the insidious onset, normal performance in the early chest X-ray,
no specific symptoms of bloody sputum and clinicians lacking
understanding of the disease, resulted in the missed diagnosis and
misdiagnosis of the disease.
Observations on a chest X-ray are usually normal, or
there may be presentations of pleural effusion and pneumothorax;
thus, diagnosis may be difficult in the early stages of the
disease. HRCT imaging has the greatest diagnostic value and is
necessary to perform for confirmation of diagnosis. In a previous
study, the diagnostic accuracy of LAM was found to be 72% by CT
alone (14). The diagnosis of LAM
requires a HRCT scan that demonstrates diffuse, round or ovoid,
thin-walled cysts, which vary in size between a few millimeters and
3 cm, and are surrounded by normal pulmonary parenchyma, while in
advanced stages of the disease, total replacement of the lung
tissue may be observed (5). A number
of HRCT features characteristic of LAM were outlined in the
European Respiratory Society (ERS) criteria, including multiple
(>10), thin-walled, round, well-defined, air-filled cysts with
preserved or increased lung volume, with no other significant
pulmonary involvement, specifically no interstitial lung disease,
with the exception of possible features of multifocal micronodular
pneumocyte hyperplasia in patients with TSC. HRCT features are
compatible with pulmonary LAM when only a small number of cysts
(>2 and ≤10), as described, are present (1). The ERS criteria indicate that
confirmation of LAM with a lung biopsy is not always necessary when
a HRCT scan exhibits the typical LAM image and there is evidence of
one of the following: Angiomyolipomas, chylous effusion, probable
or definite TSC or lymphangioleiomyomas (1). In the present case, a chest CT scan
performed on February 26 2011 revealed a scattered low-density
cystic lesion; however, the patient was misdiagnosed with another
disease. The main differential diagnosis is diffuse lung disease
that exhibits multiple cysts in the parenchyma, such as idiopathic
pulmonary fibrosis, Langerhans cell histiocytosis, emphysema,
cystic bronchiectasis and cystic fibrosis of lung. Patients should
be suspected of having LAM when they present with typical HRCT
changes, pneumothorax or chylothorax in order to avoid misdiagnosis
(5). Pulmonary function tests can
show an obstructive or restrictive pattern, and the condition is
often accompanied with hypoxemia (15). The lung function and blood gas
analysis are in line with these changes. Previous studies have
shown that the forced expiratory flow in 1 sec (FEV1) and the
carbon monoxide lung diffusing capacity (DLCO) often reflect the
dynamic changes in disease progression, and are associated with the
abnormalities observed in CT and histological examinations in
patients with LAM (16–18). Abnormalities in DLCO are more common
than in FEV1; thus, DLCO may be a more sensitive indicator of early
lesions, while DLCO and FEV1 are likely to be the best current
indicators of disease progression and survival (1).
Pathological examination is very important for the
diagnosis of LAM. Two lesions characterize LAM, namely cysts and a
multifocal nodular proliferation of immature smooth muscle and
perivascular epithelioid cells (LAM cells) (1). LAM cells are smooth muscle cell
precursors that stain positive for several smooth muscle markers,
including α-SMA and desmin, as well as melanocytic markers, such as
HMB-45, human melanosome-associated antigen-1 and Melan-A (5,19). LAM
cells may also express ER and PR. When the pathological features of
LAM are not completely clear, immunohistochemistry of SMA and
HMB-45 should be performed to assist in the diagnosis.
Immunohistochemistry analysis of SMA, desmin and HMB-45 is an
important adjunct to diagnosis (20,21).
Since the samples obtained by transbronchial biopsy are generally
small, analysis of the HMB-45 marker is particularly important for
diagnosis (22). A transbronchial
biopsy may be a safe and effective method for establishing the
diagnosis of LAM, obviating the requirement for surgical lung
biopsy in more than half of LAM patients (23). In the present study, a diagnosis was
unable to be confirmed in the patient using a transbronchial lung
biopsy; however, the patient subsequently underwent video-assisted
thoracoscopic surgery to perform a lung tissue resection.
