Introduction
Gemcitabine is a well-established anticancer agent
and has been FDA approved either as a single agent for the
treatment of pancreatic cancer (1) or in combination with another agent
(carboplatin, cisplatin or paclitaxel) for the treatment of ovarian
(2) and metastatic breast cancer
(3). Cisplatin is indicated as
first-line therapy for inoperable, locally advanced (stage IIIA or
IIIB) or metastatic (stage IV) non-small cell lung cancer (NSCLC)
(4,5), and has also been studied in other
solid tumours such as malignant pleural mesothelioma (MPM)
(6,7) and prostate cancer (8,9).
Gemcitabine is considered to act as a
pyrimidine-type anti-metabolite. The agent is a prodrug and
requires intracellular conversion to two active metabolites,
gemcitabine diphosphate and gemcitabine triphosphate, which can
function in two ways: by binding to and irreversibly inhibiting
ribonucleotide reductase (RNR) and by replacing one of the building
blocks of nucleic acids, in this case cytosine, during DNA
replication (10).
Aberrant epigenetic regulation of gene expression is
a frequent event in cancer (11).
Several of the proteins which regulate histone post-translational
modifications have now been shown to play a role in resistance to
various cancer therapies including cisplatin (12), gefitinib (13), etoposide (14) and tamoxifen (15).
DNA CpG methylation is another epigenetic
modification that is linked to loss of gene expression in cancer
(16). Inhibitors targeting DNA
methyltransferases have been developed. Of these 5-azacytidine
(Vidaza®) and 5-aza-2-deoxycytidine
(decitabine/Dacogen®) have gained FDA approval for the
treatment of myelodysplastic syndrome subtypes (17,18).
Notably, the chemotherapy agent doxorubicin has been
shown to inhibit the DNA methyltransferase DNMT1 (19) and indicates that other
chemotherapy drugs may potentially inhibit the epigenetic enzymatic
machinery. Since decitabine and gemcitabine are cytosine analogues,
and share similar structural features (Fig. 1), we reasoned that gemcitabine may
also be able to act as a DNA methyltransferase inhibitor
(DNMTi).
Materials and methods
Cell lines
The A549 (adenocarcinoma), SKMES1 (squamous cell
carcinoma), H-28 (MPM), 22Rv1 (prostate), LNCaP (prostate) and
Du145 (prostate) cell lines were purchased from the ATCC (LGC
Promochem). Cells were cultured in appropriate media and maintained
at 37°C in a humidified atmosphere containing 5%
CO2.
Cell line treatments
Decitabine was purchased from Merck and dissolved in
methanol. Cell cultures were treated for 48 h at a final
concentration of 5 μM, with the media and drug replaced
every 24 h as previously described (20).
Gemcitabine was supplied by Eli Lilly and dissolved
in phosphate-buffered saline (PBS) at a final concentration of 38
mg/ml (120.14 mM). Cell cultures were treated for 48 h at final
concentrations of 0.2, 0.5 or 1 μM with the media and drug
replaced every 24 h. We chose to use this concentration based on
literature searches in Pubmed.
RNA isolation and RT-PCR
amplification
Total RNA was extracted using TRI
Reagent® (MRCgene) according to the manufacturer’s
instructions. One microgram of total RNA was used to generate cDNA
using M-MLV-reverse transcriptase (Promega) according to the
manufacturer’s instructions.
Expression of VEGFR1, VEGFR2, sFRP4, RASSF1A and
β-actin was examined by RT-PCR, using primers and PCR conditions
outlined in Table I. Each PCR was
carried out for 35 cycles. Ten microlitres of the experimental
RT-PCR product and 2 μl of the β-actin RT-PCR product were
loaded onto a 1% agarose gel.
 | Table IPrimers and annealing temperatures
for RT-PCR. |
Table I
Primers and annealing temperatures
for RT-PCR.
| Primer Set | Sequence | Annealing Temp
(°C)/PCR cycles |
|---|
| VEGFR1 | F:
5′-CAAGTGGCCAGAGGCATGGAGTT-3′ | 56°C/35 cycles |
|
R:5′-GATGTAGTCTTTACCATCCTGTTG-3′ | |
| VEGFR2 | F:
5′-GAGGGCCTCTCATGGTGATTGT-3′ | 56°C/35 cycles |
| R:
5′-TGCCAGCAGTCCAGCATGGTCTG-3′ | |
| sFRP4 | F:
5′-TCTATGACCGTGGCGTGTGC-3′ | 56°C/35 cycles |
| R:
5′-ACCGATCGGGGCTTAGGCGTTTAC-3′ | |
| RASSF1A | F:
5′-CAGATTGCAAGTTCACCTGCCACTA-3′ | 56°C/35 cycles |
| R:
5′-GATGAAGCCTGTGTAAGAACCGTCCT | |
| β-actin | F:
5′-TGTTTGAGACCTTCAACACCC-3′ | 56°C/35 cycles |
| R:
5′-AGCACTGTGTTGGCGTACAG-3′ | |
Quantitative PCR
RNA was isolated from the cell lines using a miRVana
miRNA isolation kit (Applied Biosystems, UK) according to the
manufacturer’s guidelines. Total RNA (1 μg) was reverse
transcribed using the high capacity cDNA Archive kit. Expression of
GSTP1 (TaqMan® gene expression assay ID: Hs00168310_m1)
and IGFBP3 (TaqMan gene expression assay ID: Hs00181211_m1) was
quantified in the cell lines by qRT-PCR. Human phosphoglycerate
kinase 1 (PGK1) was used as an endogenous control (TaqMan gene
expression assay ID: Hs99999906_m1). TaqMan PCR reactions were
performed in triplicate on an ABI Prism 7900 sequence detection
system. Gene expression was calculated relative to the untreated
cell lines, using SDS RQ Manager 1.2 software, which automatically
determines relative quantities (RQ), by applying the arithmetic
formula 2−ΔΔCT. All equipment and reagents were supplied
by Applied Biosystems, Foster City, CA.
