Introduction
Traumatic brain injury (TBI) is one of the leading
causes of death in the world and is considered to be a major cause
of adult disability (1). It is
reported that approximately 5 million new cases of TBI occur in the
United States every year, with an estimated annual cost of $60
billion (2,3). After TBI, the primary mechanical
injury leads to various pathological changes comprising the
secondary brain injury, such as blood-brain barrier disruption,
cerebral edema and increase of intracranial pressure, resulting in
long-term and even permanent disability (4). The mechanisms leading to cell death
after TBI are still not fully understood, and no proven
pharmacological treatment exists (5,6).
Glutamate is an extensively distributed, primarily
excitatory neurotransmitter in the mammalian nervous system.
Excessively high extracellular glutamate concentrations, which are
frequently found in the central nervous system (CNS) after TBI,
appear to have an important role in secondary brain injury
(7,8). The glutamate excitotoxicity is
mediated by several glutamate receptor types, including ionotropic
(ligand-gated cation channels) and metabotropic (G-protein coupled)
receptors. The contribution of ionotropic glutamate receptors
(iGluRs) to traumatic brain injury has been widely investigated
(9), and several pharmacological
antagonists of iGluRs show considerable neuroprotective effects in
experimental investigations (10–12). Unfortunately, however, none of
these candidate neuroprotective agents could translate the
theoretical advantage into a real therapeutical benefit for TBI
therapy in clinic, partly because these compounds also alter vital
homeostatic functions that are modulated by the widely distributed
iGluRs. With a more limited distribution and high concentrations in
the CNS, the more recently discovered metabotropic glutamate
receptors (mGluRs) may provide a better option to regulate
excitatory neurotransmission without causing undesired side effects
(13). The mGluRs are classified
on the basis of amino acid sequence homologies, signal transduction
pathways and pharmacological sensitivities into the following three
groups: group I (mGluR1 and 5), group II (mGluR2 and 3) and group
III (mGluR4, 6, 7, 8). Group I mGluRs are typically
postsynaptically localized in somatodendritic domains and coupled
to phosphoinositide (PI) hydrolysis and intracellular
Ca2+ mobilization through phospholipase C (PLC). Several
previous studies have demonstrated that antagonists of these
receptors reduce neuronal damage after TBI (14–16), and the protective effects may be
predominantly mediated by blockage of mGluR1 (17,18). In contrast to mGluR1, most studies
of mGluR5 mainly focus on neurodegenerative diseases, and its role
in neuronal cell death is controversial because of contradictory
results obtained in different disease models (19–21). In the present study, a selective
mGluR5 agonist (R,S)-2-chloro-5-hydroxyphenylglycine (CHPG), which
does not activate mGluR1, was used to examine the specific
contribution of mGluR5 to neuronal damage after TBI.
After traumatic brain injury, both pro-survival and
pro-death pathways are triggered and the balance between these
pathways determines the destination of injured cells and influences
their functional recovery. A critical strategy for the treatment of
TBI is to find compounds that can activate pro-survival signaling
and inhibit pro-death mechanisms. On activation by phosphorylation,
Akt and extracellular signal-regulated kinase (ERK), two
well-characterized pro-survival molecules, are demonstrated to
contribute to protective effects of many neuroprotectants (22,23). Furthermore, it has been suggested
that mGluR can activate Akt and ERK in different models (24,25), but to the best of our knowledge no
investigations of their relationship on TBI have been conducted.
Thus, we examined the effects of CHPG on the activation of Akt and
ERK, and by using specific inhibitors the potential mechanism of
CHPG-induced neuroprotection against TBI was investigated in in
vitro and in vivo models.
Materials and methods
Animals
Adult Sprague-Dawley male rats weighing 280–320 g
were obtained from the Laboratory Animal Center of the Fourth
Military Medical University. The animals had continuous access to
food and water and were housed in cages in a room maintained at
20–22˚C with a 12 h light/12 h dark cycle. All experimental
protocols and animal handling procedures were performed in
accordance with the National Institutes of Health (NIH) guidelines
for the use of experimental animals and approved by the
Institutional Animal Care and Use Committee of the Fourth Military
Medical University.
Drug treatments
PD98059 and LY294002 (Cell Signaling Technology,
Ozyme, France) were dissolved in DMSO and diluted in saline (1%
final DMSO concentration). CHPG (Sigma, Saint Louis, MO, USA) was
dissolved in saline. For the in vitro experiments, CHPG (1
mM), PD98059 (10 μM) or LY294002 (50 μM) was directly added into
the culture medium 30 min before traumatic injury was induced. For
the in vivo experiments, vehicle (1% DMSO in saline), CHPG
(250 nM), PD98059 (5 nM) or LY294002 (15 nM) was injected in a
volume of 5 μl into right lateral ventricle (anteroposterior, 0.8
mm; lateral, 1.5 mm; depth, 3.5 mm from bregma) 30 min before
TBI.
Primary cultures of cortical neurons
Cortical neurons were cultured from Sprague-Dawley
rats using a modified method that has been previously described
(26). Briefly, cortical tissue
was removed from embryos at 16–18 days, and maintained in PBS at
4˚C during dissection. Tissues were dissociated by 0.25% trypsin
digestion for 15 min at 37˚C and gentle trituration. Neurons were
resuspended and plated onto poly-D-Lysine-coated (50 μg/ml) 60 mm
culture dishes at a density of 3×105
cells/cm2. The neurons were cultured in neurobasal
medium (Gibco, Gaithersburg, MD, USA) containing 2% B27, 0.5 mM
L-glutamine and 100 U/ml penicillin at 37˚C in a humidified 5%
CO2 incubator and half of the culture medium was changed
every other day. Cultures were utilized for experiments at 8–10
days when more than 95% of cells were cortical neurons as
determined by immunofluorescence staining of neurofilament 200
(data not shown).