Pathological analysis and immunohistochemical staining combined
with the CT examination and clinical manifestations were used to
confirm the diagnosis of LAM. In approximately half of LAM cases,
the ER and/or PR can be detected by immunohistochemistry (24). Therefore, tests for PR and ER may aid
diagnosis, although staining for the ER and PR in the present case
was negative.
In the ERS criteria, the gold standard for the
diagnosis of LAM is the clinical and histopathological basis of
LAM. In the present case, the clinical manifestations of the
patient were blood in the sputum, progressive dyspnea on exertion,
right chylothorax and hypoxemia. Lung function assessment revealed
a mild obstructive dysfunction and decreased diffusion capacity. In
such cases, a correlation with clinical features and the CT scan is
essential to increase the confidence level of diagnosis. In
addition, a lung biopsy fitting the pathological criteria for LAM
can further confirm the diagnosis, since the disease can occur
sporadically without the presence of TSC. Substantial chylothorax
was evident following the video-assisted thoracoscopy in the
present case. However, surgery may aggravate the condition, in
addition to considering the original pathogenesis.
In the past, LAM was difficult to diagnose and the
disease had a mortality rate of 100% after 10 years; however, more
recent statistics show there is a 71% survival rate after 10 years
(25,26). At present, there is no effective
treatment for LAM (27). The disease
affects females of a reproductive age and deteriorates during
pregnancy or therapy with exogenous estrogens and contraceptive
pills (28). It is hypothesized that
pregnancy in patients with LAM is associated with an increased risk
of pneumothorax and chylothorax. These observations have led to
several antiestrogenic therapies, including the use of
progesterone, gonadotropin-releasing-hormone analogs, such as
triptorelin and goserelin, and oophorectomy, which appear to
stabilize and improve the disease; however, the effects are
imprecise (5,29–32).
There are no randomized placebo-controlled trials that have
confirmed the efficacy of progesterone and hormonal treatment
(33). The molecular basis of LAM
has been extensively characterized over the past decade, resulting
in the development of a targeted therapy. Recent progress in the
understanding of the molecular pathogenesis of LAM and muscle cell
biology has provided a foundation for the development of novel
therapeutic strategies (27).
Inhibitors of mammalian target of rapamycin (mTOR), matrix
metalloproteinases and angiogenesis are the most promising areas of
research (27). Inherited mutations
of the TSC-1 or TSC-2 genes cause constitutive activation of the
mTOR pathway in LAM. Clinical trials using mTOR inhibitors, such as
sirolimus, for the treatment of LAM have been conducted (34), and patients should be encouraged to
participate in clinical trials using sirolimus, according to the
2010 ERS criteria. Sirolimus is an immunosuppresant approved by the
FDA that functions to inactivate the mTOR complex by imitating
tuberin, and as a result inhibits cell proliferation (35). Recent studies have shown a reduction
of chylous effusions and improvement of the lung function during
sirolimus treatment in lymphangioleiomyomas.
Sirolimus may be considered on an individual basis
in patients with a rapid decline in lung function or symptoms,
after careful evaluation of the risk/benefit ratio in an
experienced center. When sirolimus is used, the effect of therapy
should be carefully monitored for tolerance and effect on lung
function at 3 monthly intervals. The Multicenter International LAM
Efficacy of Sirolimus study demonstrated that therapy with
rapamycin for one year induced stabilization in the FEV1 and forced
vital capacity, a reduction in the serum levels of vascular
endothelial growth factor-D, and an improvement of symptoms and
patient quality of life. However, following discontinuation of the
treatment, the benefits of rapamycin were reversed after 24 months
(36). An open-label,
non-randomized, within-subject, dose escalation, safety,
tolerability and efficacy study of everolimus (a second generation
mTOR inhibitor) in females with sporadic or TSC-LAM is ongoing
(27). Patients with severe or
rapidly progressing LAM may benefit from a lung transplantation.
LAM recurring in the transplanted lung following a single or
bilateral lung transplant is rare and generally asymptomatic.