DNA methyltransferase assay
Analysis of DNA methyltransferase activity was
carried out using the EpiQuik™ DNA methyltransferase
activity/inhibition assay kit (Epigentek) using both recombinant
DNMTs (Epigentek) and nuclear extracts isolated with the EpiQuik
Nuclear Extraction kit II (Epigentek) according to the
manufacturer’s instructions. Following consultation with the
manufacturers, the recombinant proteins were incubated with the
various concentrations of decitabine or gemcitabine for 90 min at
room temperature prior to conducting the methyltransferase
assay.
Genomic DNA isolation, bisulfite
conversion and MS-PCR analysis
Genomic DNA was isolated from the cell lines using a
solution containing 0.5% SDS and 100 μg/ml Proteinase K
(21).
Genomic DNA from the cell lines (500 ng) was
bisulfite modified using the EpiTect Bisulfite kit (Qiagen, UK),
following the manufacturer’s guidelines. EpiTect methylated DNA and
unmethylated DNA were used as controls.
Promoter hypermethylation of the GSTP1 promoter was
analysed by methylation-specific PCR (MS-PCR) in the LNCaP and
22Rv1 cell lines using the GoTaq HotStart enzyme (Promega). PCR
primer sets complementary to both modified, methylated DNA (M) and
modified, unmethylated DNA (U) were designed for GSTP1: GSTP1 MF2,
5′-TTCGGGGGTGTA GCGGTCGTC-3′; GSTP1 MR1, 5′-CCAACGAAAACCTCGC
GACCTCCG-3′ (expected product, 145 bp); GSTP1 UF2,
5′-GATGTTTGGGGTGTAGTGGTTGTT-3′; GSTP1 UR1,
5′-AAACTCCAACAAAAACCTCACAACCTCCA-3′ (expected product, 154 bp).
Bisulfite pyrosequencing
Pyrosequencing methylation analysis (PMA) of the
LINE-1.2 element, the RASSF1A promoter and the VEGFR gene
promoters, was performed as previously described (22–24). Genomic DNA (500–1,000 ng) was
bisulfite treated, using the EpiTect Bisulfite kit or the EZ DNA
Methylation™ kit (ZymoResearch). The PyroMarkAssay Design software
was used to design the primers for amplification and sequencing to
cover a number of CpG sites as shown in Table II. PCR amplification products were
cleaned and subjected to pyrosequencing on either a PyroMark Q96 ID
pyrosequencer using PyroMark Gold Q96 SQA reagents (all were from
Qiagen), or on a Q24 pyrosequencer according to the manufacturer’s
protocol (Biotage). The methylation of C in each analysed CpG site
was quantified from 0 to 100%, using the PyroMark software
(Qiagen).
| Table IIPrimers for bisulfite
pyrosequencing. |
Table II
Primers for bisulfite
pyrosequencing.
| LINE1.2 (6
CpGs) |
| LINE-1 fwd:
5′-BIO-TAGGGAGTGTTAGATAGT GG-3′, |
| LINE-1 rev:
5′-AACTCCCTAACCCCTTAC-3′, |
| LINE-1 seq:
5′-CAAATAAAACAATACCTC-3′. |
| RASSF1A (9
CpGs) |
| RASSF1 methF:
5′-AGTATAGTAAAGTTGGTTTTTAGAAA-3′ |
| RASSF1 methR:
5′-CCCTTCCTTCCCTCCTT-3′ |
| RASSF1 methPSEQ:
5′-AAGTTGGTTTTTAGAAATA-3′ |
| VEGFR1 (15
CpGs) |
| VEGFR1 PyroF:
5′-AGGAGGGGTAAGGGTAAGAG-3′ |
| VEGFR1 PyroR:
5′-TCCCCACCTACCCTCTTCTT-3′ |
| VEGFR1 PyroSEQ:
5′-GGGAGAGGAGTAAAGATTTTGAATT-3′ |
Western blot analysis
Protein lysates were extracted from cell cultures
using RIPA buffer [50 mM Tris HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA,
1% (v/v) Triton X-100, 0.1% (w/v) SDS], supplemented with 10
μl phenylmethylsulfonyl fluoride (87 mg/ml in 96% EtOH) and
100 μl protease inhibitor cocktail (2 mM AEBSF, 1 mM EDTA,
130 μM bestatin, 14 μM E-64, 1 μM leupepin,
0.3 μM aprotinin). Lysates were separated by SDS/PAGE and
subsequently transferred onto pre-activated polyvinylidene fluoride
nitrocellulose membranes (PVDF). Membranes were blocked for 1 h at
room temperature (RT) in TBST [10 mM Tris-HCl (pH 7.5), 10 mM NaCl,
0.1% (v/v) Tween 20] containing 5% nonfat dry milk powder.
