Introduction
Airway narrowing is the final common pathway leading
to symptoms and physiological changes in asthma. Airway smooth
muscle contraction in response to multiple bronchoconstrictor
mediators and neurotransmitters is the predominant mechanism of
airway narrowing. The lung has the capacity to locally generate
angiotensin II (Ang II) (1,2).
Under the pathological conditions of asthma, with antigen
sensitization and challenge, the airway renin-angiotensin system
(RAS) is triggered, thereby elevating the Ang II concentration,
which then contributes to bronchoconstriction and bronchial
hyperactivity (3,4). New constituents of RAS,
angiotensin-(1-7) [Ang-(1-7)] and its main inducer, angiotensin I
converting enzyme 2 (ACE2), have been discovered. As opposed to Ang
II, Ang-(1-7) has been shown to suppress heart or vascular muscle
remodeling, proliferation, migration, inflammation and fibrosis
(5–7). Ang-(1-7) acting via its receptor,
Mas (8,9), is considered a protective factor,
with beneficial effects as opposed to the Ang II-angiotensin type 1
receptor (AT1R), particularly in acute lung injury induced by
severe acute respiratory syndrome (SARS) (10,11). As previously reported, AT1R
expression increased in patients with asthma associated with airway
remodeling and dysfunction; however, valsartan, an AT1R antagonist,
inhibits AT1R expression and partially inhibits structural airway
changes in chronic ovalbumin-exposed rats (12).
Ang II activates the small guanosine tripho-sphatase
(GTPase), RhoA, and its downstream effector, Rho-associated
coiled-coil containing protein kinase 2 (ROCK2), which plays a main
role in the intensity and persistence of vascular smooth muscle
cell contraction and vasoconstriction (13,14). According to recent research, Ang
II acts via AT1R in vascular smooth muscle cells (15,16), hepatic stellate cells (17) and airway smooth muscle cells
(18,19). The RhoA/ROCK2 signaling pathway
also plays a crucial role in biological functions, including
contraction, migration and immunoregulation. Rho kinase activation
is associated with the maintance of the contractive phenotype of
human airway smooth muscle cells (HASMCs) (18).
In the present study, we investigated the possible
effects of Ang-(1-7) on HASMC contraction. We hypothesized that
Ang-(1-7) may inhibit Ang II-induced airway smooth muscle cell
contraction via the Mas receptor. We further hypothesized that
Ang-(1-7) treatment may suppress the Ang II-induced activation of
the RhoA/ROCK signaling pathway, a possible mechanism for its
inhibitory effect on smooth muscle cell contraction.
Materials and methods
Ethics statement
All experimental procedures and protocols in this
study were approved by the Ethics Committee of Nanfang Hospital,
Southern Medical University (NFEC-201109-K1). Prior to the
experiments, the patients were informed of the objectives and
provided written informed consent to participate in the study.
Cell isolation and culture
HASMCs were isolated from the lobar or main bronchus
obtained from lung resection donors, approved by the Division of
Thoracic Surgery. The cells were maintained as primary culture in
Dulbecco’s modified Eagle’s medium (DMEM; Hyclone) with 10% fetal
bovine serum (FBS; Hyclone), 100 U/ml penicillin and 100 U/ml
streptomycin. The morphology and phenotype of the cells was
identified, with a purity of ≥95%. Cells from passages 3–8 were
grown to confluence and were harvested by trypsin digestion and
used for the experiments. To evaluate whether Ang II affects HASMCs
in a time-dependent manner, Ang II was used to stimulate the cells
for 5, 15, 30 or 60 min, at a concentration of 10−7 M
under serum-free conditions. In all other experiments,
10−7 mol/l Ang II with or without Ang-(1-7) was added to
the cell cultures followed by incubation for 15 min. Y-27632 (a
ROCK-2 inhibitor) and irbesartan (IRB, an AT1R inhibitor) were used
at a concentration of 10−5 M, 0.5 h prior to the
addition of Ang II (10−7 M).
Gel contraction assay
HASMCs were treated with IRB (10−5 M) or
Y-27632 (10−5 M) for 30 min, then Ang II
(10−7 M) with or without Ang-(1-7) (10−7 M)
was added to the culture medium and the cells were then added to
the collagen suspension. Buffer without Ang II was used as the
control. Type 1 rat-tail collagen suspension (5 mg/ml; Shengyou
Biotechnology Co., Ltd.) was prepared according to the
manufacturer’s instructions. The final collagen suspension (1
mg/ml) containing the HASMCs in 0.76 ml (1x105 cells)
with or without pre-treatment, was cast in 35-mm culture plates and
allowed to polymerize (20 min, 37°C). Once polymerized, the gels
were carefully detached from the culture plates and filled with
serum-free medium. The gels were equilibrated overnight after
detachment to avoid the initial contraction. The surface area of
the collagen gels was measured using ImageJ analysis software. All
experiments were performed in triplicate. The relative maximum was
expressed by the formula: [(gel surface area of control − gel
surface area of test substance)/gel surface area of control]
x100%.