In vitro trauma model
Our in vitro trauma model was based somewhat
on the mechanical injury model described previously (17,27). Briefly, each 60 mm dish confluent
culture was manually scratched with a sterile plastic pipette tip
following a 20×20-square grid (with 3 mm spacing between the
lines). These cuts caused immediate death to cells directly under
the blades, followed by progressive secondary injury of neurons at
a distance from these cuts. After injury, the cultures were washed
with PBS to remove cellular debris, and then incubated for further
24 h at 37˚C in a humidified 5% CO2 incubator.
Lactate dehydrogenase (LDH)
measurement
The release of LDH, a cytoplasmic enzyme released
from neurons with ruined cell membranes, was used as a marker of
neuronal damage and was assessed 24 h after traumatic injury. The
amount of LDH released into the medium was measured using a
diagnostic kit according to the manufacturer's instructions
(Jiancheng Bioengineering Institute, Nanjing, China). Pyruvate and
reduced form of nicotinamide-adenine dinucleotide (NADH) were added
into the medium samples from each group, and after 15 min of
incubation at 37˚C, the reaction was stopped by adding 0.4 mol/l
NaOH. The absorbance of the sample was read at 490 nm, and the
results were expressed as a percentage of LDH release from the
sample vs. the maximal value, which was determined by treating
control cultures with 1% Triton X-100 for 60 min to lyse all
cells.
Identification of apoptotic neurons
Neuronal apoptosis was analyzed by staining the
nuclear chromatin with Hoechst 33342 (Molecular Probes, USA). In
brief, 24 h after TBI the culture medium was removed and neurons
were washed with PBS. Hoechst 33342 (5 μg/ml) was added, and then
neurons were maintained for 15 min at 37˚C in a CO2
incubator. Finally, these labeled neurons were observed using a
Leica fluorescence microscope (B-251, Berlin, Germany), and the
number of apoptotic cells with nuclear condensation and
fragmentation were counted. Apoptotic rate is presented as the
percentage of the total number of neurons.
Traumatic brain injury in vivo
Traumatic brain injury in vivo was induced as
previously described (28). In
brief, rats were anesthetized using 2% isoflurane in oxygen and
placed in the stereotaxic frame. A craniotomy was performed using a
portable drill and a 5 mm trephine over the right parietotemporal
cortex. The resulting bone flap was removed and the dura remained
intact. To induce injury, a pneumatic piston impactor device with a
5 mm diameter and rounded tip (Biomedical Engineering Facility,
Virginia Commonwealth University, Richmond, VA) was used to impact
the brain at a depth of 2 mm for 250 ms. After injury, the bone
flap was replaced and sealed with bone wax. Sham animals underwent
similar anesthetic and surgical interventions, including
craniotomy, but did not receive the TBI application. Core body
temperature was continuously monitored with a rectal probe and
maintained at 37˚C with a thermostatically controlled heating pad
during surgery.
TUNEL staining
Apoptosis in brain sections was detected by the
TUNEL assay, a method used to observe DNA strand breaks in nuclei.
In brief, after being washed in Tris-HCl (pH 7.7) three times,
sections were treated with proteinase K solution (20 μg/ml) for 10
min at room temperature to permeabilize tissues. Sections were then
labelled with fluorescein TUNEL reagent mixture for 60 min at 37˚C
according to the manufacturer's suggested protocol (Promega
Corporation, Madison, USA). The reactions were terminated by
immersing the sections in 2X SSC buffer (0.3 M NaCl, 30 mM
Na3C6H5O7, pH 7.0) for
15 min at room temperature. After that, sections were examined by
fluorescence microscopy and the number of TUNEL-positive
(apoptotic) cells was counted in five fields in each section.
Lesion volume assay
Lesion volume was measured 7 days after TBI. Rats
were anesthetized with 4% isoflurane in oxygen and decapitated. The
brains were rapidly removed and cooled in iced saline for 10 min.
At each 500 μm interval, 30 μm sections were mounted on slides and
stained with 0.2% cresyl violet solution (Sigma Chemical, St.
Louis, MO) to visualize lesions. The areas of the lesions were
integrated, and the results are presented as percentage of
control.
Western blot analysis
For Western blot analysis, cortical neurons and
tissue samples were homogenized in a lysis buffer containing
protease inhibitor 1 mM PMSF and phosphatase inhibitors 10 mM
glycerophosphate, 10 mM NaF and 0.3 mM
Na3Vo4. The lysates were sonicated and
centrifuged, and the protein concentration was determined using a
BCA protein assay kit (Jiancheng Bioengineering Institute).
Equivalent amounts of protein (40 μg/lane) were loaded and
separated on 10% SDS-PAGE gels, and transferred to polyvinylidene
difluoride (PVDF) membranes. Membranes were blocked with 5% nonfat
milk solution in Tris-buffered saline with 0.1% Triton X-100 (TBST)
for 1 h, and then incubated overnight at 4˚C with the following
primary antibody dilutions in TBST: anti-p-ERK1/2, ERK1/2, 1:800;
p-Akt and Akt, 1:1,000 (Cell Signaling Technology, Danvers, MA).
After that the membranes were washed and incubated with secondary
antibody for 1 h at room temperature. The analysis software ImageJ
was used to quantify the optical density of each band. The
activation of Akt and ERK1/2 is presented as the ratio of
phosphorylated kinase bands to the total kinase bands.