Membranes were immunoblotted overnight at 4°C in TBST with 5%
nonfat dry milk powder with DNMT1 (supplier) with HDAC1 (Cell
Signalling Technologies) used as a loading control as appropriate.
All secondary antibodies were HRP-labelled and bound antibody
complexes were detected using the Supersignal West Pico
Chemiluminescent kit (Pierce, Rockford, IL, USA).
Results
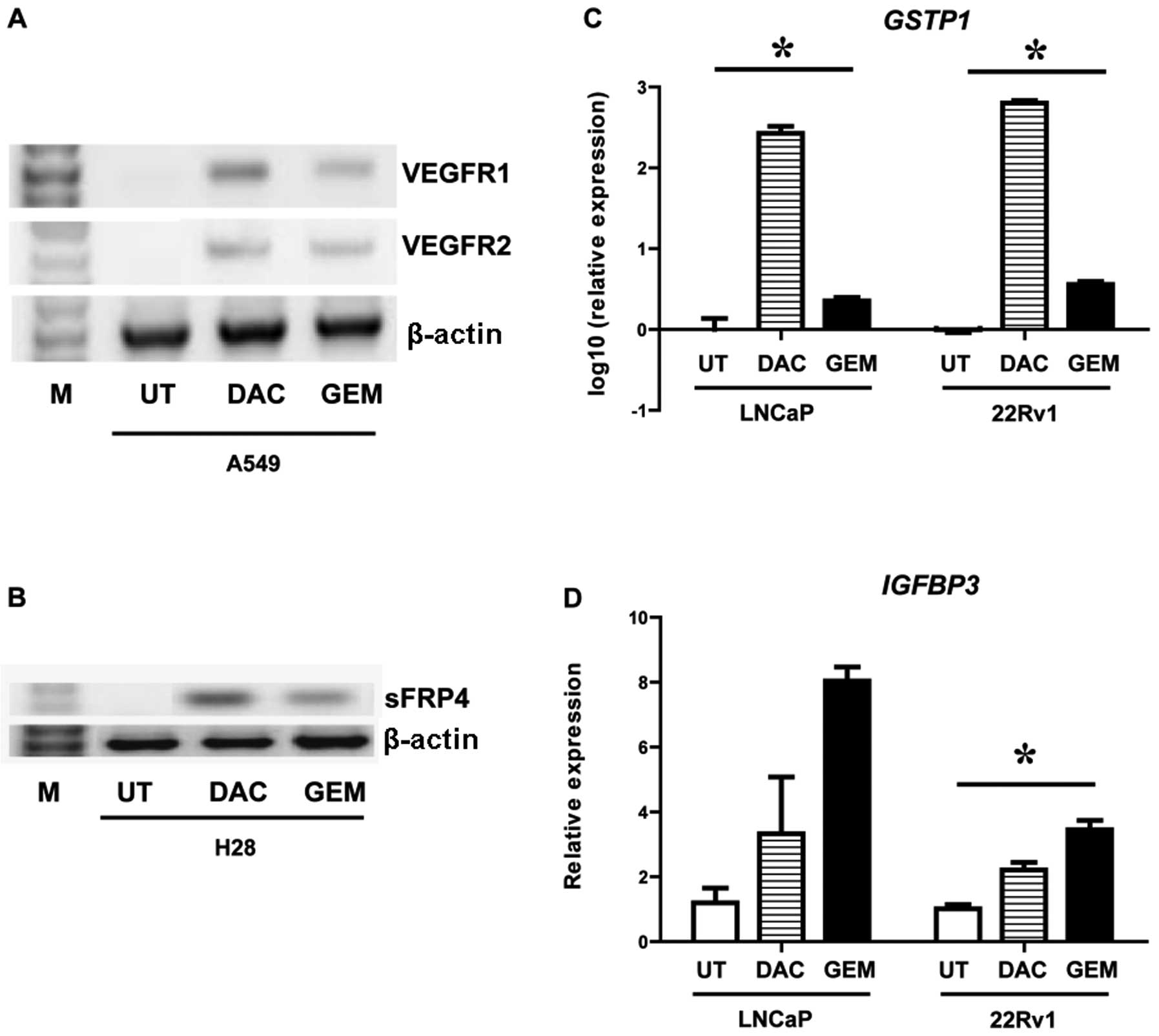
Gemcitabine reactives mRNA expression of
epigenetically silenced genes
A549 (NSCLC), H28 (MPM), LNCaP (prostate) and 22Rv1
(prostate) cells were treated with either decitabine or gemcitabine
and the effects of these drugs on gene expression were examined. In
A549 cells decitabine and gemcitabine induced both VEGFR1 and
VEGFR2 (Fig. 2A). In the H28 cell
line a gene previously shown to be epigenetically silenced in this
cell line by DNA CpG methylation (25,26), sFRP4, was reactivated by both
drugs (Fig. 2B). Both GSTP1 and
IGFBP3 have been shown to be epigenetically silenced by DNA CpG
methylation in prostate cancer by us and others (27–29). Using quantitative PCR we measured
the effect of decitabine and gemcitabine on these genes in two
prostate cancer cell lines (LNCaP and 22Rv1). Both drugs were shown
to significantly induce expression of GSTP1 (Fig. 2C) and IGFBP3 (Fig. 2D).