Immunofluorescence
HASMCs were cultured on slides at a density of
5x104 cells/cm2 and incubated for 24 h to
permit cell attachment. Under serum-free conditions, the cells were
treated with various reagents as described in the section ‘Cell
isolation and culture’. Subsequently, the cells were fixed with
3.7% paraformaldehyde for 10 min. After washing with
phosphate-buffered saline (PBS), the cells were treated with 0.1%
Triton X-100 for 5 min to permeabilize them. To visualize F-actin
stress fibers, the cells were stained with 2 μg/ml of
TRITC-conjugated phalloidin (Sigma) for 20 min at room temperature
and then restained with 4′-6-diamidino-2-phenylindole (DAPI,
BioTime) for the visualization of the nuclei. After mounting,
images were obtained using an inverted fluorescence microscope
(Olympus-FL 500; Tokyo, Japan).
Real-time PCR
Total RNA was isolated from the HASMCs by using the
RNAiso Plus kit. The cDNA was generated by the
PrimeScript® RT reagent kit (from Takara Biotechnology
Co., Ltd.). In order to analyze messenger RNA (mRNA), we
synthesized the template cDNA for the subsequent PCR analyses.
Real-time PCR analysis of the rat genes was performed by the use of
SYBR-Green-based assays with the ABI 7500 Real-Time PCR System
(Applied Biosystems, Foster City, CA, USA), using SYBR®
Premix Ex Taq™ II (from Takara Biotechnology Co., Ltd.). The human
RhoGEF (NM-198997), RhoAGTP (NM-001664.2) and ROCK2 (NM-021804.2)
primers were designed by Primer 5.0 software (Table I). The primers were used at a
concentration of 0.4 μM in each reaction. Cycling conditions
were as follows: step 1, 30 sec at 95°C; step 2, 5 sec at 95°C and
34 sec at 60°C; step 3, 15 sec at 95°C, 1 min at 60°C and 15 sec at
95°C, with repetition of step 2 for 35 times. Data from the
reaction were collected and analyzed by the 7500 software v2.0.4,
using a standard curve. The relative quantification of gene
expression was normalized to GAPDH.
 | Table IPrimers used in the real-time PCR
analyses. |
Table I
Primers used in the real-time PCR
analyses.
| Gene name | Accession no. | Primers | Size (bp) |
|---|
| ARHGEF1 | NM_198997.1 |
5′-AACCAAGCCGTGCGTGAC-3′
5′-TGAACTCGCTCAGCATAGGG-3′ | 109 |
| RhoAGTP | NM_001664.2 |
5′-AGATATGGCAAACAGGATT-3′
5′-TTTCACAAGACAAGGCAC-3′ | 150 |
| ROCK2 | NM_021804.2 |
5′-TATGCGGATTCACTTGTAG-3′
5′-AGTCGTACCTCCCTATCTG-3′ | 137 |
Western blot analysis
Total protein was extracted using the
radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime
Biotech, Haimen, China), containing protease inhibitors (Roche,
Basel, Switzerland) or phosphatase inhibitors (Sigma). Protein
samples (20 μg) were heated at 100°C for 7 min before
loading and separation on 10–12% sodium dodecyl sulfate
polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were
transferred onto a polyvinylidene fluoride (PVDF) membranes
(Bio-Rad, Hercules, CA, USA). Subsequently, the membranes were
blocked with 5% bovine serum albumin in TBST buffer (20 mM Tris,
500 mM NaCl and 0.1% Tween-20) for 1 h at room temperature. The
membranes were then incubated at 4°C overnight with various primary
antibodies at a 1:1,000 dilution in 5% bovine serum albumin buffer.
The following primary antibodies were used: rabbit
anti-phosphorylated myosin light chain (P-MLC), rabbit
anti-phospho-moesin, rabbit anti-MLC and rabbit anti-moesin (all
from Cell Signaling Technology, Danvers, MA, USA). To visualize the
positive protein bands, horseradish peroxidase-linked secondary
antibodies and the ECL system (Thermo, USA) were used. The density
of the individual bands developed on radiographic film was then
quantified using a densitometric scanner with Gel-Pro Analyzer 4
software.
