Introduction
Exposure to arsenic as an environmental pollutant is
known to increase the incidence of a variety of diseases, including
cancer and cardiovascular disease (1,2).
At the same time, arsenic is known to disturb the functions of
various types of immune cells (3,4),
and this disturbance in immunological functions may play a role not
only in the etiology of arsenic-associated infectious diseases, but
also in cancer and cardio-vascular disease. Although the effects of
arsenic exposure on the functions of T cells and macrophages have
been examined (4,5), little information is available on
the effects of arsenic on mast cells. We previously carried out a
microarray analysis by using rat RBL-2H3 mast cells after chronic
exposure to inorganic arsenite [As(III)] for 4 weeks. It was found
that some S100 proteins, including S100A9, S100A10, S100A6 and
S100A13, were upregulated in RBL-2H3 cells after chronic exposure
to As(III). Among S100 proteins, the mRNA levels of S100A8 and
S100A9 determined by real-time RT-PCR were highly upregulated
following a 1- and 2-week exposure to As(III) (6).
S100A8 and S100A9, which comprise a complex called
calprotectin, are highly expressed in neutrophils, monocytes and
activated macrophages. Accumulating evidence has shown that S100A8
and S100A9 are strongly associated with infection, autoimmunity,
cardiovascular disease, and cancer (7,8).
The S100A8 and S100A9 proteins possess intracellular and
extracellular functions. Intracellularly, the heterocomplex of
S100A8 and A9 supports the activation of NADPH oxidase through the
interaction with p67phox and Rac2, leading to the
enhancement of reactive oxygen species (ROS) formation (9). On the other hand, the excreted
calprotectin acts as a ligand for Toll-like receptor 4 (TLR4),
leading to the development of autoreactive lymphocytes, and as a
ligand for the receptor for advanced glycation endproducts (RAGE)
(10,11), leading to the disturbance in
vascular functions.
Transcription NF-E2-related factor 2 (Nrf2) is one
of the major cellular defense mechanisms against oxidative and/or
electrophilic stress (12). The
Nrf2 recognizes the antioxidant response element (ARE) in the
promoter region of the genes and regulates the basal and inducible
expression of numerous antioxidant and detoxifying genes. Kumagai
and Sumi (14) reported that
arsenicals such as As(III) activate nuclear Nrf2 accumulation,
leading to an upregulation of antioxidant enzymes such as heme
oxygenase-1 and phase II detoxifying enzymes such as glutathione
S-transferase (13). However,
whether Nrf2 is involved in trans-activation of the S100A8 and
S100A9 genes has not been investigated.
In this study, we found that S100A8 and S100A9
expression are upregulated after exposure to As(III) in skin
keratinocyte HaCaT cells, leukemic monocyte U937 cells, and bladder
urothelial UROtsa cells. In addition, the results of a study using
the S100A9 promoter-dependent luciferase showed that S100A9
upregulation is dependent on the activation of Nrf2.
Materials and methods
Materials
Sodium arsenite (90% purity) was purchased from Wako
Pure Chemicals (Osaka, Japan). Antibody for Nrf2 was purchased from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA), and
anti-β-actin was purchased from Cell Signaling Technology (Beverly,
MA, USA). Anti-S100A8 antibody was purchased from Acris Antibodies
GmbH (Herford, Germany). Anti-S100A9 antibody was purchased from
Novus Biologicals (Littleton, CO, USA). All other reagents and
chemicals used were of the highest grade available.
Cell culture
Human skin keratinocyte HaCaT cells were obtained
from Dr Norbert E. Fusenig (15).
Human leukemic monocyte U937 cells were purchased from American
Type Culture Collection (Manassas, VA, USA). Human urothelial
UROtsa cells were obtained from Dr Mary Ann Sens (16). HaCaT and UROtsa cells were
cultured at 37°C in a humidified atmosphere of 5% CO2
using Dulbecco’s modified Eagle’s medium (DMEM) containing 10%
fetal calf serum, penicillin (100 U/ml) and streptomycin (100
μg/ml). U937 cells were cultured at 37°C in a humidified atmosphere
of 5% CO2 using RPMI-1640 (all were from Wako Pure
Chemicals) containing 10% fetal calf serum, penicillin (100 U/ml)
and streptomycin (100 μg/ml).
Semi-quantitative reverse
transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from cells using Isogen
reagents (Wako Pure Chemicals). A reaction mixture (20 μl)
containing RT buffer (Fermentas, Burlington, ON, Canada), dNTPs,
oligo(dT)15 primer, RNase inhibitor (Toyobo, Osaka, Japan), M-MuLV
Reverse Transcriptase (Fermentas), and 1 μg of total RNA was
incubated at 37°C for 90 min followed by inactivation of the enzyme
at 65°C for 5 min. For the PCR amplification of cDNA, a 25 μl
mixture containing Premix Taq (Takara Bio, Inc., Shiga, Japan), 2
μl cDNA, and the specific PCR primers was prepared. The primer
sequences are given in Table I.