In conclusion, women of a child-bearing age with
symptoms of pneumothorax, hemoptysis, progressive dyspnoea and
chylous pleural effusions may be suffering from LAM. A HRCT and
lung biopsy should be performed in time, and a lung biopsy can be
used to decrease the rate of misdiagnosis. In this situation, the
increased awareness of diagnosis and knowledge with regard to the
clinical presentations of LAM are key factors in ensuring an
immediate diagnosis and adequate intervention. All patients
diagnosed with LAM should be referred to a LAM treatment center to
receive improved guidance with regard to the treatment options
available (37).
References
|
1
|
Johnson SR, Cordier JF, Lazor R, et al:
Review Panel of the ERS LAM Task Force: European Respiratory
Society guidelines for the diagnosis and management of
lymphangioleiomyomatosis. Eur Respir J. 35:14–26. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Meraj R, Wikenheiser-Brokamp KA, Young LR
and McCormack FX: Lymphangioleiomyomatosis: new concepts in
pathogenesis, diagnosis, and treatment. Semin Respir Crit Care Med.
33:486–497. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pallisa E, Sanz P, Roman A, et al:
Lymphangioleiomyomatosis: pulmonary and abdominal findings with
pathologic correlation. Radiographics. 22:S185–S198. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Johnson S: Rare diseases. 1.
Lymphangioleiomyomatosis: clinical features, management and basic
mechanisms. Thorax. 54:254–264. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mavroudi M, Zarogoulidis P, Katsikogiannis
N, et al: Lymphangioleiomyomatosis: current and future. J Thorac
Dis. 5:74–79. 2013.PubMed/NCBI
|
|
6
|
Hancock E and Osborne J:
Lymphangioleiomyomatosis: a review of the literature. Respir Med.
96:1–6. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chu SC, Horiba K, Usuki J, et al:
Comprehensive evaluation of 35 patients with
lymphangioleiomyomatosis. Chest. 115:1041–1052. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Johnson SR: Lymphangioleiomyomatosis. Eur
Respir J. 27:1056–1065. 2006.PubMed/NCBI
|
|
9
|
Ansótegui Barrera E, Mancheño Franch N,
Vera-Sempere F and Padilla Alarcón J: Lymphangioleiomyomatosis.
Arch Bronconeumol. 47:85–93. 2011.[(In English and Spanish)].
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Almoosa KF, Ryu JH, Mendez J, et al:
Management of pneumothorax in lymphangioleiomyomatosis: effects on
recurrence and lung transplantation complications. Chest.
129:1274–1281. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Johnson SR and Tattersfield AE: Clinical
experience of lymphangioleiomyomatosis in the UK. Thorax.
55:1052–1057. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ryu JH, Doerr CH, Fisher SD, et al:
Chylothorax in lymphangioleiomyomatosis. Chest. 123:623–627. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Juvet SC, McCormack FX, Kwiatkowski DJ, et
al: Molecular pathogenesis of lymphangioleiomyomatosis: lessons
learned from orphans. Am J Respir Cell Mol Biol. 36:398–408. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Koyama M, Johkoh T, Honda O, et al:
Chronic cystic lung disease: diagnostic accuracy of high-resolution
CT in 92 patients. AJR Am J Roentgenol. 180:827–835. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Taveira-DaSilva AM, Stylianou MP, Hedin
CJ, et al: Decline in lung function in patients with
lymphangioleiomyomatosis treated with or without progesterone.
Chest. 126:1867–1874. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Johnson SR and Tattersfield AE: Decline in
lung function in lymphangioleiomyomatosis: relation to menopause
and progesterone treatment. Am J Respir Crit Care Med. 160:628–633.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Avila NA, Chen CC, Chu SC, et al:
Pulmonary lymphangioleiomyomatosis: correlation of
ventilation-perfusion scintigraphy, chest radiography, and CT with
pulmonary function tests. Radiology. 214:441–446. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Taveira-DaSilva AM, Hedin C, Stylianou MP,
et al: Reversible airflow obstruction, proliferation of abnormal
smooth muscle cells, and impairment of gas exchange as predictors
of outcome in lymphangioleiomyomatosis. Am J Respir Crit Care Med.