Statistical analyses
All data are presented as the means ± SEM, based on
experiments repeated in triplicate, unless otherwise specified.
Multiple comparisons were analyzed using one-way analysis of
variance (ANOVA) with the Statistical Package for Social Sciences
(SPSS) 13.0 (Statistical Software for Social Sciences, Chicago,
IL). Differences were considered to be statistically significant
when P<0.05.
Results
Cell culture
Primary cultured HASMCs displayed the typical ‘hill
and valley’ appearance when confluenced under an inverted light
microscope. HASMCs were initially characterized by positive
immunostaining for α-smooth muscle actin (α-SMA). There they were
identified by their typical morphology and phenotype, degree of
purity ≥95% (Fig. 1).
Ang II induction of collagen gel
contraction
In order to validate whether Ang II induces the
contraction of HASMCs embedded in collagen gel, a hydrated collagen
gel assay was utilized. Fig. 2
displays a representative experiment with collagen lattice
contraction. As shown in Fig. 2
(upper panel), the control group exposed to gel buffer only,
demonstrated a minimal decrease in the collagen surface area.
Compared with the control cells, Ang II at 10−7 M caused
an obvious decrease in surface gel area. The cells co-treated with
Ang II and Ang-(1-7) demonstrated a slight decrease in the collagen
surface area compared with Ang II, and A779 reversed the effect of
Ang-(1-7) by blocking the Mas receptor. The addition of the
inhibitors, Y-27632 and IRB, to the HASMCs for 0.5 h prior to a
60-min exposure to Ang II, resulted in significant loss of gel
contraction compared with Ang II alone (Fig. 2, upper panel). The quantification
of percentage gel contraction is shown in Fig. 2 (bottom panel).
 | Figure 2Mean maximal contraction of collagen
gels with various stimuli, compared with the initial surface area.
Upper panel: representative images of gel contraction in response
to various stimuli. Lower panel: After incubation with buffer, the
cells were treated with 10−7 M Ang II and Ang-(1-7) with
or without pre-treatment with 10−5 M A779,
10−5 M irbesartan (IRB) or 10−5 M Y-27632 (Y,
a ROCK-2 inhibitor) followed treatment with 10−7 M Ang
II for 60 min. A, Control; B, Ang II; C, Ang II + Ang-(1-7); D, Ang
II + Ang-(1-7) + A779; E, Ang II + IRB; F, Ang II + Y-27632
(*P<0.05 vs. the other groups; #P<0.05
vs. the other groups). |
Ang II induction of increased actin
stress fiber formation
Primary cultured HASMCs developed a fusiform shape,
with some quantity and a regular arrangement of actin stress
fibers. The Fig. 3 showed
cytoskeletal reorganizations triggered by Ang II (10−7
M, 60 min) compared with the other groups in sparsely cultivated
HASMCs typically stained with fluorophore-conjugated phalloidin
with increased mass and stress fibers. Within 30 min of
co-treatment with Ang II and Ang-(1-7), staining of the F-actin
stress fibers showed a significant decrease in stress fibers in the
HASMCs, compared with treatment with Ang II alone. A779 blocked the
effect of Ang-(1-7), and the Ang II-induced increase in actin
stress fiber formation was partially reversed by treatment with
Y-27632 or IRB (Fig. 3).
 | Figure 3Cytoskeletal reorganizations
triggered by the Ang II treatment (10−7 M, 60 min) of
HASMCs. The staining HASMCs with TRITC-phalloidin showed
cytoskeletal changes in the corresponding group F-actin (red) and
DAPI (blue). After a 60-min exposure to 10−7 M Ang II,
the cells had increased numbers of stress fibers and developed
fiber-positive projections. Following costimulation with Ang II and
Ang-(1-7), there were fewer stress fibers. Furthermore, the cells
were pre-incubated for 30 min with A779, an inhibitor of Mas,
irbesartan (IRB), or Y-27632 (Y) at a concentration of
10−5 M and then treated with Ang II or co-treated with
Ang II and Ang-(1-7) for 60 min. The Ang II-induced increase in
stress fiber formation was partially inhibited by Ang-(1-7), IRB
and Y-27632 (Y). In addition, the effect of Ang-(1-7) was partially
reversed by A779. All scale bars, 50 μm. |
Ang II activates the RhoA/ROCK-2
signaling pathway
Ang II induces various pathological effects through
AT1R, such as contraction, migration, cell growth or hypertrophy,
via different signaling pathways (20). However, the mechanisms behind the
effect of Ang II on HASMCs and the role of the RhoA/ROCK2 signaling
pathway remain unknown. Therefore, we performed real-time PCR in
ordr to evaluate the expression of the genes, ARHGEF1, RhoAGTP and
ROCK2, which are key molecules of the RhoA/ROCK2 signaling pathway.