The PCR reactions were carried out as follows: for S100A8 and
S100A9, 1 cycle of 94°C for 5 min, followed by 27 cycles of 94°C
for 30 sec, 58°C for 30 sec, and 72°C for 1 min, and a final cycle
at 72°C for 7 min; for β-actin, 1 cycle of 94°C for 5 min, followed
by 35 cycles of 94°C for 30 sec, 55°C for 30 sec, and 72°C for 1
min, and a final cycle of 72°C for 7 min. The amplified products
were resolved by 2% agarose gel electrophoresis.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Gene name | Sequence | Size (bp) |
|---|
| S100A8 | F:
5′-TGTCAGCCTGCTTTCAGAAG-3′ | 286 |
| R:
5′-ACGCCCATCTTTATCACCAG-3′ | |
| S100A9 | F:
5′-GGGAATTCAAAGAGCTGGTG-3′ | 267 |
| R:
5′-CACTGTGATCTTGGCCACTG-3′ | |
| β-actin | F:
5′-CATGGATGACGATATCGCT-3′ | 571 |
| R:
5′-CATGAGGTAGTCTGTCAGGT-3′ | |
Western blotting
The total cell lysates were used for western
blotting of the proteins. Samples for each analysis were separated
by sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE). Gels were transferred to an Immun-Blot PVDF membrane
and then placed in a blocking solution consisting of TBST [10 mM
Tris (pH 8.0), 150 mM NaCl, and 0.05% Tween-20] and 5% skim milk
for 1 h. The blotted membranes were incubated with the appropriate
antibody, washed with TBST, and incubated with HRP-conjugated
secondary antibody. The bound IgG was visualized with Western
Blotting Detection Reagents (Thermo Scientific, Rockford, IL, USA)
according to the manufacturer’s protocol.
Plasmid construction for the S100A9
promoter (−1000/+429)- luciferase
Genomic DNA from HaCaT cells was prepared with a
Wizard Genome DNA purification kit (Promega, Madison, WI, USA). For
the PCR amplification of the S100A9 promoter region (−1000/+429), a
50 μl mixture containing PrimeSTAR™ Max DNA Polymerase (Takara Bio,
Inc.), 200 pg genomic DNA, and the PCR primers 5′-ATGGTACC
ATCACTGTGGAGTAGGGGAAGGGCACT-3′ and 5′-GGA
GATCTCGTCTTGCACTCTGTCTGTGTAATGGA-3′ (underlined letters
indicate the recognition site for the restriction enzymes
KpnI and BglII, respectively) was prepared. The PCR
reaction consisted of 35 cycles of 98°C for 10 sec, 55°C for 5 sec,
and 72°C for 10 sec. Amplified cDNA was cloned using a StrataClone
Blunt PCR cloning kit (Agilent Technologies, Palo Alto, CA, USA).
The sequenced cDNA of the S100A9 promoter was subcloned into
pGL3-Basic (Promega).
Truncation of two ARE sites on the S100A9
promoter to construct ΔARE1 and ΔARE2
To construct deletion mutants of each ARE site on
the S100A9 promoter on pGL3-Basic (ΔARE1 and ΔARE2), PCR was
carried out with the following primers: for ΔARE1,
5′-GAAGCTGGCCGGACGCCATTC CAAGAGG-3′ and 5′-CCTCTTGGAATGGCGTCCGGCCA
GCTTC-3′; and for ΔARE2, 5′-ACTGCTCACCTGTGAA GAAGCAATAGACAGGGCCA-3′
and 5′-TGGCCCTGTCTA TTGCTTCACAGGTGAGCAGT-3′. The deletion mutants
were amplified with a QuickChange II Site-Directed Mutagenesis kit
(Agilent Technologies) according to the manufacturer’s
protocol.
Transient transfection and luciferase
assay
HaCaT cells (1.0×105) were seeded in
12-well plates. Transfection was performed with Lipofectamine 2000
(Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s
instructions. HaCaT cells were transfected with 1.6 μg S100A9
promoter-luciferase cDNA and 0.16 μg pRL-TK for 24 h. Following
exposure to As(III), HaCaT cells were washed with PBS and lysed
with Passive Lysis Buffer (Promega). Luciferase activities in the
cellular extracts were measured with a Dual-Luciferase Reporter
Assay System (Promega). Renilla reniformis-derived
luciferase activity was used for the correction of transfection
efficiency.
Statistical analysis
Data were obtained from three separate experiments.
The values are shown as the means ± SD. Statistical significance
was assessed with Student’s t-test. Differences between groups were
considered statistically significant at P<0.05.
