Introduction
Prostate carcinoma is the most common type of cancer
in men in Western countries. At the time of diagnosis, two thirds
of carcinoma cases are limited to the prostate and can be treated
successfully (1). However, the
last third cannot be cured by surgery due to dissemination of the
tumor.
The therapy of choice for these patients is a
complete deprivation of androgens, resulting in a remission which
is, however, limited to 2–4 years, followed by the development of a
castration-resistant prostate carcinoma, finally leading to
mortality (2).
A key role in promoting the castration-resistant
prostate carcinoma is the androgen receptor. This transcription
factor in its activated state forms a stable complex with several
cofactors and facilitates transcription of distinct genes, which
have been shown to be crucial for cell growth and survival
(3,4). Androgen deprivation abolishes this
activation, leading to cell death or cell cycle arrest. However,
treatment with anti-androgens does not result in a permanent
regression and cure of the disease. After 2–4 years, increased
serum levels of prostate specific antigen (PSA) indicate that the
androgen receptor becomes transcriptionally active again (4).
Different pathways are known to reactivate the
androgen receptor complex. First, intensified expression of the
androgen receptor results in markedly increased protein levels,
compensating for the decrease of androgen ligands upon castration.
A different possibility is a broadening of the receptor’s ligand
spectrum by mutations in its coding gene. Finally, altered
phosphorylation pathways for the androgen receptor can enhance the
receptor activity and even the synthesis of tumor-produced ligands
has been described (4).
Therefore, new therapeutic approaches are needed to
interfere with carcinoma-specific signal cascades in order to cure
this disease.
Various studies investigated the role of
histone-deacetylases (HDACs) in prostate carcinoma, which have been
shown to be upregulated in these cells, mediating a decreased
expression of tumor suppressors (5–8).
Blocking HDACs with specific inhibitors leads to an increase in the
protein levels of crucial tumor suppressors.
The drug valproate, which has been used for decades
in the treatment of epilepsy, has also been shown to have
inhibitory effects on HDACs (9).
The treatment of prostate carcinoma cell lines with
valproate resulted in an increase in histone acetylation.
Furthermore, expression of the tumor suppressor p21 was elevated
and the proliferation rate of the treated cells was significantly
reduced (10). In previous
studies, we showed that the treatment of prostate carcinoma cells
(LNCaP) with valproate leads to the induction of apoptosis and an
altered gene expression in genes essential for proliferation or
apoptosis (11). Functional
analyses showed that the increased expression of the estrogen
receptor β (ERβ) is responsible for these effects (12).
A further pathway which is involved in cell growth,
metabolism and survival is the mammalian target of rapamycin (mTOR)
signal cascade. The activity of the mTOR pathway is regulated by
various positive and negative signals. Enhancement of mTOR activity
is, for instance, mediated by growth factors such as insulin-like
growth factor 1 (IGF1) and its receptor (IGF1-R), which activates
phosphoinositide-3-kinase (PI3K). PI3K, in turn, is inhibited by
the tumor suppressor phosphatase and tensin homolog (PTEN), an
essential mediator of apoptosis (13). Mutations in PTEN have been found
in various cancer specimens and facilitate proliferation and tumor
growth (14–16). PTEN is downregulated in advanced
stages of prostate carcinoma (17).
The amount of stimulating IGF1 is controlled by
IGF-binding protein-3 (IGFBP-3). IGFBP-3 binds IGF1 and thereby
prevents its association with its receptor, resulting in decreased
cell proliferation (18).
Moreover, IGFBP-3 also exhibits IGF-independent antiproliferative
and pro-apoptotic capacities (19,20). Therefore, drugs that increase the
expression of IGFBP-3 may potentially inhibit tumor cell
proliferation and growth. In this study, we investigated whether a
combination of the HDAC-inhibitor valproate and the mTOR inhibitor
temsirolimus show synergistic effects on cell proliferation and
tumor growth in vitro and in vivo.