As all these genes were examined separately in
different cell lines we reviewed the literature to find whether
there was a common gene frequently silenced by DNA CpG methylation
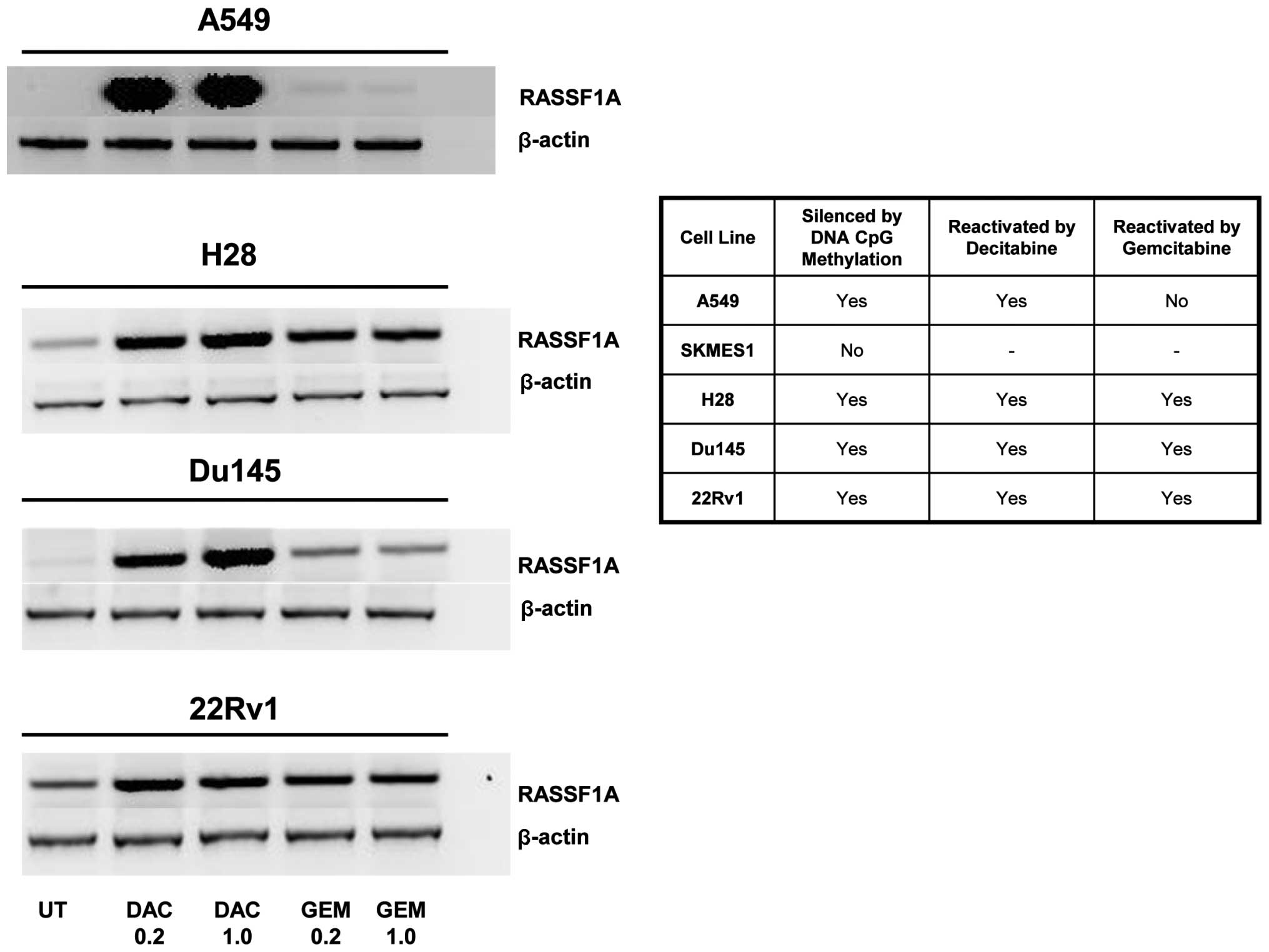
in lung, MPM and prostate cell lines. From this analysis we chose
the gene RASSF1A which was shown to be silenced by methylation in
several of our cell lines (30–32). Gemcitabine was able to
reactivate/upregulate RASSF1A in 3 out of the 4 cell lines tested
whereas decitabine reactived RASSF1A in 4 out of the 4 cell lines
at all concentrations tested (Fig.