164:1072–1076. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Krymskaya VP: Smooth muscle-like cells in
pulmonary lymphangioleiomyomatosis. Proc Am Thorac Soc. 5:119–126.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu J, Astrinidis A, Howard S, et al:
Estradiol and tamoxifen stimulate LAM-associated angiomyolipoma
cell growth and activate both genomic and nongenomic signaling
pathways. Am J Physiol Lung Cell Mol Physiol. 286:L694–L700. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gao J, Zhu P, Zhang S, et al: A
clinicopathological analysis of pulmonary lymphangioleiomyomatosis.
Zhongguo Fei Ai Za Zhi. 14:378–382. 2011.[(In Chinese)]. PubMed/NCBI
|
|
22
|
Bonetti F, Chiodera PL, Pea M, et al:
Transbronchial biopsy in lymphangiomyomatosis of the lung. HMB45
for diagnosis. Am J Surg Pathol. 17:1092–1102. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Meraj R, Wikenheiser-Brokamp KA, Young LR,
et al: Utility of transbronchial biopsy in the diagnosis of
lymphangioleiomyomatosis. Front Med. 4:395–405. 2012. View Article : Google Scholar
|
|
24
|
Glassberg MK, Elliot SJ, Fritz J, et al:
Activation of the estrogen receptor contributes to the progression
of pulmonary lymphangioleiomyomatosis via matrix
metalloproteinase-induced cell invasiveness. J Clin Endocrinol
Metab. 93:1625–1633. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Riojas RA, Bahr BA, Thomas DB, et al: A
case report of lymphangioleiomyomatosis presenting as spontaneous
pneumothorax. Mil Med. 177:477–480. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Matsui K, Beasley MB, Nelson WK, et al:
Prognostic significance of pulmonary lymphangioleiomyomatosis
histologic score. Am J Surg Pathol. 25:479–484. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Harari S, Torre O and Moss J:
Lymphangioleiomyomatosis: what do we know and what are we looking
for? Eur Respir Rev. 20:34–44. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yano S: Exacerbation of pulmonary
lymphangioleiomyomatosis by exogenous oestrogen used for
infertility treatment. Thorax. 57:1085–1086. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Johnson SR and Tattersfield AE: Clinical
experience of lymphangioleiomyomatosis in the UK. Thorax.
55:1052–1057. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fujimoto M, Ohara N, Sasaki H, et al:
Pregnancy complicatedwith pulmonary lymphangioleiomyomatosis: case
report. Clin Exp Obstet Gynecol. 32:199–200. 2005.PubMed/NCBI
|
|
31
|
Brunelli A, Catalini G and Fianchini A:
Pregnancy exacerbating unsuspected mediastinal
lymphangioleiomyomatosis and chylothorax. Int J Gynaecol Obstet.
52:289–290. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
McLoughlin L, Thomas G and Hasan K:
Pregnancy and lymphangioleiomyomatosis: anaesthetic management. Int
J Obstet Anesth. 12:40–44. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Casanova A and Ancochea J:
Lymphangioleiomyomatosis: new therapeutic approaches. Arch
Bronconeumol. 47:579–580. 2011.[(In Spanish)]. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bissler JJ, McCormack FX, Young LR, et al:
Sirolimus for angiomyolipoma in tuberous sclerosis complex or
lymphangioleiomyomotosis. N Engl J Med. 358:140–151. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Casanova A, María Girón R, Acosta O,
Barrón M, Valenzuela C and Ancochea J: Lymphangioleiomyomatosis
treatment with sirolimus. Arch Bronconeumol. 47:470–472. 2011.[(In
Spanish)]. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
McCormack FX, Inoue Y, Moss J, et al:
National Institutes of Health Rare Lung Diseases Consortium; MILES
Trial Group: Efficacy and safety of sirolimus in
lymphangioleiomyomatosis. N Engl J Med. 364:1595–1606. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kinder B and McCormack FX: Clinical trials
for rare lung diseases; lessons from lymphangioleiomyomatosis.
Lymphat Res Biol. 8:71–79. 2010. View Article : Google Scholar : PubMed/NCBI
|