Materials and methods
Cell culture treatment of human tumor
cells
LNCaP cells were maintained in RPMI medium,
supplemented with 10% fetal calf serum, 2% amino acid solution and
1% L-glutamine. For HDAC-inhibitor stimulation, 100,000 cells were
seeded in 6-well petri dishes. After 24 h, cells were treated with
1 mmol/l sodium valproate (Sigma, Taufkirchen, Germany). After 48
h, an additional 1 mmol/l sodium valproate was added. Temsirolimus
(Pfizer) treatment was carried out 72 h after cell seeding with a
final concentration of 1 mmol/l. Combined treatment was carried out
similarly. Five days after seeding, RNA was extracted as described
below.
Cell vitality was estimated by the Alamar Blue assay
(Biosource, Solingen, Germany), and cell proliferation was analyzed
by BrdU-ELISA (Roche Diagnostics GmbH, Mannheim, Germany).
Animal experiments
In vivo experiments were approved by the
local animal protection committee (Reference
33.9-42502-04-10/0111).
Eight-week-old male athymic nude
NMRInu/nu mice were purchased from Janvier Laboratory,
Le Genest Saint Isle, France. For acclimatization, mice were kept
for 2 weeks in standard cages with air filter hoods. All animals
received a subcutaneous inoculation dorsal of the forelegs of
106 LNCaP cells resuspended in 100 μl PBS mixed
with 100 μl Matrigel (BD Biosciences, Heidelberg, Germany)
through a 26-gauge needle. Subcutaneous tumors were measured twice
a week with calipers. Tumor volumes were calculated by the formula:
large diameter × (smaller diameter)2 ×0.5 (21). Treatment of the animals was
initiated as soon as the tumor volume had reached 120
mm3. In four different groups, mice were left untreated
(control group), or were treated with 800 mg/kg/day valproate (via
drinking water), with 0.3 mg/kg temsirolimus intravenously once a
week, or with a combination of both drugs by their individual
application. Tumor volume was calculated over the period of 7
weeks. Subsequently, animals were sacrificed and tumor samples were
excised and stored in RNAlater (Qiagen, Hilden, Germany) for mRNA
measurements or in 4% formaldehyde for histology.
mRNA-expression analysis
Total cellular RNA from LNCaP cells was extracted
with the Quick-RNA™MiniPrep (Zymo Research, Freiburg, Germany).
Total cellular RNA from tumor biopsies was extracted by peqGold
TriFast™ (Peqlab, Erlangen, Germany). RNA integrity and quantity
were assessed on an Agilent 2100 Bioanalyzer with a RNA 6000 Nano
LabChip kit (Agilent Technologies, Waldbronn, Germany). Reverse
transcription of 500 ng total cellular RNA with random hexamer
primers was performed with an Omniscript RT kit (Qiagen).
Expression of acidic ribosomal protein (ARP), PSA, IGF1, IGF1-R and
IGFBP-3 was assayed on an iCycler iQ real-time detection system
(Bio-Rad, Munich, Germany) with SsoFast EvaGreen supermix,
(Bio-Rad). The 20 μl reaction from the kit was supplemented
with 2 μl cDNA, 0.6 μM gene-specific primers (IBA,
Göttingen, Germany). The following primers were used: IGF1,
forward, 5′-TGGATGC TCTTCAGTTCGTG-3′ and reverse, 5′-AGGGGTGC
GCAATACATCT-3′; IGF1-R, forward, 5′-CCGAAGGTCTG TGAGGAAGA-3′ and
reverse, 5′-AATGGCGGATCTT CACGTAG-3′; IGFBP-3, forward,
5′-GAACTTCTCCTCCGA GTCCAA-3′ and reverse, 5′-CTGGGACTCAGCACATTG
AG-3′; PSA, forward, 5′-TGAACCAGAGGAGTTCTTGAC-3′ and reverse,
5′-CCCCAGAATCACCCGAGCAG-3′; ARP, forward,
5′-CGACCTGGAAGTCCAACTAC-3′ and reverse,
5′-ATCTGCTGCATCTGCTTG-3′.