Data analysis
Statistical analysis was performed using SPSS 16.0,
a statistical software package. All data are presented as mean ±
SD. Statistical evaluation of the data was performed by one-way
ANOVA followed by Student-Newman-Keuls test (SNK test) for
comparison of differences between the two groups by ANOVA. A value
of P<0.05 was considered statistically significant. All
apoptosis measures were analyzed by observers that were blinded to
treatment grouping.
Results
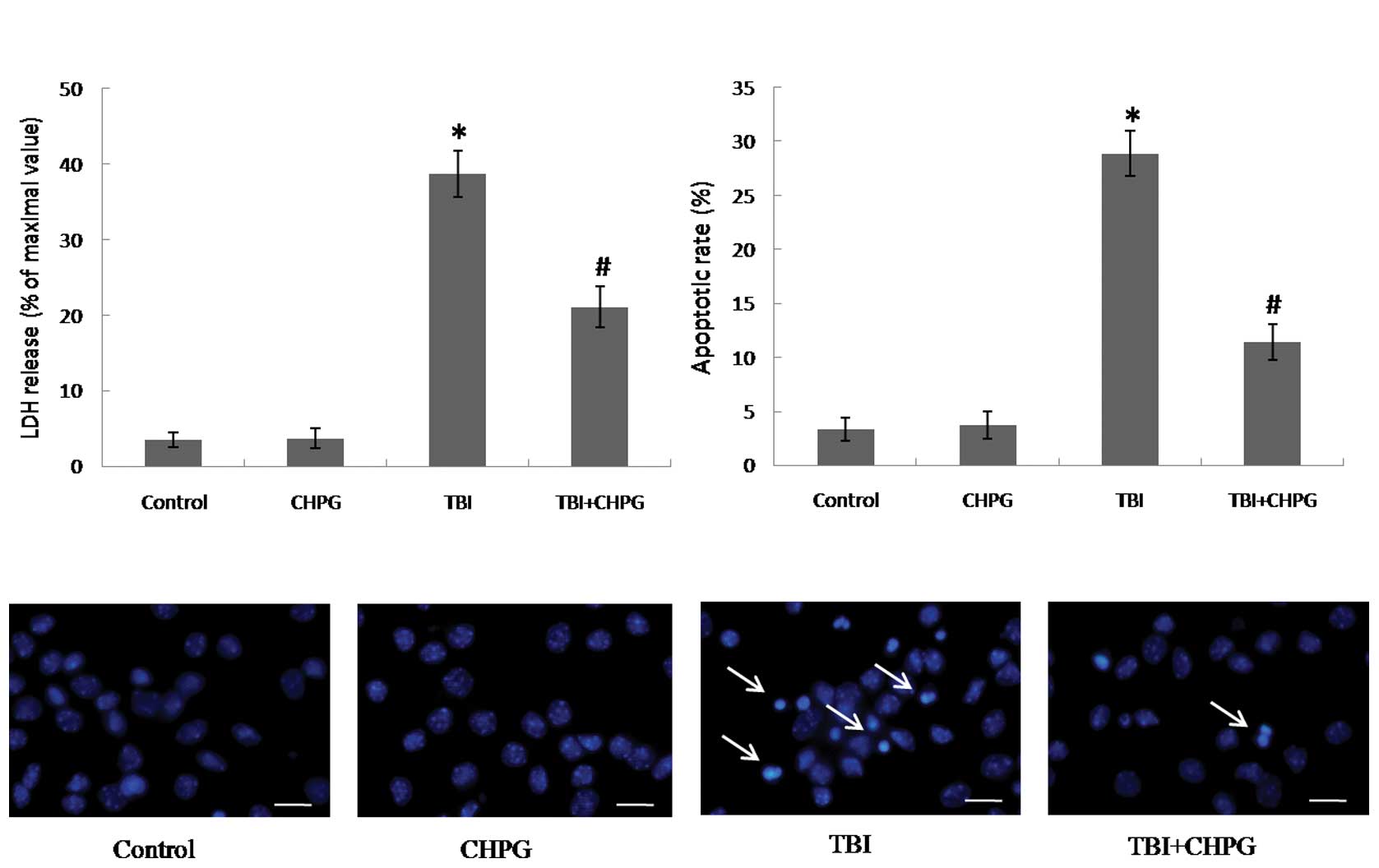
CHPG attenuates neuronal damage in
vitro
To determine the potential protective effects of
CHPG in an in vitro trauma model, LDH release and Hoechst
33342 staining were measured 24 h after mechanical injury (Fig. 1). In this model, injury to neuron
cultures markedly increased LDH release, and this increase was
attenuated by the addition of 1 mM CHPG. CHPG significantly reduced
the neuronal apoptotic rate from 28.9±1.1% in the TBI group to
11.4±1.0% in the TBI+CHPG group (Fig.
1B and C). The results in the control group and the CHPG group
were not apparently different, suggesting that CHPG in all
concentrations used have no cytotoxicity.
CHPG protects against TBI in vivo
To assess the efficacy of CHPG in an in vivo
model of TBI, rats were randomly divided into the following four
groups, the vehicle group (which received saline but did not
undergo TBI application), the CHPG group (which was treated with
CHPG but did not undergo TBI application), the TBI group (which
underwent TBI application) and the TBI+CHPG group (which was
treated with CHPG and underwent TBI application). Rats in the first
two groups underwent similar surgical procedure, but TBI was not
induced. There were no obvious TUNEL-positive cells in the vehicle
group or the CHPG group (Fig. 2).