3).
Decitabine functions in vitro and in vivo
to inhibit DNA methyltransferases
A DNA methyltransferase activity/inhibition assay
was used to measure the effects of gemcitabine on DNMT enzymatic
activity, with decitabine used as a positive inhibitor control.
Using recombinant DNMT protein mixture we demonstrated that both
decitabine and gemcitabine inhibited DNMT activity (Fig. 4A and B). Furthermore, DNMT
activity was also inhibited in nuclear extracts from cells exposed
to either decitabine or gemcitabine (Fig. 4C and D), indicating that
gemcitabine inhibits DNA methyltransferase activity.
Gemcitabine affects DNMT1 protein
stability
Decitabine has been shown to selectively degrade
DNMT1 protein levels by a proteasomal-based pathway (33). To examine whether gemcitabine also
affects the levels of DNMT1 protein, western blot analyses were
carried out on extracts from cells treated with various
concentrations of gemcitabine or decitabine. Gemcitabine was found
to affect DNMT1 protein levels to different degrees for all the
cell lines examined (Fig.
4E).
Gemcitabine does not alter global DNA CpG
methylation or at the promoters of reactivated genes
Schafer and colleagues published data demonstrating
that gemcitabine affects DNA methylation in cancer cells by
inhibiting nucleotide excision repair (NER)-mediated DNA
demethylation. Intriguingly, the authors showed that gemcitabine
treatments did not affect global levels of DNA methylation but
induced hypermethylation and silencing of MLH1 (34).
Using pyrosequencing of long interspersed nucleotide
element (LINE-1) sequences, we examined global methylation in our
cell lines. In agreement with Schafer et al (34), we did not observe any gross
alterations in the global DNA CpG methylation following treatment
with gemcitabine (Table III). For
the lower concentration of decitabine tested (0.2 μM) we
noted a decrease in CpG methylation which did not occur in the
cells treated at the higher concentration (1 μM) in
agreement with effects reported by others (35).
| Table IIIPercentage of methylation for LINE-1
and RASSF1A as determined by pyrosequencing. |
Table III
Percentage of methylation for LINE-1
and RASSF1A as determined by pyrosequencing.
| Cell line | Treatment | LINE-1 | RASSF1 |
|---|
| A549 | UT | 55.8 | 85.2 |
| DAC 0.2 | 32.8 | 45.3 |
| DAC 1.0 | 51.3 | 79.2 |
| Gem 0.2 | 54.7 | 85.8 |
| Gem 1.0 | 55.7 | 86.2 |
| SKMES-1 | UT | 53.1 | 7.3 |
| DAC 0.2 | 45.2 | 4.4 |
| DAC 1.0 | 52.7 | 6.5 |
| Gem 0.2 | 51.2 | 5.2 |
| H28 | UT | 68.0 | 94.9 |
| DAC 0.2 | 47.8 | 65.8 |
| DAC 1.0 | 63.8 | 88.1 |
| Gem 0.2 | 69.6 | 94.6 |
| Gem 1.0 | 68.2 | 94.5 |
| Du145 | UT | 65.2 | 71.2 |
| DAC 0.2 | 45.8 | 51.0 |
| DAC 1.0 | 56.5 | 64.0 |
| Gem 0.2 | 64.3 | 71.6 |
| Gem 1.0 | 66.0 | 71.9 |
| 22Rv1 | UT | 50.0 | 83.8 |
| DAC 0.2 | 37.7 | 53.3 |
| DAC 1.0 | 42.5 | 61.2 |
| Gem 0.2 | 50.4 | 82.6 |
| Gem 1.0 | 52.0 | 74.3 |
Next, we examined the effect of decitabine and
gemcitabine on CpG methylation within RASSF1A. Again decitabine
caused alterations to DNA CpG methylation levels at the lower
concentration, yet there was no effect on methylation at the higher
concentration (Table III). For
the most part gemcitabine did not alter DNA CpG methylation for
either concentration examined (Table
III). Similar results were observed for VEGFR1 (data not
shown).