Following treatment with Ang II, the expression of ARHGEF1, RhoAGTP
and ROCK2 in the HASMCs increased, indicating the activation of the
RhoA/ROCK2 pathway (Fig. 4).
Ang-(1-7) partially reversed the increase in ARHGEF1, RhoAGTP and
ROCK2 expression, similar to IRB and Y-37632.
Ang II enhances the level of
phosphorylated moesin (P-moesin) and P-MLC
In order to elucidate the mechanism by which Ang II
induces HASMC contraction, we then determined how the Ang
II-treated HASMCs respond to Ang-(1-7), IRB and Y-27632
co-treatment compared to the untreated HASMCs. Once the RhoA/ROCK
pathway was activated, increasing RhoAGTP levels activated the
ROCK2 synthesis, which was reflected by the phosphorylation of
moesin, as shown by western blot analysis in Fig. 5. Furthermore, moesin [a member of
the ezrin/radixin/moesin (ERM) family of proteins], a substrate of
ROCK2, provides a crucial link with the F-actin cytoskeleton and
membrane proteins at the cell periphery (21). The P-moesin protein was assayed to
elucidate how ERM proteins affect the cell cytoskeleton and
contraction function.
Furthermore, to evaluate the effect of Ang II on MLC
expression, we exposed the HASMCs to Ang II for different amounts
of time, as described in Materials and methods, then measured P-MLC
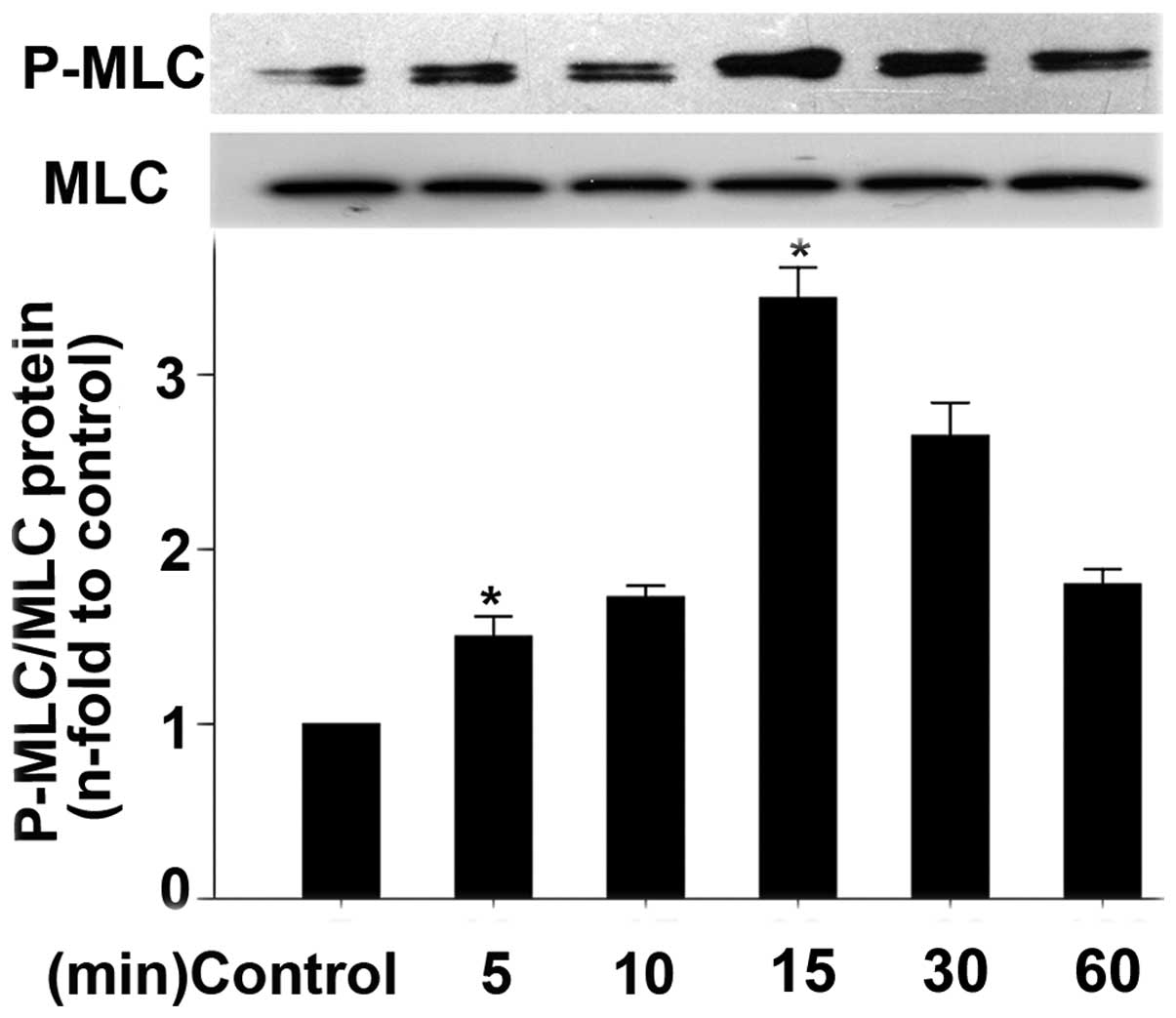
protein expression by western blot analyses. Fig. 6 shows the optical time of the Ang
II stimulation of HASMCs, which is in accordance with a previous
study (22). The Ang II
stimulatin of HASMCs occurred in a time-dependent manner. As shown,
Ang II induced an increase in P-MLC expression as early as 5 min
and this increase peaked at 15 min.