However, 24 h after TBI the number of TUNEL-positive cells
significantly increased (117.0±5.0 for the TBI group).
Pre-treatment with 250 nM of CHPG attenuated this increase to
63±7.0 for the TBI+CHPG group. Cresyl violet staining, as a measure
of cerebral lesion volume, was done 7 days after TBI, and the
results demonstrated that the lesion volume of the TBI+CHPG group
was significantly smaller than that of the TBI group
(P<0.05).
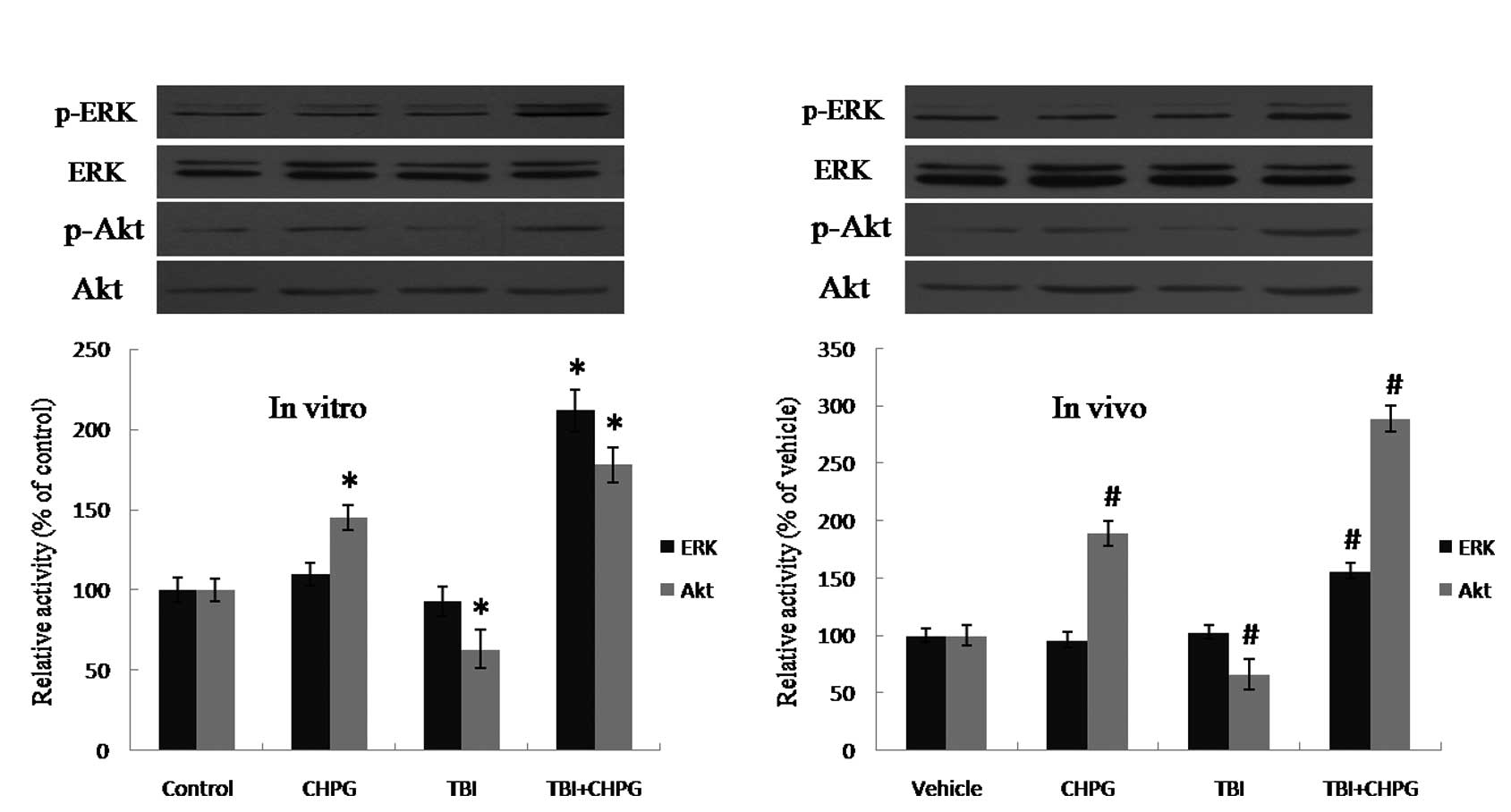
CHPG enhances the activation of ERK and
Akt
To explore possible mechanisms of CHPG-induced
neuroprotection, the expression levels of total and phosphorylated
ERK and Akt, two pro-survival molecules downstream of mGluR5, were
examined by Western blot analysis. CHPG induced an up-regulation
(212±13% in vitro and 156±7% in vivo) of phosphor-ERK
(p-ERK) after traumatic injury, but with no effect on control
neurons or in the non-injured animals (Fig. 3). The expression of phsopho-Akt
(p-Akt) was reduced following traumatic injury. CHPG treatment
alone increased p-AKT levels (145±8% in vitro and 189±11%
in vivo), an effect which was augmented following traumatic
injury (178±9% in vitro and 289±10% in vivo).