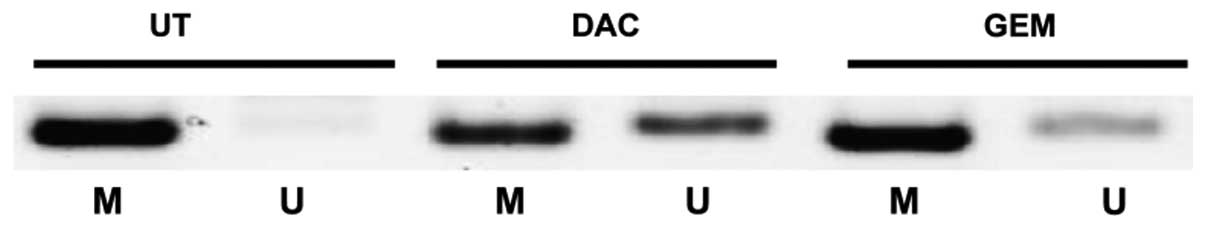
GSTP1 has been previously shown to be downregulated
by DNA CpG methylation (27–29). Given that we were able to
demonstrate that both gemcitabine and decitabine reactivated the
expression of GSTP1 in prostate cancer cell lines (Fig. 2), using MS-PCR, we examined
whether or not there is a loss of methylation at the GSTP1 promoter
in the 22Rv1 cell line. Our results showed a clear increase in the
levels of unmethylated DNA at the GSTP1 promoter following
treatment with either drug, indicating that there may be some
changes to DNA CpG methylation occurring in cells treated with
gemcitabine (Fig. 5).
Discussion
Gemcitabine is a well-established chemotherapy agent
used as a single-agent treatment in pancreatic cancer, or in
combination with various other agents in the treatment of many
solid tumours. Its mode of action is generally considered to be
that of an anti-metabolite acting to either inhibit ribonucleotide
reductase (RNR) or by replacing cytidine during DNA
replication.
Aberrant epigenetic regulation through DNA CpG
methylation is a frequent event in cancer and reactivation of the
target of methylation induced silencing 1 (TMS1) by DNA
methyltransferase inhibitors (DNMTi) was found to enhance
sensitivity to gemcitabine in pancreatic cancer cells (36).
We noted that gemcitabine shares structural
similarity with the FDA-approved DNMTi decitabine (Dacogen)
(Fig. 1) and tested whether or
not it has the ability to affect DNA CpG methylation. We showed
that gemcitabine actively induces expression of epigenetically
silenced genes in lung, mesothelioma (MPM) and prostate cancer cell
lines (Figs. 2 and 3). We showed that gemcitabine directly
inhibits DNA methyltransferases (Fig.
4). In addition, we also showed that gemcitabine depletes
levels of DNMT1 similar to decitabine (Fig. 4). However, despite gemcitabine’s
ability to inhibit DNMTs, alter cellular DNMT1 protein levels and
reactivate epigenetically silenced genes, there were few if any
changes to DNA CpG methylation levels. We provide some evidence
that active demethylation of an epigenetically silenced gene can
occur (Fig. 5), but another
mechanism may have to be invoked.
Schafer et al (34) published additional data
demonstrating that gemcitabine affects DNA methylation in cancer
cells by inhibiting nucleotide excision repair (NER)-mediated DNA
demethylation. The authors showed that gemcitabine treatments did
not affect global levels of DNA methylation but induced
hypermethylation and silencing of MLH1 (34). In agreement with the authors we
also found that global levels of DNA CpG methylation were
unaffected. In addition, methylation at the promoters of genes
which could be reactivated following treatments with either
decitabine or gemcitabine did not demonstrate any major changes in
methylation status. This may in part be due to the concentrations
of drugs used. For example, it has been established that high
concentrations of decitabine have less effect on DNA methylation
due to cytostatic effects, and our results using pyrosequencing
confirm this demonstrating that changes in DNA methylation only
occurred at the lower concentration of drug used (Table III). Using MTT assays we examined
cellular proliferation across a range of concentrations (decitabine
200–5,000 nM; gemcitabine 50–1,000 nM) in A549 and SKMES1 cells.
While decitabine did not affect cellular proliferation to any
appreciable effect at any concentration, gemcitabine caused
significant decreases (of the order 25–50%) in cellular
proliferation (data not shown), which was not unexpected given the
other known mechanisms of action of this drug.
Schafer et al (34) used a range of concentrations (34,
67 or 134 nM), whereas we used higher concentrations (0.2–1
μM). It must be noted, however, that in gemcitabine infusion
studies of patients with solid tumours, blood concentrations of
gemcitabine at Day 1 of infusion varied between 18 and 77 μM
and at Day 15 between 13 and 90 μM (37). As such the amount of gemcitabine
used in our analysis was still below the levels utilised in
patients.
The concentration of gemcitabine may become a
critical determinant in how this chemotherapeutic agent is used in
the clinic. For instance, in a study using MPM cells, gemcitabine
inhibited the secretion of the proinflammatory cytokine IL-6 at
drug concentrations that produced only small decreases in cell
viability, whereas at higher doses a surge of IL-6 release was
noted (38). We also observed an
increase in the protumourigenic cytokine IL-23 in a NSCLC cell line
treated with gemcitabine (Gray and O’Byrne, unpublished data).
How then does gemcitabine induce or reactivate
epigenetically silenced genes. The nitrogen substitution of carbon
5 in the cytosine ring (Fig. 1)
is involved in the mechanism of DNMT inactivation. As this nitrogen
is not present in gemcitabine it may be that some sort of steric
hindrance is involved affecting DNMTs and or methyl-binding
proteins. Conceivably by inhibiting binding of methyl-binding
protein complexes to DNA CpG sites this may relax chromatin and
result in the re-expression of silenced genes.