Results
Effects of inorganic arsenite on S100A8
and S100A9 expression in nine lines of human-derived cells
We previously showed that expression of S100A8 and
S100A9 is upregulated by chronic exposure to As(III) in rat RBL-2H3
cells (6). In the present study,
we first examined whether As(III) also induces S100A8 and S100A9
expression in other human-derived cell lines, including lung
epithelial A549 cells, kidney carcinoma Caki-2 cells, immortalized
skin keratinocytic HaCaT cells, embryonic kidney HEK293 cells,
liver carcinoma HepG2 cells, immortalized T lymphocyte Jurkat
cells, normal human epidermal keratinocytes (NHEK), leukemic
monocyte lymphoma U937 cells, and immortalized bladder urothelial
UROtsa cells. These cells were exposed to 0.3 and 1.0 μM As(III)
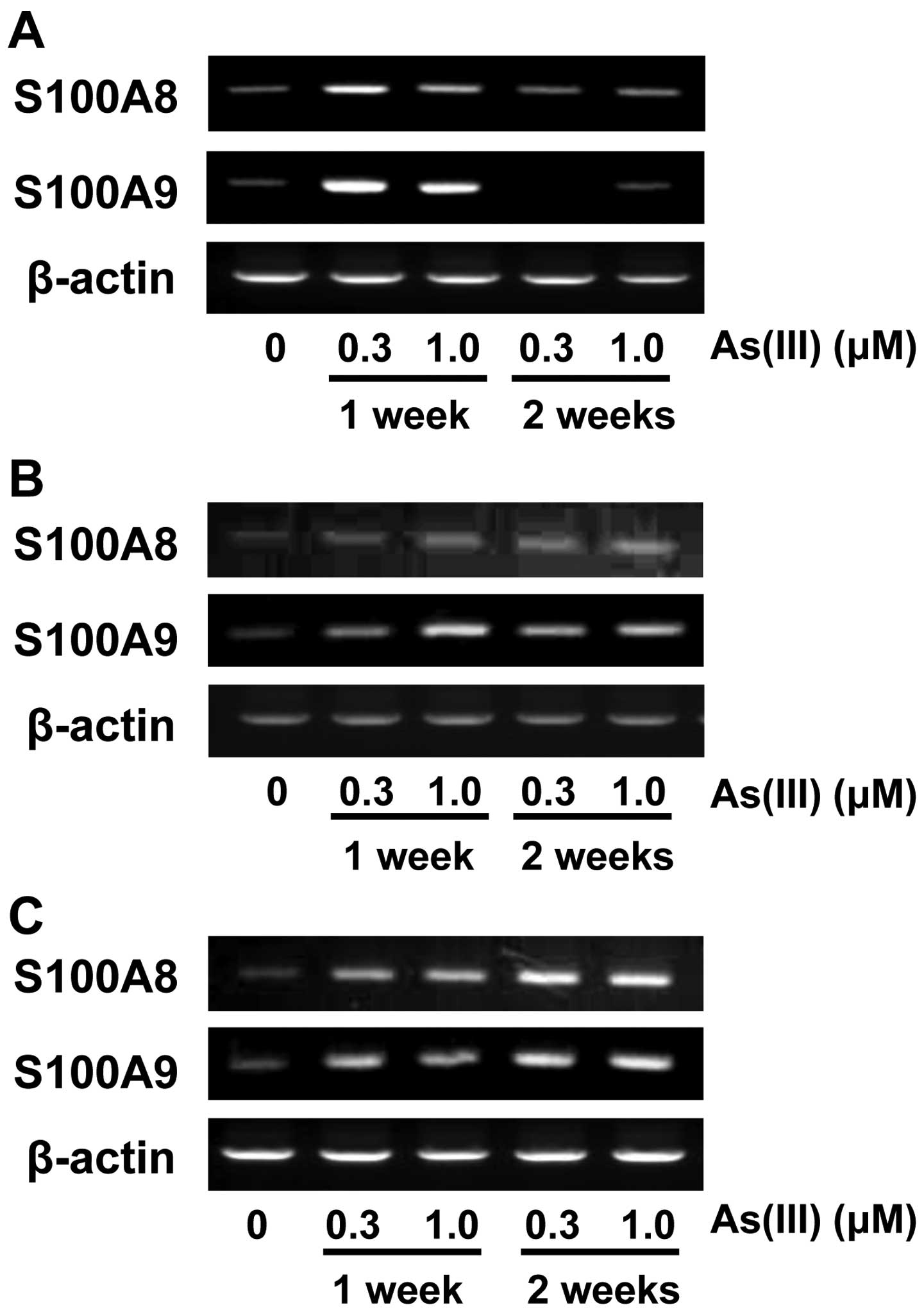
for 1 and 2 weeks. Increased levels of S100A8 and S100A9 mRNA were
observed in HaCaT (Fig. 1A),
UROtsa (Fig. 1B) and U937 cells
(Fig. 1C), but not in the other
cell types. Since HaCaT cells showed a marked increase in mRNA
levels of S100A8 and S100A9 after a 1-week exposure to As(III), we
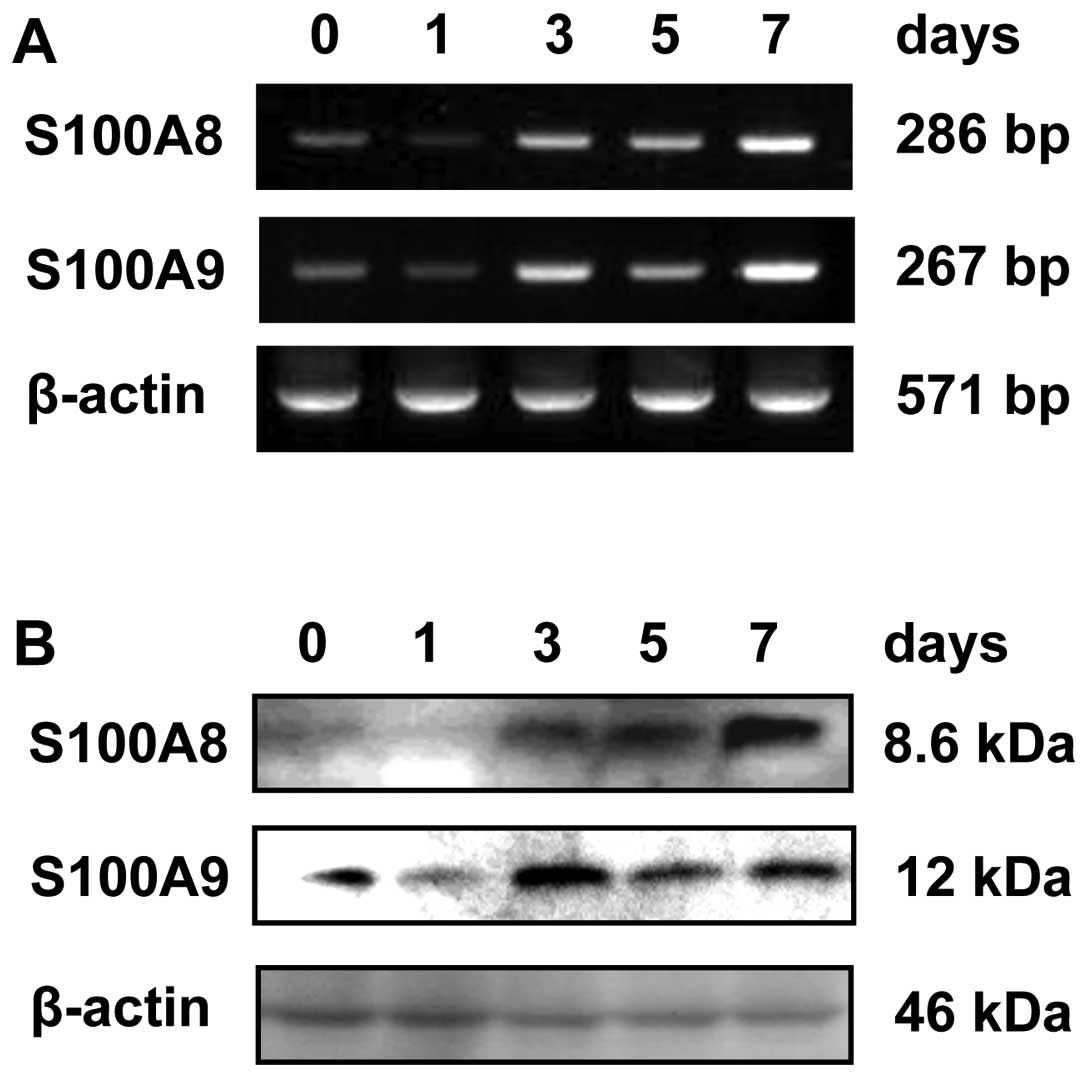
used HaCaT cells in the subsequent experiments. We next examined
whether exposure of HaCaT cells to As(III) for 1, 3, 5 and 7 days
induces S100A8 and S100A9 expression. The results showed that
increased levels of S100A8 and S100A9 mRNA and protein were
observed 3, 5 and 7 days after exposure to As(III) (Fig. 2).
Inorganic arsenite enhances the S100A9
promoter activity in HaCaT cells
The expression of S100A8 and S100A9 genes could be
regulated in either harmonized or independent ways. Some studies
have suggested a more important role of S100A9 than S100A8
(17,18), and cis-elements for
trans-activation on the human S100A9 gene have been well documented
(19,20). Thus, we focused on the mechanisms
underlying S100A9 trans-activation in HaCaT cells exposed to
As(III). To investigate whether As(III) activates transcription of