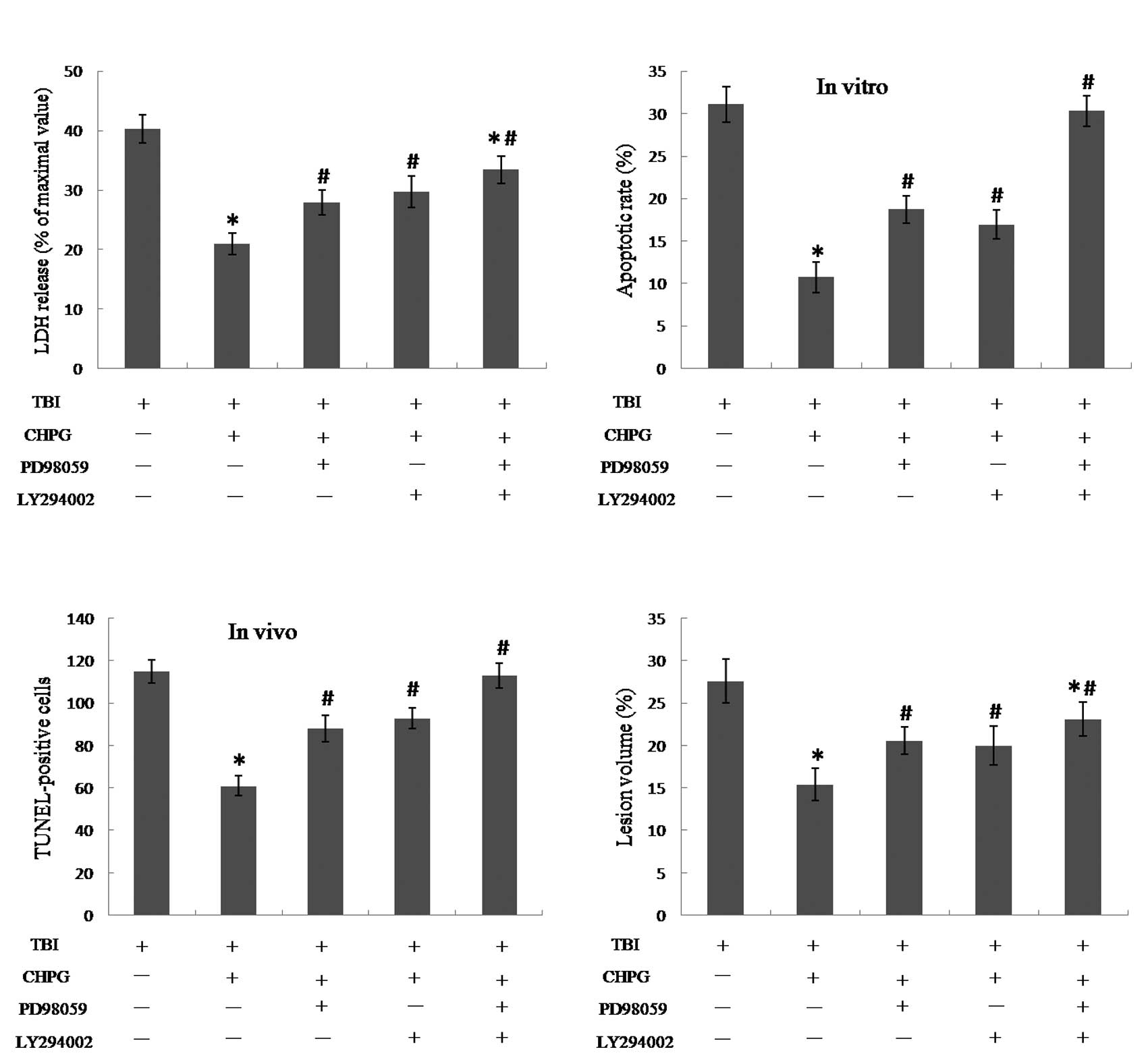
The inhibition of ERK and Akt partially
blocked the protective effects of CHPG
To further elucidate the mechanism of
neuroprotection by CHPG, two antagonists PD98059 and LY294002 were
used in both in vitro and in vivo models of TBI to
block ERK and Akt activation, respectively. CHPG reduced the
TBI-induced LDH release and neuronal apoptosis, and these effects
were diminished by application of either PD98059 or LY294002
(Fig. 4A and B). Similar results
were obtained in the in vivo model of TBI. Specifically,
pretreatment with either PD98059 or LY294002 significantly
increased the number of TUNEL-positive cells and the lesion volume
compared to the TBI+CHPG group (Fig.
4C and D), suggesting that neuroprotection was partially
reversed. When PD98059 and LY294002 were used together, the
CHPG-induced reduction of TBI-induced apoptosis both in
vitro and in vivo was abolished (Fig. 4B and C). LDH release and lesion
volume were increased following co-application of PD98059 and
LY294002 as compared to the TBI+CHPG group, but still lower than
TBI group (P<0.05), suggesting that CHPG-induced protection was
attenuated, but not totally reversed.
Discussion
The major findings of the present study are: i) The
selective mGluR5 agonist CHPG attenuated neuronal damage after
traumatic injury in vitro; ii) CHPG reduced neuronal
apoptosis and lesion volume in an in vivo model of TBI; iii)
CHPG enhanced the expression of p-ERK and p-Akt after traumatic
brain injury; iv) Activated ERK and Akt both contribute to the
protective effects of CHPG against TBI.