One of the major consequences of the activation of
the RhoA/ROCK2 pathway is the phosphorylation of MLC, which is
critically involved in the contraction of HASMCs. As demonstrated
in Fig. 7, the exposure of the
activated HASMCs to Ang II led to increased amounts of P-MLC. These
effects were significantly inhibited by co-treatment with
Ang-(1-7), IRB and Y-27632. Similarly, pre-treatment with A779
attenuated the inhibitory effect of Ang-(1-7) in response to Ang II
stimulation. The above results suggest that Ang-(1-7) negatively
regulates the contractile response of HASMCs to Ang II via the
inhibition of the RhoA/ROCK2 pathway.
Discussion
The results from the present study demonstrated that
Ang II (10−7 M) caused the significant contraction of
primary cultured HASMCs in embedded in collagen gels. This effect
was inhibited by IRB and Y-27632. Ang-(1-7) suppressed the
contraction of HASMCs caused by Ang II and the inhibitory effects
of Ang-(1-7) were partially reversed by the Ang-(1-7) receptor
antagonist, A799. The difference in contraction among the groups
was confirmed by immunofluorescence. Ang II, a key peptide fragment
of RAS, was originally described as an important regulator of blood
pressure and electrolytic balance, while Ang-(1-7) has demonstrated
cardiovascular protection. Ang II is considered to induce the
contraction of airway smooth muscle and bronchoconstriction
directly and potentiates methacholine (Mch)-induced
bronchoconstriction in mild asthmatic patients (23). A previous meta-analysis suggested
that the insertion/deletion (I/D) polymorphism of the ACE gene, a
key gene in Ang II synthesis, is a risk factor for asthma (24). In the current study, we
demonstrate the role of Ang II in HASMC contraction and
investigated the potential mechanism involved.
Collagen gel contraction assay is a common method
used to examine several constrictors, such as cysteinyl
leukotrienes (cysLTD4), and has been used to measure Mch-induced
contractile capacity in established human bronchial smooth muscle
cells (25,26). It had been used to examine the
contractile capacity of HASMCs between asthmatic and non-asthmatic
patients (27). In our study, we
found out that Ang II as a vasoconstrictor, induced an increase in
the contractility of HASMCs. LTD4 and Mch had a similar effect.
Ang II and Ang-(1-7) are considered as the ‘devil
and angel’ in RAS (6), one
counteracting the effects of the other. Ang-(1-7) plays a
protective role through the Mas receptor by inhibiting AT1R.
However, it is not clear which intracellular signaling pathway is
invovled. Several factors, such as extracellular signal-regulated
kinase (ERK) 1/2 phosphorylation, ERK Akt phosphorylation and
mitogen-activated protein (MAP) kinase inhibition seem to be
involved in this signaling pathway (28–30). A previous study indicated that the
chronic hypotensive effects of losartan in normal rats were
mediated in part through the action of Ang-(1-7) (31). Another study showed that the Mas
receptor is essential in mediating the endothelium-dependent
relaxation response induced by perivascular adipose tissue, thus
highlighting the important role of Ang-(1-7) (32). In the lung, Ang-(1-7) inhibits the
apoptosis of alveolar epithelial cells (AECs) through Mas and ACE-2
regulates the AEC survival by balancing pro-apoptotic Ang II and
its anti-apoptotic degradation product, Ang-(1-7) (33). Further studies are required to
clarify the roles of Ang-(1-7) in vasodilatation, natriuresis,
anti-proliferation and the increase in the bradykinin-nitric oxide
(NO) system, counteracting the effects of Ang II. Therefore, we
investigated whether Ang-(1-7) has an inhibitory effect on the Ang
II-induced contraction of HASMCs.