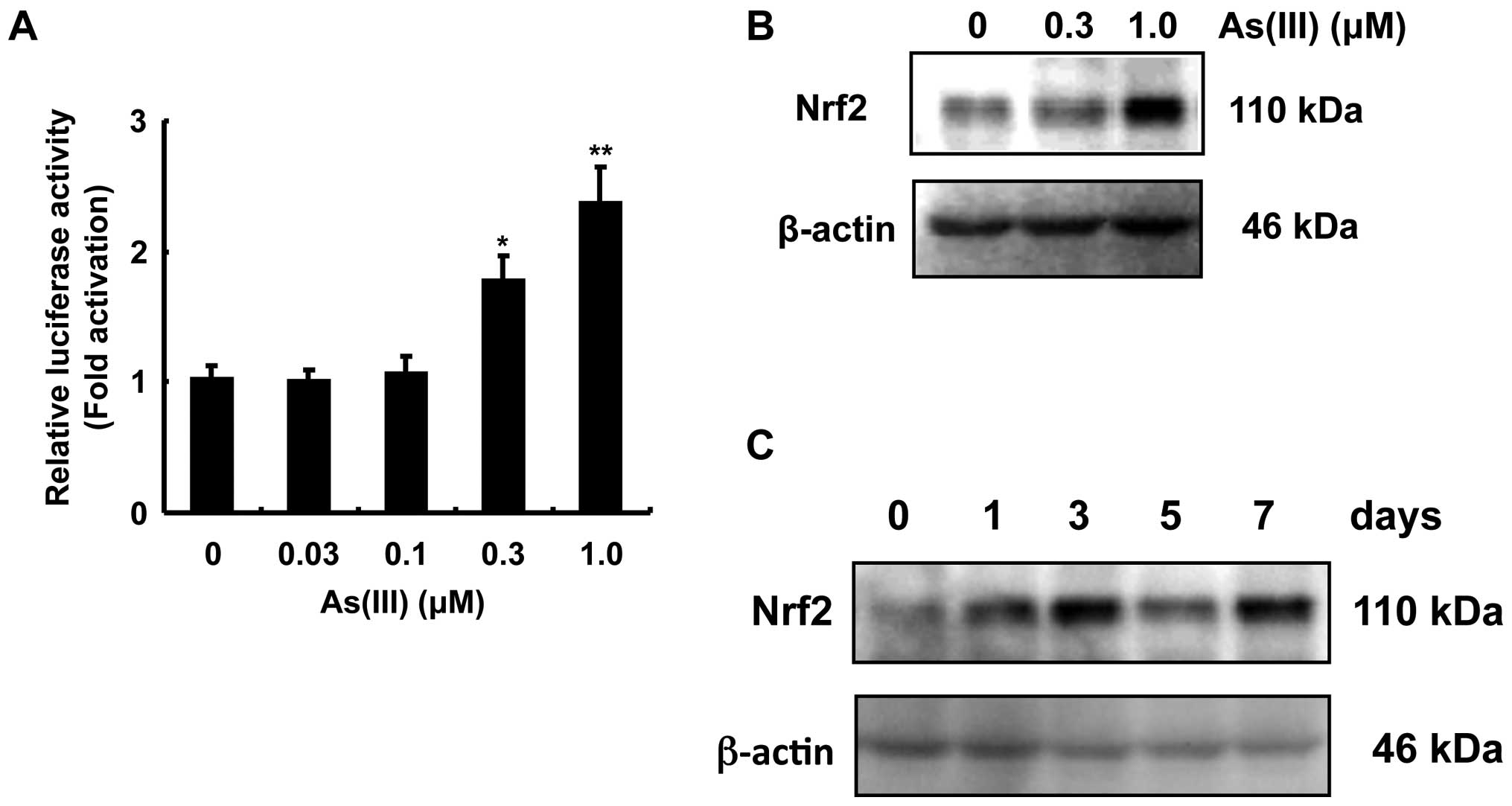
the S100A9 gene in HaCaT cells, we measured the activity of
luciferase in HaCaT cells transfected with the cDNA of the S100A9
promoter (−1000/+429)-fused to luciferase. As(III) significantly
enhanced S100A9 promoter-dependent luciferase activity (Fig. 3A). Since the transcription factor
Nrf2 is known to be activated by exposure to As(III), we examined
Nrf2 accumulation after exposure of HaCaT cells to As(III). Nrf2
accumulation was detected after exposure to 0.3 and 1.0 μM As(III)
for 3 days (Fig. 3B), and 1.0 μM
As(III) for the indicated number of days (Fig. 3C).
Nrf2 activation by inorganic arsenite
enhances the S100A9 expression in HaCaT cells
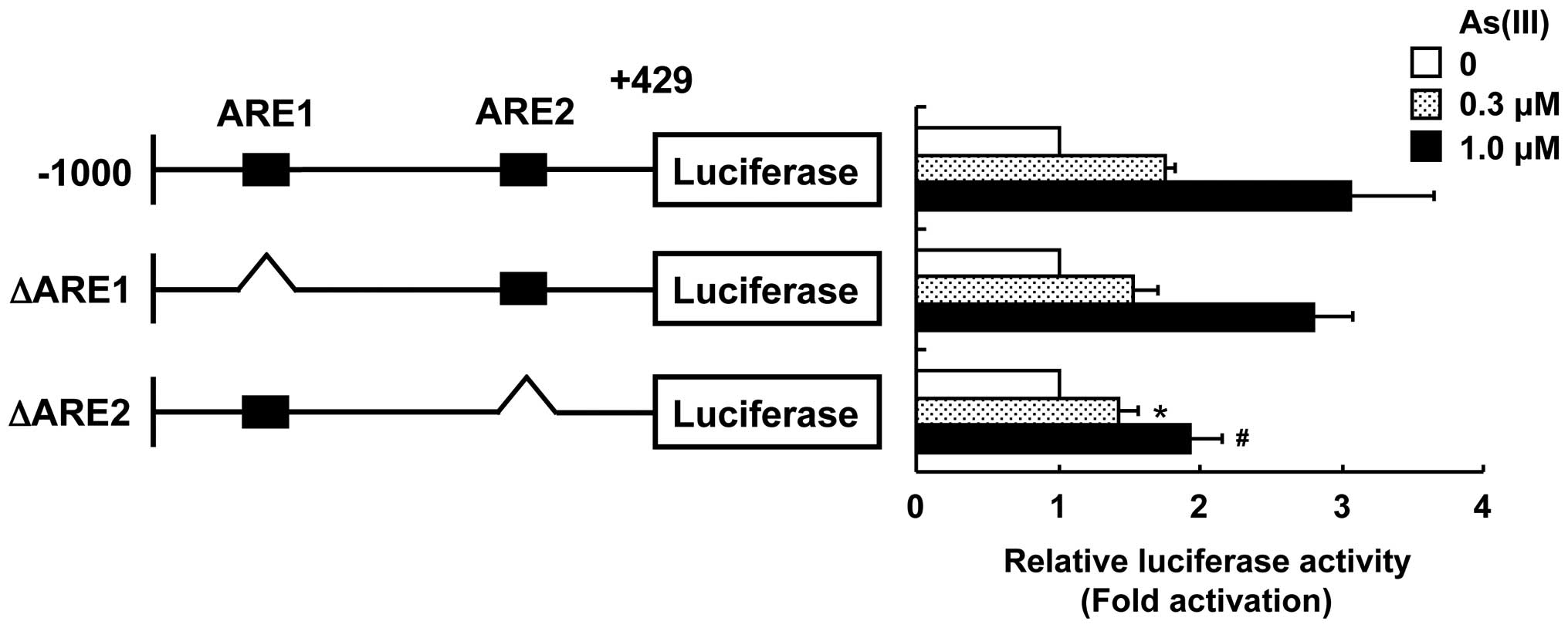
We found that two AREs, 5′-ACAGGCAGGG-3′ from −897 to −887
(ARE1) and 5′-ATCTTCCGGAG-3′ from −78 to −67
(ARE2), were present in the S100A9 promoter region from −1,000 to
+429. To examine the direct correlation between Nrf2 activation and
S100A9 promoter activation in response to As(III), the underlined
nucleotides from each ARE region were deleted from the S100A9
promoter (−1,000/+429)-fused luciferase cDNA to construct ΔARE1 and
ΔARE2. The deletion of ARE1 did not affect the activation of the
S100A9 promoter in response to As(III) (Fig. 4). However, HaCaT cells transfected
with the ARE2-deleted S100A9 promoter-fused luciferase cDNA showed
a significant reduction in As(III)-induced S100A9 promoter
activation as compared with cells transfected with the native
S100A9 promoter. These results suggest that Nrf2 activation is
involved in the stimulation of S100A9 promoter activity and the
ARE2 region is responsible for activation of the S100A9 promoter in
response to As(III).
Discussion
We previously showed that S100A8 and S100A9 are
upregulated in rat RBL-2H3 mast cells after chronic exposure to
As(III) (6). To test whether this
induction is restricted to RBL-2H3 cells, nine human-derived cell