It has long been known that group I mGluRs have a
predominantly post-synaptic distribution and can mediate signal
transduction through the activation of Gq-protein and phospholipase
C (29). The distribution of
mGluR5 is greatest in the cortex, striatum and hippocampus, all of
which are sensitive to brain insults, including traumatic injury
(30). The results of previous
studies using general and specific antagonists of mGluR5 suggest
that these receptors play important roles in central nervous system
injury. For example, MPEP and the structurally-related selective
mGluR5 antagonist SIB-1893 significantly attenuated post-traumatic
neuronal cell death and improved functional recovery. However, the
neuroprotective effects of these compounds were mediated by their
antagonism of N-methyl-D-aspartate (NMDA) receptors, not by their
actions on mGluR5 (31). An in
vitro experiment showed that antisense oligodeoxynucleotides
directed at mGluR1 (but not at mGluR5) was neuroprotective
(18). More recently, the
neuroprotective effects of mGluR5 activation were demonstrated
using an in vitro model of β-amyloid-induced cell death
(32,33). Our findings confirm their
observations in in vitro and in vivo TBI models. We
found that potentiating the mGluR5 with the selective agonist CHPG
attenuated traumatic brain injury by inhibiting apoptosis. These
findings are consistent with a previous study in CHO cells
(34). Together, these data
provide strong evidence for the neuroprotective role of mGluR5 in
TBI and suggest for the first time that CHPG, through its
activation of mGluR5, has potential therapeutic applications for
TBI.
Extracellular signal-regulated protein kinase (ERK),
a member of the mitogen-activated protein kinase (MAPK) family, is
a potential downstream mediator of mGluR5 activity (35). ERK participates in cell survival,
and numerous studies demonstrate that ERK activation by
phosphorylation of both threonine and tyrosine residues is
neuroprotective (23,36,37). A previous study demonstrated that
administration of inhibitors of the ERK cascade reduced the
recovery of cognitive and motor deficits in rats with cortical
impact injury (38). As a
beneficial treatment, hypothermia remarkably improved functional
outcome after TBI by augmentation of ERK1/2 activation and its
downstream signalling components (39). The results of the current study
showed that CHPG increased ERK phosphorylation and reduced neuronal
damage after TBI. Furthermore, the protective effects of CHPG were
partially reversed by the selective ERK inhibitor PD98059. These
findings indicate that the neuroprotective effects of CHPG are
associated with an up-regulation of ERK1/2 activation.
Akt, also known as protein kinase B (PKB), is a
serine/threonine kinase and plays a critical role in the modulation
of cell death and survival in the adult brain (40,41). It is well-known that activation of
Akt is dependent upon PI3-K, and the activation of a G
protein-coupled receptor (GPCR) is required to activate PI3-K
(42). As a GPCR, mGluR5 can
activate the Akt pathway through PI3-K (24). In our study, the selective mGluR5
agonist CHPG significantly increased the activation of Akt and
attenuated cell damage induced by TBI. Recent studies have
indicated that active Akt can inactivate several pro-apoptotic
target molecules such as the initiator caspase, caspase-9 (43), the proapoptotic protein Bad and
the transcription factor FKHRL-1 (44,45). Another investigation shows that
Akt can phosphorylate and activate the transcription factor cAMP
response element (CRE)-binding protein (CREB), which is implicated
in the transcription of the anti-apoptotic bcl-2 gene (46). In this study, blocking the
activation of AKT by application of the selective inhibitor
LY294002 partially reversed the anti-apoptotic and neuroprotective
effects of CHPG. This finding suggests that the neuroprotective
effects of CHPG are also mediated by Akt activation.
Cell death is divided into at least two categories,
apoptosis (programmed) and necrosis (mostly non-programmed). In
general, the nature of cell death is dependent on the cell types
and the extent of exposure to an insult, though both forms can
simultaneously occur in a tissue (47). Although we did not discriminate
necrosis from apoptosis in our research, traumatic brain injury can
cause neuronal damage through both forms (48), and both forms contribute to the
increase of LDH release and lesion volume. Interestingly, the
present work found that the anti-apoptotic effect of CHPG was
abolished by inhibitors of ERK and Akt, whereas co-application of
the two inhibitors did not completely reverse the CHPG-induced
neuroprotective effects (as compared to that observed in the TBI
group). Thus, the inhibition of CHPG's anti-apoptotic activity by
co-application of ERK and Akt inhibitors does not completely
reverse its neuroprotective effects. These results suggest that
other mechanisms, such as anti-necrotic pathways, may also be
involved in the CHPG-induced neuroprotection, a possibility which
requires further studies.
In conclusion, our results provide evidence that the
selective mGluR5 agonist CHPG has anti-apoptotic and
neuroprotective effects in in vitro and in vivo
models of TBI. The possible mechanisms through which CHPG provides
neuroprotection are by activating ERK and Akt. Therefore, compounds
that selectively activate mGluR5 may be promising candidates for
the treatment of traumatic brain injury.
Acknowledgements
This study was financially supported by the National
Natural Science Foundation of China (nos. 30670796, 30930093 and
81071034).