In our study, the receptor inhibitors, IRB and Mas,
were used to elucidate the potential mechanism of cell contraction,
stress fiber formation and corresponding protein changes. Our
results confirmed that the contraction of HASMCs in response to Ang
II is mediated through the well-characterized RhoA/ROCK2 signal
pathway.
As previously reported, in vascular cells,
endogenous ACE2 counteracts the Ang II-mediated cellular response
partly by the upregulation of Ang-(1-7) through Mas (30). However, in our study, we proved
that Ang-(1-7) inhibits Ang II-mediated cellular contraction via
the downregulation of the RhoA/ROCK2 signaling pathway. ROCK2, a
cytosolic small GTPase Rho kinase, plays a predominant role in
vascular smooth muscle cell contractility (14). The activation of RAS also leads to
the phosphorylation of moesin downstream of Rho and MLC. Moreover,
previous studies have proven that Ang II activates the RhoA/ROCK-2
signaling pathway in vascular smooth muscle cells (34,35).
However, to date, no information is available on the
mechanisms causing the contraction induced by Ang II in HASMCs at
the cellular level. In our study, the inhibition of the RhoA/ROCK2
signaling pathway was clearly demonstrated by the inhibition of the
contraction of HASMCs in the group pre-treated with Y-27632, as the
phosphorylation of MLC downstream of Rho was decreased
significantly.
From the above results, we hypothesized that
Ang-(1-7) may weaken the effects of Ang II by downregulating the
RhoA/ROCK2 signaling pathway. The activation of key molecules,
including RhoGEFs, RhoAGTP and ROCK2, was associated with the
contraction of HASMCs and cytoskeletal rearrangement. Y-27632, as a
ROCK2 inhibitor, demonstrated a significant inhibitory effect on
the increased contraction by inhibiting the increase in stress
fibers. This correlated with the dephosphorylation of moesin.
Traction forces generated by the increase in stress fibers induced
by the addition of Ang II leads to a significant increase in cell
contractility (36,37).
In conclusion, our study reveals a function of RAS
in regulating airway narrowing through its induction of intense
HASMC contraction. Further studies are required to better
understand the underlying mechanism; however, the RhoA/ROCK2
signaling pathway plays a key role in the Ang II/Ang-(1-7)-mediated
regulation of HASMC contraction.
Acknowledgements
This study was supported by the
Presidential Foundation of Nanfang hospital (grant no. 2009B007).
We would like to thank the Department of Thoracic Surgery of our
hospital for its valuable contribution of the lung specimens.
References
|
1.
|
MI PhillipsEA SpeakmanB KimuraLevels of
angiotensin and molecular biology of the tissue renin angiotensin
systemsRegul Pept43120199310.1016/0167-0115(93)90403-U8426906
|
|
2.
|
RP MarshallP GohlkeRC ChambersDC HowellSE
BottomsT UngerRJ McAnultyGJ LaurentAngiotensin II and the
fibroproliferative response to acute lung injuryAm J Physiol Lung
Cell Mol
Physiol286L156L164200410.1152/ajplung.00313.200212754187
|
|
3.
|
E CojocaruIL DumitriuG BogdanM
CostuleanuSM SlatineanuG PetrescuInvolvement of angiotensin on
adenosine-induced bronchial hyperreactivityPneumologia5819232009(In
Romanian)
|
|
4.
|
A VeerappanAC ReidR EstephanN O’ConnorM
Thadani-MuleroM Salazar-RodriguezR LeviRB SilverMast cell renin and
a local renin-angiotensin system in the airway: role in
bronchoconstrictionProc Natl Acad Sci
USA10513151320200810.1073/pnas.070973910518202178
|
|
5.
|
J ZhongR BasuD GuoFL ChowS ByrnsM
SchusterH LoibnerXH WangJM PenningerZ KassiriGY
OuditAngiotensin-converting enzyme 2 suppresses pathological
hypertrophy, myocardial fibrosis and cardiac
dysfunctionCirculation122717728201010.1161/CIRCULATIONAHA.110.95536920679547
|
|
6.
|
M IwaiM HoriuchiDevil and angel in the
renin-angiotensin system: ACE-angiotensin II-AT1 receptor axis vs.