lines were exposed to As(III). We found that HaCaT, UROtsa and U937
cells have the ability to increase the levels of S100A8 and S100A9
mRNAs upon exposure to As(III) (Fig.
1). This result suggests that the induction of S100A8 and
S100A9 expression by As(III) exposure may have a significant role
in some of the target cells of arsenic toxicity. Since the
keratinocyte-derived HaCaT cells showed a marked increase in mRNA
levels of S100A8 and S100A9 after a 1-week exposure to As(III), we
used HaCaT cells in the subsequent experiments on
trans-activation.
Although the functions of the hetero-dimerized form
of S100A8 and S100A9 have been well documented, recent findings
have shown that S100A9 rather than S100A8 appears to have a
predominant role in immunological responses and inflammation.
Indeed, the differentiation of dendritic cells and macrophages was
inhibited in S100A9 transgenic mice (17), and anti-S100A9 antibody but not
anti-S100A8 antibody reduced trans-endothelial migration of
monocytes (18). Thus, we
examined the mechanisms underlying S100A9 trans-activation in HaCaT
cells exposed to As(III).
It has been reported that exposure to As(III) and
inorganic arsenate [As(V)] caused Nrf2 activation and subsequent
upregulation of downstream proteins in MC-3T3E osteoblast and HaCaT
cells (13,21). Not only inorganic arsenicals, but
also organic arsenicals such as monomethylarsonous acid caused Nrf2
activation in RAW264.7 and UROtsa cells (14,22). In Nrf2-deficient cells, the
adaptive response to arsenicals was profoundly impaired, leading to
the augmentation of arsenic-induced cytotoxicity and oxidative
stress (14,23). However, the role of Nrf2 in the
expression of S100A8 and S100A9 has not been investigated.
Therefore, we explored the involvement of Nrf2 in the activation of
the S100A9 gene by exposure to As(III). As a result, Nrf2
activation was detected by accumulation of Nrf2 protein in HaCaT
cells exposed to As(III) (Fig. 3B and