The results presented by us and Schafer et al
(34) demonstrate a novel
indication for gemcitabine and may have important implications in
oncology as DNA methylation effects are potentially useful as
biomarkers to either monitor response to therapy (39), or may have predictive value
(40). In particular, the ability
of gemcitabine to reactivate epigenetically silenced genes
indicates a potential means to monitor and identify those patients
who actually respond to gemcitabine treatment.
There are exciting new avenues which could be
exploited, for example the use of low dose gemcitabine regimens
combined with other epigenetic targeting drugs such histone
deacetylase inhibitors. In addition low dose regimens may have the
ability to sensitise resistant cancer to other cancer therapies.
Further study will be required to fully delineate these
possibilities.
Acknowledgements
Funding support for this project was
provided by the Irish Cancer Society to Antoinette Perry.
References
|
1.
|
M HidalgoPancreatic cancerN Engl J
Med36216051617201010.1056/NEJMra0901557
|
|
2.
|
VT DeVita JrA step in the right
directionNat Clin Pract Oncol3641200610.1038/ncponc066917139311
|
|
3.
|
KH TkaczukReview of the contemporary
cytotoxic and biologic combinations available for the treatment of
metastatic breast cancerClin
Ther3122732289200910.1016/j.clinthera.2009.11.01120110041
|
|
4.
|
J GoffinC LacchettiPM EllisYC UngWK
EvansFirst-line systemic chemotherapy in the treatment of advanced
non-small cell lung cancer: a systematic reviewJ Thorac
Oncol5260274201010.1097/JTO.0b013e3181c6f035
|
|
5.
|
S NagelR CalifanoN ThatcherF
BlackhallGemcitabine and carboplatin in combination for the
treatment of advanced, metastatic, non-small cell lung cancerExpert
Opin
Pharmacother832653275200710.1517/14656566.8.18.326518035969
|
|
6.
|
DA FennellG GaudinoKJ O’ByrneL MuttiJ van
MeerbeeckAdvances in the systemic therapy of malignant pleural
mesotheliomaNat Clin Pract
Oncol5136147200810.1038/ncponc103918227828
|
|
7.
|
M RayHL KindlerMalignant pleural
mesothelioma: an update on biomarkers and
treatmentChest136888896200910.1378/chest.08-266519736192
|
|
8.
|
P JantscheffN EsserR GraeserLiposomal
gemcitabine (GemLip)-efficient drug against hormone-refractory
Du145 and PC-3 prostate cancer
xenograftsProstate6911511163200910.1002/pros.2096419399788
|
|
9.
|
P JantscheffV ZiroliN EsserAnti-metastatic
effects of liposomal gemcitabine in a human orthotopic LNCaP
prostate cancer xenograft modelClin Exp
Metastasis26981992200910.1007/s10585-009-9288-119784785
|
|
10.
|
W PlunkettP HuangYZ XuV HeinemannR
GrunewaldV GandhiGemcitabine: metabolism, mechanisms of action, and
self-potentiationSemin Oncol22Suppl 1131019957481842
|
|
11.
|
MW LawlessS NorrisKJ O’ByrneSG
GrayTargeting histone deacetylases for the treatment of diseaseJ
Cell Mol
Med13826852200910.1111/j.1582-4934.2008.00571.x19175682
|
|
12.
|
KJ O’ByrneMP BarrSG GrayThe role of
epigenetics in resistance to cisplatin chemotherapy in lung
cancerCancers314261453201124212667
|
|
13.
|
D CaiDS ShamesMG RasoSteroid receptor
coactivator-3 expression in lung cancer and its role in the
regulation of cancer cell survival and proliferationCancer
Res7064776485201010.1158/0008-5472.CAN-10-000520663904
|
|
14.
|
N HajjiK WallenborgP VlachosJ FullgrabeO
HermansonB JosephOpposing effects of hMOF and SIRT1 on H4K16
acetylation and the sensitivity to the topoisomerase II inhibitor
etoposideOncogene2921922204201010.1038/onc.2009.50520118981
|
|
15.
|
W ZhaoQ ZhangX KangS JinC LouAIB1 is
required for the acquisition of epithelial growth factor
receptor-mediated tamoxifen resistance in breast cancer
cellsBiochem Biophys Res
Commun380699704200910.1016/j.bbrc.2009.01.15519285025
|
|
16.
|
SB BaylinPA JonesA decade of exploring the
cancer epigenome - biological and translational implicationsNat Rev
Cancer11726734201110.1038/nrc313021941284
|
|
17.
|
SD GoreC JonesP KirkpatrickDecitabineNat
Rev Drug Discov5891892200610.1038/nrd218017117522
|
|
18.
|
E KaminskasA FarrellS AbrahamApproval
summary: azacitidine for treatment of myelodysplastic syndrome
subtypesClin Cancer
Res1136043608200510.1158/1078-0432.CCR-04-213515897554
|
|
19.