References
|
1
|
JA LangloisW Rutland-BrownMM WaldThe
epidemiology and impact of traumatic brain injury: a brief
overviewJ Head Trauma
Rehabil21375378200610.1097/00001199-200609000-0000116983222
|
|
2
|
S YuY KanekoE BaeSeverity of controlled
cortical impact traumatic brain injury in rats and mice dictates
degree of behavioral deficitsBrain
Res1287157163200910.1016/j.brainres.2009.06.06719573519
|
|
3
|
A MammisTK McIntoshAH
ManikerErythropoietin as a neuroprotective agent in traumatic brain
injury ReviewSurg
Neurol71527531200910.1016/j.surneu.2008.02.04018789503
|
|
4
|
JW FinniePC BlumbergsTraumatic brain
injuryVet Pathol39679689200210.1354/vp.39-6-679
|
|
5
|
TW McAllisterPsychopharmacological issues
in the treatment of TBI and PTSDClin
Neuropsychol2313381367200910.1080/1385404090327728919882475
|
|
6
|
RK NarayanME MichelB AnsellClinical trials
in head injuryJ
Neurotrauma19503557200210.1089/08977150275375403712042091
|
|
7
|
R BullockA ZaunerJJ WoodwardFactors
affecting excitatory amino acid release following severe human head
injuryJ Neurosurg89507518199810.3171/jns.1998.89.4.05079761042
|
|
8
|
R BullockA ZaunerJS MyserosA MarmarouJJ
WoodwardHF YoungEvidence for prolonged release of excitatory amino
acids in severe human head trauma. Relationship to clinical
eventsAnn NY Acad
Sci765290298199510.1111/j.1749-6632.1995.tb16586.x7486616
|
|
9
|
AI FadenP DemediukSS PanterR VinkThe role
of excitatory amino acids and NMDA receptors in traumatic brain
injuryScience244798800198910.1126/science.25670562567056
|
|
10
|
J SchumannGA AlexandrovichA BiegonR
YakaInhibition of NR2B phosphorylation restores alterations in NMDA
receptor expression and improves functional recovery following
traumatic brain injury in miceJ
Neurotrauma25945957200810.1089/neu.2008.0521
|
|
11
|
L YurkewiczJ WeaverMR BullockLF
MarshallThe effect of the selective NMDA receptor antagonist
traxoprodil in the treatment of traumatic brain injuryJ
Neurotrauma2214281443200510.1089/neu.2005.22.142816379581
|
|
12
|
L BelayevOF AlonsoY LiuTalampanel, a novel
noncompetitive AMPA antagonist, is neuroprotective after traumatic
brain injury in ratsJ
Neurotrauma1810311038200110.1089/0897715015269372811686490
|
|
13
|
H HomayounB MoghaddamGroup 5 metabotropic
glutamate receptors: role in modulating cortical activity and
relevance to cognitionEur J
Pharmacol6393339201010.1016/j.ejphar.2009.12.04220371231
|
|
14
|
BG LyethQZ GongS ShieldsJP MuizelaarRF
BermanGroup I metabotropic glutamate antagonist reduces acute
neuronal degeneration and behavioral deficits after traumatic brain
injury in ratsExp Neurol169191199200110.1006/exnr.2001.7643
|
|
15
|
QZ GongTM DelahuntyRJ HammBG
LyethMetabotropic glutamate antagonist, MCPG, treatment of
traumatic brain injury in ratsBrain
Res700299302199510.1016/0006-8993(95)01081-68624726
|
|
16
|
Z FeiX ZhangHM BaiXF JiangXL
WangMetabotropic glutamate receptor antagonists and agonists:
potential neuroprotectors in diffuse brain injuryJ Clin
Neurosci1310231027200610.1016/j.jocn.2005.11.04217113985
|
|
17
|
AI FadenDM O'LearyL FanW BaoPG MullinsVA
MovsesyanSelective blockade of the mGluR1 receptor reduces
traumatic neuronal injury in vitro and improves outcome after brain
traumaExp Neurol167435444200110.1006/exnr.2000.757711161632
|
|
18
|
A MukhinL FanAI FadenActivation of
metabotropic glutamate receptor subtype mGluR1 contributes to
post-traumatic neuronal injuryJ Neurosci166012602019968815884
|
|
19
|
N MorinL GregoireB Gomez-MancillaF
GaspariniT Di PaoloEffect of the metabotropic glutamate receptor
type 5 antagonists MPEP and MTEP in parkinsonian
monkeysNeuropharmacology58981986201010.1016/j.neuropharm.2009.12.02420074579
|
|
20
|
A CopaniG CasabonaV BrunoThe metabotropic
glutamate receptor mGlu5 controls the onset of developmental
apoptosis in cultured cerebellar neuronsEur J
Neurosci1021732184199810.1046/j.1460-9568.1998.00230.x9753103
|
|
21
|
A CopaniV BrunoG BattagliaActivation of
metabotropic glutamate receptors protects cultured neurons against
apoptosis induced by beta-amyloid peptideMol
Pharmacol4789089719957746277
|
|
22
|
X XuCC ChuaJ GaoNeuroprotective effect of
humanin on cerebral ischemia/reperfusion injury is mediated by a
PI3K/Akt pathwayBrain
Res12271218200810.1016/j.brainres.2008.06.01818590709
|
|
23
|
B GuerraM DiazR AlonsoR MarinPlasma
membrane oestrogen receptor mediates neuroprotection against
beta-amyloid toxicity through activation of Raf-1/MEK/ERK cascade
in septal-derived cholinergic SN56 cellsJ
Neurochem9199109200410.1111/j.1471-4159.2004.02695.x
|
|
24
|
L HouE KlannActivation of the
phosphoinositide 3-kinase-Akt-mammalian target of rapamycin
signaling pathway is required for metabotropic glutamate
receptor-dependent long-term depressionJ
Neurosci2463526361200410.1523/JNEUROSCI.0995-04.200415254091
|
|
25
|
ES ChoeJQ WangGroup I metabotropic