C). In addition, we found two putative ARE sites (ARE1 and
ARE2) for the binding of Nrf2 in the promoter region of the human
S100A9 gene. The experiments using ARE deletion mutants showed that
ARE2 is the responsive site for As(III)-induced promoter activation
of the S100A9 gene (Fig. 4).
These results are the first to demonstrate that the induction of
S100A9 expression by exposure to As(III) is dependent on the ARE2
region in the S100A9 gene with concomitant activation of Nrf2.
It has been reported that the C/EBP and NF-κB
binding sites are the putative cis-elements for
trans-activation on the human S100A9 gene (20). These two elements may also be
involved in As(III)-induced trans-activation. Lin et al
(24) reported that exposure of
HaCaT cells to As(III) enhanced the expression of RTP801, which is
currently known as a DNA-damage-inducible transcript 4, through the
activation of C/EBPβ. On the other hand, NF-κB activation has been
reported in aortic endothelial cells exposed to As(III) (25,26). Since our data showed that the ARE2
deletion does not completely block the induction of
S100A9-dependent promoter activity by As(III) exposure, C/EBP
and/or NF-κB in addition to Nrf2 might also be involved in the
As(III)-induced S100A9 transcription. A study with human mature
monocytic Mono Mac 6 cells, which exhibit high S100A9 expression
under steady-state conditions (20), showed that expression of S100A9
was dependent on the two regions in the promoter of the S100A9
gene, from 153 to 361 (first intron) and from −400 to −374
(promoter region). Additionally, the Kruppel-related zinc finger
protein and the transcriptional intermediary factor 1β (TIF1β) were
shown to bind to the region from −400 to −374 in the promoter of
the S100A9 gene, resulting in the upregulation of S100A9 expression
(19,20). Further studies are required to
clarify whether the Kruppel-related zinc finger protein and TIF1β
are activated by As(III) in HaCaT cells.