|
T YokochiKD RobertsonDoxorubicin inhibits
DNMT1, resulting in conditional apoptosisMol
Pharmacol6614151420200410.1124/mol.104.00263415340041
|
|
20.
|
SG GrayN Al-SarrafAM BairdMC CathcartE
McGovernKJ O’ByrneRegulation of EP receptors in non-small cell lung
cancer by epigenetic modificationsEur J
Cancer4530873097200910.1016/j.ejca.2009.09.00619818596
|
|
21.
|
PW LairdA ZijderveldK LindersMA RudnickiR
JaenischA BernsSimplified mammalian DNA isolation procedureNucleic
Acids Res194293199110.1093/nar/19.15.42931870982
|
|
22.
|
A DaskalosS LogothetiS MarkopoulouGlobal
DNA hypomethylation-induced DeltaNp73 transcriptional activation in
non-small cell lung cancerCancer
Lett3007986201110.1016/j.canlet.2010.09.00920926182
|
|
23.
|
A DaskalosG NikolaidisG
XinarianosHypomethylation of retrotransposable elements correlates
with genomic instability in non-small cell lung cancerInt J
Cancer1248187200910.1002/ijc.2384918823011
|
|
24.
|
A DaskalosU OleksiewiczA
FiliaUHRF1-mediated tumor suppressor gene inactivation in nonsmall
cell lung cancerCancer11710271037201110.1002/cncr.2553121351083
|
|
25.
|
B HeAY LeeS DadfarmaySecreted
frizzled-related protein 4 is silenced by hypermethylation and
induces apoptosis in beta-catenin-deficient human mesothelioma
cellsCancer Res65743748200515705870
|
|
26.
|
AY LeeB HeL YouExpression of the secreted
frizzled-related protein gene family is downregulated in human
mesotheliomaOncogene2366726676200410.1038/sj.onc.120788115221014
|
|
27.
|
WG NelsonAM De MarzoS
YegnasubramanianEpigenetic alterations in human prostate
cancersEndocrinology15039914002200910.1210/en.2009-057319520778
|
|
28.
|
AS PerryH LiyanageM LawlerK
WoodsonDiscovery of DNA hypermethylation using a DHPLC screening
strategyEpigenetics24349200710.4161/epi.2.1.388217965617
|
|
29.
|
AS PerryB LoftusR MorooseIn silico mining
identifies IGFBP3 as a novel target of methylation in prostate
cancerBr J Cancer9615871594200710.1038/sj.bjc.660376717453001
|
|
30.
|
H EndohY YatabeS ShimizuRASSF1A gene
inactivation in non-small cell lung cancer and its clinical
implicationInt J Cancer1064551200310.1002/ijc.1118412794755
|
|
31.
|
I KuzminJW GillespieA ProtopopovThe
RASSF1A tumor suppressor gene is inactivated in prostate tumors and
suppresses growth of prostate carcinoma cellsCancer
Res6234983502200212067994
|
|
32.
|
S ToyookaHI PassN ShivapurkarAberrant
methylation and simian virus 40 tag sequences in malignant
mesotheliomaCancer Res6157275730200111479207
|
|
33.
|
K GhoshalJ DattaS
Majumder5-Aza-deoxycytidine induces selective degradation of DNA
methyltransferase 1 by a proteasomal pathway that requires the KEN
box, bromo-adjacent homology domain, and nuclear localization
signalMol Cell
Biol2547274741200510.1128/MCB.25.11.4727-4741.2005
|
|
34.
|
A SchaferL SchomacherG BarretoG DoderleinC
NiehrsGemcitabine functions epigenetically by inhibiting repair
mediated DNA demethylationPLoS
One5e14060201010.1371/journal.pone.001406021124914
|
|
35.
|
HC TsaiH LiL Van NesteTransient low doses
of DNA-demethylating agents exert durable antitumor effects on
hematological and epithelial tumor cellsCancer
Cell21430446201210.1016/j.ccr.2011.12.02922439938
|
|
36.
|
K RamachandranH MillerE GordianC
Rocha-LimaR SingalMethylation-mediated silencing of TMS1 in
pancreatic cancer and its potential contribution to
chemosensitivityAnticancer Res3039193925201021036703
|
|
37.
|
SM de LangeK van der BornJR KroepNo
evidence of gemcitabine accumulation during weekly
administrationEur J Clin Pharmacol61843849200516283278
|
|
38.
|
BR McLarenBW RobinsonRA LakeNew
chemotherapeutics in malignant mesothelioma: effects on cell growth
and IL-6 productionCancer Chemother
Pharmacol45502508200010.1007/s00280005102610854139
|
|
39.
|
VV LevensonDNA methylation as a universal
biomarkerExpert Rev Mol
Diagn10481488201010.1586/erm.10.1720465502
|
|
40.
|
F SalazarMA MolinaM
Sanchez-RoncoFirst-line therapy and methylation status of CHFR in
serum influence outcome to chemotherapy versus EGFR tyrosine kinase
inhibitors as second-line therapy in stage IV non-small-cell lung
cancer patientsLung
Cancer728491201110.1016/j.lungcan.2010.07.00820705357
|