ACE2-angiotensin-(1-7)-Mas receptor axisHypertens
Res32533536200910.1038/hr.2009.7419461648
|
|
7.
|
JL GrobeAP MeccaM LingisV ShenoyTA
BoltonJM MachadoRC SpethMK RaizadaMJ KatovichPrevention of
angiotensin II-induced cardiac remodeling by angiotensin-(1-7)Am J
Physiol Heart Circ
Physiol292H736H742200710.1152/ajpheart.00937.200617098828
|
|
8.
|
V EstebanS Heringer-WaltherA Sterner-KockR
de BruinS van den EngelY WangS MezzanoJ EgidoHP SchultheissM
Ruiz-OrtegaT Waltherangiotensin-(1-7) and the g protein-coupled
receptor MAS are key players in renal inflammationPLoS
One4e5406200910.1371/journal.pone.000540619404405
|
|
9.
|
RA SantosAC Simoes e SilvaC MaricDM
SilvaRP MachadoI de BuhrS Heringer-WaltherSV PinheiroMT LopesM
Baderangiotensin-(1-7) is an endogenous ligand for the G
protein-coupled receptor MasProc Natl Acad Sci
USA10082588263200310.1073/pnas.143286910012829792
|
|
10.
|
W LiMJ MooreN VasilievaJ SuiSK WongMA
BerneM SomasundaranJL SullivanK LuzuriagaTC
GreenoughAngiotensin-converting enzyme 2 is a functional receptor
for the SARS
coronavirusNature426450454200310.1038/nature0214514647384
|
|
11.
|
I HammingW TimensML BulthuisAT LelyGJ
NavisH van GoorTissue distribution of ACE2 protein, the functional
receptor for SARS coronavirus. A first step in understanding SARS
pathogenesisJ Pathol203631637200410.1002/path.157015141377
|
|
12.
|
T WangKS YinKY LiuGJ LuYH LiJD ChenEffect
of valsartan on the expression of angiotensin II receptors in the
lung of chronic antigen exposure ratsChin Med J
(Engl)12123122319200819080339
|
|
13.
|
N KogataRM TribeR FasslerM WayRH
AdamsIntegrin-linked kinase controls vascular wall formation by
negatively regulating Rho/ROCK-mediated vascular smooth muscle cell
contractionGenes Dev2322782283200910.1101/gad.53540919797768
|
|
14.
|
Y WangXR ZhengN RiddickM BrydenW BaurX
ZhangHK SurksROCK isoform regulation of myosin phosphatase and
contractility in vascular smooth muscle cellsCirc
Res104531540200910.1161/CIRCRESAHA.108.18852419131646
|
|
15.
|
K KimuraS EguchiAngiotensin II type-1
receptor regulates RhoA and Rho-kinase/ROCK activation via multiple
mechanisms. Focus on ‘Angiotensin II induces RhoA activation
through SHP2-dependent dephosphorylation of the RhoGAP p190A in
vascular smooth muscle cells’Am J Physiol Cell
Physiol297C1059C1061200919741194
|
|
16.
|
H OhtsuM MifuneGD FrankS SaitoT InagamiS
Kim-MitsuyamaY TakuwaT SasakiJD RothsteinH SuzukiSignal-crosstalk
between Rho/ROCK and c-Jun NH2-terminal kinase mediates migration
of vascular smooth muscle cells stimulated by angiotensin
IIArterioscler Thromb Vasc
Biol2518311836200510.1161/01.ATV.0000175749.41799.9b15994438
|
|
17.
|
J LiR KurubaA WilsonX GaoY ZhangS
LiInhibition of endothelin-1-mediated contraction of hepatic
stellate cells by FXR ligandPLoS
One5e13955201010.1371/journal.pone.001395521085652
|
|
18.
|
R GosensD SchaafsmaH MeursJ ZaagsmaSA
NelemansRole of Rho-kinase in maintaining airway smooth muscle
contractile phenotypeEur J
Pharmacol4837178200410.1016/j.ejphar.2003.10.02714709328
|
|
19.
|
PV LoGrassoY FengRho kinase (ROCK)
inhibitors and their application to inflammatory disordersCurr Top
Med Chem9704723200910.2174/15680260978904445219689376
|
|
20.
|
V Regitz-ZagrosekM NeussJ HolzmeisterC
WarneckeE FleckMolecular biology of angiotensin receptors and their
role in human cardiovascular diseaseJ Mol
Med74233251199610.1007/BF001965778773261
|
|
21.
|
A BretscherK EdwardsRG FehonERM proteins
and merlin: integrators at the cell cortexNat Rev Mol Cell
Biol3586599200210.1038/nrm88212154370
|
|
22.