Exposure of HaCaT cells to As(III) also increased
mRNA and protein levels of S100A8 (Figs. 1 and 2). Xu et al (27) showed that the 178-bp region of the
S100A8 promoter is responsible for the lipopolysaccharide-induced
increase in S100A8 mRNA in macrophages. Kuwayama et al
(28) revealed that the 426-bp
region of the S100A8 promoter is essential for the S100A8 induction
in differentiated leukemia cells. However, based on our database
searches, there have been no previous reports of ARE sites in the
essential S100A8 promoter. This suggests that a
trans-activator other than Nrf2 is responsible for the
S100A8 induction in HaCaT cells exposed to As(III), although
further studies will be needed to examine whether Nrf2 is
implicated in As(III)-induced S100A8 upregulation.
Kerkhoff et al (9) revealed that S100A8 and S100A9
interact with p67phox and Rac2, the components of NADPH
oxidase, resulting in an activation of NADPH oxidase activity.
Several lines of evidence have indicated that arsenicals are
capable of generating ROS via the activation of NADPH oxidase
(29,30). Benedyk et al (31) reported that the HaCaT cells in
which S100A8 and S100A9 are overexpressed showed increased NADPH
oxidase activity, resulting in an enhanced ROS production. Taken
together with these previous findings, our present results raise
the possibility that the induction of S100A8 and S100A9 expression
may be involved, at least partly, in ROS production through the
activation of NADPH oxidase in response to As(III).
The current study is the first to demonstrate that
As(III) enhanced S100A8 and S100A9 expression in several lines of
human-derived cells and that As(III)-induced S100A9 expression is
dependent on Nrf2 activation. Since S100A8/S100A9 is reported to be
associated with tumor development and cardio-vascular diseases, the
upregulation of S100A8/S100A9 upon exposure to As(III) may play a
significant role in the development of the adverse effects of
arsenic.
Acknowledgements
This study was supported by a grant-in-aid for
Scientific Research from the Ministry of Education, Science,
Culture, and Sports of Japan (no. 24310048 to D.S. and no. 22390127
to S.H.).
Abbreviations:
|
As(III)
|
inorganic arsenite
|
|
Nrf2
|
NF-E2-related factor 2
|
References
|
1
|
Engel RR, Hopenhayn-Rich C, Receveur O and
Smith AH: Vascular effects of chronic arsenic exposure: a review.
Epidemiol Rev. 16:184–209. 1994.PubMed/NCBI
|
|
2
|
Tseng WP, Chu HM, How SW, Fong JM, Lin CS
and Yeh S: Prevalence of skin cancer in an endemic area of chronic
arsenicism in Taiwan. J Natl Cancer Inst. 40:453–463.
1968.PubMed/NCBI
|
|
3
|
Kozul CD, Hampton TH, Davey JC, et al:
Chronic exposure to arsenic in the drinking water alters the
expression of immune response genes in mouse lung. Environ Health
Perspect. 117:1108–1115. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liao WT, Yu CL, Lan CC, et al:
Differential effects of arsenic on cutaneous and systemic immunity:
focusing on CD4+ cell apoptosis in patients with
arsenic-induced Bowen’s disease. Carcinogenesis. 30:1064–1072.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Banerjee N, Banerjee S, Sen R, et al:
Chronic arsenic exposure impairs macrophage functions in the
exposed individuals. J Clin Immunol. 29:582–594. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shimizu Y, Fujishiro H, Matsumoto K, Sumi
D, Satoh M and Himeno S: Chronic exposure to arsenite induces
S100A8 and S100A9 expression in rat RBL-2H3 mast cells. J Toxicol
Sci. 36:135–139. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ehrchen JM, Sunderkotter C, Foell D, Vogl
T and Roth J: The endogenous Toll-like receptor 4 agonist
S100A8/S100A9 (calprotectin) as innate amplifier of infection,
autoimmunity, and cancer. J Leukoc Biol. 86:557–566. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nacken W, Roth J, Sorg C and Kerkhoff C:
S100A9/S100A8: Myeloid representatives of the S100 protein family
as prominent players in innate immunity. Microsc Res Tech.
60:569–580. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kerkhoff C, Nacken W, Benedyk M, Dagher
MC, Sopalla C and Doussiere J: The arachidonic acid-binding protein
S100A8/A9 promotes NADPH oxidase activation by interaction with
p67phox and Rac-2. FASEB J. 19:467–469. 2005.PubMed/NCBI
|
|
10
|
Kerkhoff C, Sorg C, Tandon NN and Nacken
W: Interaction of S100A8/S100A9-arachidonic acid complexes with the
scavenger receptor CD36 may facilitate fatty acid uptake by
endothelial cells. Biochemistry. 40:241–248. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Loser K, Vogl T, Voskort M, et al: The
Toll-like receptor 4 ligands Mrp8 and Mrp14 are crucial in the
development of autoreactive CD8+ T cells. Nat Med.