glutamate receptor activation increases phosphorylation of cAMP
response element-binding protein, Elk-1, and extracellular
signal-regulated kinases in rat dorsal striatumBrain Res Mol Brain
Res947584200110.1016/S0169-328X(01)00217-0
|
|
26
|
L RedmondAH KashaniA GhoshCalcium
regulation of dendritic growth via CaM kinase IV and CREB-mediated
transcriptionNeuron349991010200210.1016/S0896-6273(02)00737-712086646
|
|
27
|
WD HuangZ FeiX ZhangTraumatic injury
induced homer-1a gene expression in cultured cortical neurons of
ratNeurosci Lett3894650200510.1016/j.neulet.2005.07.01416087291
|
|
28
|
M FukushimaSM LeeN MoroDA HovdaRL
SuttonMetabolic and histologic effects of sodium pyruvate treatment
in the rat after cortical contusion injuryJ
Neurotrauma2610951110200910.1089/neu.2008.077119594384
|
|
29
|
R LujanZ NusserJD RobertsR ShigemotoP
SomogyiPerisynaptic location of metabotropic glutamate receptors
mGluR1 and mGluR5 on dendrites and dendritic spines in the rat
hippocampusEur J
Neurosci814881500199610.1111/j.1460-9568.1996.tb01611.x8758956
|
|
30
|
C RomanoMA SesmaCT McDonaldK O'MalleyAN
Van den PolJW OlneyDistribution of metabotropic glutamate receptor
mGluR5 immunoreactivity in rat brainJ Comp
Neurol355455469199510.1002/cne.9035503107636025
|
|
31
|
VA MovsesyanDM O'LearyL FanmGluR5
antagonists 2-methyl-6-(phenylethynyl)-pyridine and
(E)-2-methyl-6-(2-phenylethenyl)-pyridine reduce traumatic neuronal
injury in vitro and in vivo by antagonizing N-methyl-D-aspartate
receptorsJ Pharmacol Exp Ther29641472001
|
|
32
|
VA MovsesyanBA StoicaAI FadenMGLuR5
activation reduces beta-amyloid-induced cell death in primary
neuronal cultures and attenuates translocation of cytochrome c and
apoptosis-inducing factorJ
Neurochem8915281536200410.1111/j.1471-4159.2004.02451.x15189356
|
|
33
|
F LiuX GongG ZhangK MarquisP ReinhartTH
AndreeThe inhibition of glycogen synthase kinase 3beta by a
metabotropic glutamate receptor 5 mediated pathway confers
neuroprotection to Abeta peptidesJ
Neurochem9513631372200510.1111/j.1471-4159.2005.03474.x16277616
|
|
34
|
AJ DohertyMJ PalmerJM HenleyGL
CollingridgeDE Jane(RS)-2-chloro-5-hydroxyphenylglycine (CHPG)
activates mGlu5, but no mGlu1, receptors expressed in CHO cells and
potentiates NMDA responses in the
hippocampusNeuropharmacology36265267199710.1016/S0028-3908(97)00001-49144665
|
|
35
|
L YangL MaoQ TangS SamdaniZ LiuJQ WangA
novel Ca2+-independent signaling pathway to
extracellular signal-regulated protein kinase by coactivation of
NMDA receptors and metabotropic glutamate receptor 5 in neuronsJ
Neurosci2410846108572004
|
|
36
|
Y KurokiK FukushimaY KandaK MizunoY
WatanabeNeuroprotection by estrogen via extracellular
signal-regulated kinase against quinolinic acid-induced cell death
in the rat hippocampusEur J
Neurosci13472476200110.1046/j.0953-816x.2000.01409.x11168553
|
|
37
|
Y ZhuGY YangB AhlemeyerTransforming growth
factor-beta 1 increases bad phosphorylation and protects neurons
against damageJ Neurosci2238983909200212019309
|
|
38
|
PK DashSA MachAN MooreThe role of
extracellular signal-regulated kinase in cognitive and motor
deficits following experimental traumatic brain
injuryNeuroscience114755767200210.1016/S0306-4522(02)00277-412220576
|
|
39
|
CM AtkinsAA Oliva JrOF AlonsoHypothermia
treatment potentiates ERK1/2 activation after traumatic brain
injuryEur J
Neurosci26810819200710.1111/j.1460-9568.2007.05720.x17666079
|
|
40
|
K FukunagaT KawanoAkt is a molecular
target for signal transduction therapy in brain ischemic insultJ
Pharmacol Sci92317327200310.1254/jphs.92.31712939516
|
|
41
|
CJ MullonkalLH Toledo-PereyraAkt in
ischemia and reperfusionJ Invest
Surg20195203200710.1080/0894193070136647117613695
|
|
42
|
ZZ ChongF LiK MaieseActivating Akt and the
brain's resources to drive cellular survival and prevent
inflammatory injuryHistol Histopathol202993152005
|
|
43
|
MH CardoneN RoyHR StennickeRegulation of
cell death protease caspase-9 by
phosphorylationScience28213181321199810.1126/science.282.5392.13189812896
|
|
44
|
SR DattaH DudekX TaoAkt phosphorylation of
BAD couples survival signals to the cell-intrinsic death
machineryCell91231241199710.1016/S0092-8674(00)80405-59346240
|
|
45
|
GM LeinningerC BackusMD UhlerSI LentzEL
FeldmanPhosphatidylinositol 3-kinase and Akt effectors mediate
insulin-like growth factor-I neuroprotection in dorsal root ganglia
neuronsFASEB J1815441546200415319368
|
|
46
|
S PugazhenthiA NesterovaC SableAkt/protein
kinase B up-regulates Bcl-2 expression through cAMP-response
element-binding proteinJ Biol
Chem2751076110766200010.1074/jbc.275.15.1076110753867
|
|
47
|
G MajnoI JorisApoptosis, oncosis, and
necrosis. An overview of cell deathAm J Pathol14631519957856735
|
|
48
|
PG SullivanJN KellerWL BussenSW
ScheffCytochrome c release and caspase activation after traumatic
brain injuryBrain
Res9498896200210.1016/S0006-8993(02)02968-212213303
|