|
RM TouyzG YaoE VielF AmiriEL
SchiffrinAngiotensin II and endothelin-1 regulate MAP kinases
through different redox-dependent mechanisms in human vascular
smooth muscle cellsJ
Hypertens2211411149200410.1097/00004872-200406000-0001515167449
|
|
23.
|
EA MillarJE NallyNC ThomsonAngiotensin II
potentiates methacholine-induced bronchoconstriction in human
airway both in vitro and in vivoEur Respir
J818381841199510.1183/09031936.95.081118388620948
|
|
24.
|
YG ZhangXB LiJ ZhangJ HuangC HeC TianY
DengH WanD ShresthaYY YangH FanThe I/D polymorphism of
angiotensin-converting enzyme gene and asthma risk: a
meta-analysisAllergy66197205201110.1111/j.1398-9995.2010.02438.x20880211
|
|
25.
|
N KitamuraO KaminumaN KobayashiA MoriA
contraction assay system using established human bronchial smooth
muscle cellsInt Arch Allergy Immunol146Suppl
1S36S39200810.1159/00012605918504405
|
|
26.
|
N KitamuraO KaminumaT OhtomoN KiyokawaN
KobayashiM SukoA MoriEvaluation of cysteinyl leukotriene-induced
contraction of human cultured bronchial smooth muscle cellsInt Arch
Allergy Immunol149Suppl 18386200910.1159/00021137819494511
|
|
27.
|
H MatsumotoLM MoirBG OliverJK BurgessM
RothJL BlackBE McParlandComparison of gel contraction mediated by
airway smooth muscle cells from patients with and without
asthmaThorax62848854200710.1136/thx.2006.07047417412779
|
|
28.
|
JF GianiMM GironacciMC MunozC PenaD
TurynFP DominiciAngiotensin-(1 7) stimulates the phosphorylation of
JAK2, IRS-1 and Akt in rat heart in vivo: role of the AT1 and Mas
receptorsAm J Physiol Heart Circ
Physiol293H1154H1163200710.1152/ajpheart.01395.200617496209
|
|
29.
|
Z SuJ ZimpelmannKD Burnsangiotensin-(1-7)
inhibits angiotensin II-stimulated phosphorylation of MAP kinases
in proximal tubular cellsKidney
Int6922122218200610.1038/sj.ki.500150916672906
|
|
30.
|
N HayashiK YamamotoM OhishiY TataraY
TakeyaA ShiotaR OguroY IwamotoM TakedaH RakugiThe counterregulating
role of ACE2 and ACE2-mediated angiotensin 1–7 signaling against
angiotensin II stimulation in vascular cellsHypertens
Res3311821185201020703229
|
|
31.
|
JP CollisterMD HendelThe role of Ang (1-7)
in mediating the chronic hypotensive effects of losartan in normal
ratsJ Renin Angiotensin Aldosterone
Syst4176179200310.3317/jraas.2003.02814608523
|
|
32.
|
RM LeeM BaderN AleninaRA SantosYJ GaoC
LuMas receptors in modulating relaxation induced by perivascular
adipose tissueLife
Sci89467472201110.1016/j.lfs.2011.07.01621820449
|
|
33.
|
BD UhalX LiA XueX GaoA
Abdul-HafezRegulation of alveolar epithelial cell survival by the
ACE-2/angiotensin 1–7/Mas axisAm J Physiol Lung Cell Mol
Physiol301L269L274201121665960
|
|
34.
|
C Cario-ToumaniantzD Ferland-McColloughG
ChadeufG ToumaniantzM RodriguezJP GalizziB LockhartA BrilE
ScalbertG LoirandP PacaudRhoA guanine exchange factor expression
profile in arteries: Evidence for a Rho kinase-dependent negative
feedback in angiotensin II-dependent hypertensionAm J Physiol Cell
Physiol302C1394C1404201210.1152/ajpcell.00423.2011
|
|
35.
|
C SavoiaF TabetG YaoEL SchiffrinRM
TouyzNegative regulation of RhoA/Rho kinase by angiotensin II type
2 receptor in vascular smooth muscle cells: role in angiotensin
II-induced vasodilation in stroke-prone spontaneously hypertensive
ratsJ Hypertens2310371045200510.1097/01.hjh.0000166845.49850.39
|
|
36.
|
CD GildnerAL LernerDC HockingFibronectin
matrix polymerization increases tensile strength of model tissueAm
J Physiol Heart Circ
Physiol287H46H53200410.1152/ajpheart.00859.200315001442
|
|
37.
|
DC HockingJ SottileKJ
LangenbachStimulation of integrin-mediated cell contractility by
fibronectin polymerizationJ Biol
Chem2751067310682200010.1074/jbc.275.14.1067310744764
|