16:713–717. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Itoh K, Tong KI and Yamamoto M: Molecular
mechanism activating Nrf2-Keap1 pathway in regulation of adaptive
response to electrophiles. Free Radic Biol Med. 36:1208–1213. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Aono J, Yanagawa T, Itoh K, et al:
Activation of Nrf2 and accumulation of ubiquitinated A170 by
arsenic in osteoblasts. Biochem Biophys Res Commun. 305:271–277.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kumagai Y and Sumi D: Arsenic: signal
transduction, transcription factor, and biotransformation involved
in cellular response and toxicity. Annu Rev Pharmacol Toxicol.
47:243–262. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fusenig NE and Boukamp P: Multiple stages
and genetic alterations in immortalization, malignant
transformation, and tumor progression of human skin keratinocytes.
Mol Carcinog. 23:144–158. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rossi MR, Masters JR, Park S, et al: The
immortalized UROtsa cell line as a potential cell culture model of
human urothelium. Environ Health Perspect. 109:801–808. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cheng P, Corzo CA, Luetteke N, et al:
Inhibition of dendritic cell differentiation and accumulation of
myeloid-derived suppressor cells in cancer is regulated by S100A9
protein. J Exp Med. 205:2235–2249. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Eue I, Pietz B, Storck J, Klempt M and
Sorg C: Transendothelial migration of 27E10+ human
monocytes. Int Immunol. 12:1593–1604. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kerkhoff C, Hofmann HA, Vormoor J, et al:
Binding of two nuclear complexes to a novel regulatory element
within the human S100A9 promoter drives the S100A9 gene expression.
J Biol Chem. 277:41879–41887. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Melkonyan H, Hofmann HA, Nacken W, Sorg C
and Klempt M: The gene encoding the myeloid-related protein 14
(MRP14), a calcium-binding protein expressed in granulocytes and
monocytes, contains a potent enhancer element in the first intron.
J Biol Chem. 273:27026–27032. 1998. View Article : Google Scholar
|
|
21
|
Pi J, Qu W, Reece JM, Kumagai Y and
Waalkes MP: Transcription factor Nrf2 activation by inorganic
arsenic in cultured keratinocytes: involvement of hydrogen
peroxide. Exp Cell Res. 290:234–245. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang XJ, Sun Z, Chen W, Li Y, Villeneuve
NF and Zhang DD: Activation of Nrf2 by arsenite and
monomethylarsonous acid is independent of Keap1-C151: enhanced
Keap1-Cul3 interaction. Toxicol Appl Pharmacol. 230:383–389. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang XJ, Sun Z, Chen W, Eblin KE, Gandolfi
JA and Zhang DD: Nrf2 protects human bladder urothelial cells from
arsenite and monomethylarsonous acid toxicity. Toxicol Appl
Pharmacol. 225:206–213. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin L, Stringfield TM, Shi X and Chen Y:
Arsenite induces a cell stress-response gene, RTP801, through
reactive oxygen species and transcription factors Elk-1 and
CCAAT/enhancer-binding protein. Biochem J. 392:93–102. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Barchowsky A, Dudek EJ, Treadwell MD and
Wetterhahn KE: Arsenic induces oxidant stress and NF-kappa B
activation in cultured aortic endothelial cells. Free Radic Biol
Med. 21:783–790. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Barchowsky A, Roussel RR, Klei LR, et al:
Low levels of arsenic trioxide stimulate proliferative signals in
primary vascular cells without activating stress effector pathways.
Toxicol Appl Pharmacol. 159:65–75. 1999. View Article : Google Scholar
|
|
27
|
Xu K, Yen T and Geczy CL: IL-10
upregulates macrophage expression of the S100 protein S100A8. J
Immunol. 166:6358–6366. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kuwayama A, Kuruto R, Horie N, Takeishi K
and Nozawa R: Appearance of nuclear factors that interact with
genes for myeloid calcium binding proteins (MRP-8 and MRP-14) in
differentiated HL-60 cells. Blood. 81:3116–3121. 1993.PubMed/NCBI
|
|
29
|
Shen SC, Yang LY, Lin HY, Wu CY, Su TH and
Chen YC: Reactive oxygen species-dependent HSP90 protein cleavage
participates in arsenical As(+3)- and MMA(+3)-induced apoptosis
through inhibition of telomerase activity via JNK activation.
Toxicol Appl Pharmacol. 229:239–251. 2008.PubMed/NCBI
|
|
30
|
Smith KR, Klei LR and Barchowsky A:
Arsenite stimulates plasma membrane NADPH oxidase in vascular
endothelial cells. Am J Physiol Lung Cell Mol Physiol.
280:L442–L449. 2001.PubMed/NCBI
|
|
31
|
Benedyk M, Sopalla C, Nacken W, et al:
HaCaT keratinocytes overexpressing the S100 proteins S100A8 and
S100A9 show increased NADPH oxidase and NF-kappaB activities. J
Invest Dermatol. 127:2001–2011. 2007. View Article : Google Scholar : PubMed/NCBI
|