Data analysis was performed according to the
ΔΔCt-method (22). In cell
culture and in animal experiments, ARP served as an internal
control.
PSA secretion in serum from mice was measured with
PSA Enzyme Immunoassay Test kit (GenWay Biotech, Inc., San Diego,
CA, USA).
Histology
Sections of tumors were fixed with 4% formaldehyde
in PBS for 72 h. The samples were processed according to standard
procedures with a Leica TP1020 automatic tissue processor (Leica
Mikrosysteme Vertrieb GmbH, Wetzlar, Germany). Paraffin-embedded
tumor samples were dissected and stained with hematoxylin and eosin
(HE) and immunostained against Ki-67 (23,24). For the calculation of the
proliferation index, Ki-67 positive cells in correlation with the
total cell number were counted in 10 fields of vision at ×400
magnification.
Results
Valproate and temsirolimus reduce
proliferation of LNCaP cells and act synergistically
In order to test whether the HDAC-inhibitor
valproate or the mTOR-inhibitor temsirolimus or the combination of
both drugs influence cell proliferation or vitality, we performed
an Alamar Blue Assay and BrdU test with treated cells. In the
Alamar Blue Assay, no significant difference to the untreated
control was observed, indicating that cell vitality is not
immediately affected by treatment with valproate or temsirolimus
(Fig. 1).
By contrast, incubation of LNCaP cells with
valproate resulted in a decreased cell proliferation by 64.6%
(Fig. 2). Similarly, the
stimulation with temsirolimus diminished cell proliferation by
23.9%. Furthermore, the combined treatment of LNCaP cells with both
drugs resulted in a reduction of proliferation by 81.8% compared
with untreated cells.
Expression changes in LNCaP cells
following treatment with valproate, temsirolimus or a combination
of both
To elucidate if the reduced cell proliferation of
treated LNCaP cells was associated with altered expression of key
regulators of cell proliferation, we analyzed the mRNA expression
of PSA, IGF1, IGF1-R and IGFBP-3 using real-time RT-PCR.
Incubation with valproate led to decreased
expression of PSA, IGF1 and IGF1-R (Fig. 2). By contrast, treatment with
temsirolimus or with both drugs combined did not influence the
expression of these genes.
Consistent with a decreased proliferation, IGFBP-3
expression was elevated 7.5-fold upon valproate treatment. This
effect was markedly enhanced by the combination of valproate and
temsirolimus, resulting in a 53-fold increase in IGFBP-3 expression
(Fig. 3).
Influence of valproate and temsirolimus
on tumor growth in the mouse model
Next, we investigated whether the observed effect on
cultured prostate carcinoma cells is also observed in vivo
using LNCaP-derived tumors in a nude mouse model.
Therefore, 106 LNCaP cells were implanted
subcutaneously into immune-suppressed nude mice. Of the 80 mice,
only 31 (38.8%) developed tumors.
After reaching a distinct tumor volume (120
mm3), mice were treated with valproate (800 mg/kg/day
per os), temsirolimus (0,3 mg/kg weekly intravenously) or with a
combination of both. A fourth group was left untreated and served
as control.
Tumor growth (calculated by weekly measurements of
tumor volume) did not show any significant differences between the
distinct groups (Fig. 4).
Variation in tumor growth within each group was rather high. Only
the temsirolimus treated group exhibited, with one exception, a
homogenous growth. Similarly, serum levels of human PSA, which was
produced by the LNCaP cells of the tumors, did not differ between
the groups (data not shown). The amount of secreted PSA serves as
an indicator for the transcriptional activity of the androgen
receptor.
Combined therapy of valproate and
temsirolimus leads to a decreased proliferation capacity in
vivo
Although we detected no difference in tumor growth
(measured by the increase in tumor volume), histology preparations
showed that the combined application of valproate and temsirolimus
resulted in a significantly decreased proliferation capacity (49%)
compared to untreated control animals (80%) or mice treated with
valproate (81%) or temsirolimus (77%) alone (Figs. 5–8).
Expression changes in tumor cells
following treatment with valproate, temsirolimus or a combination
of both
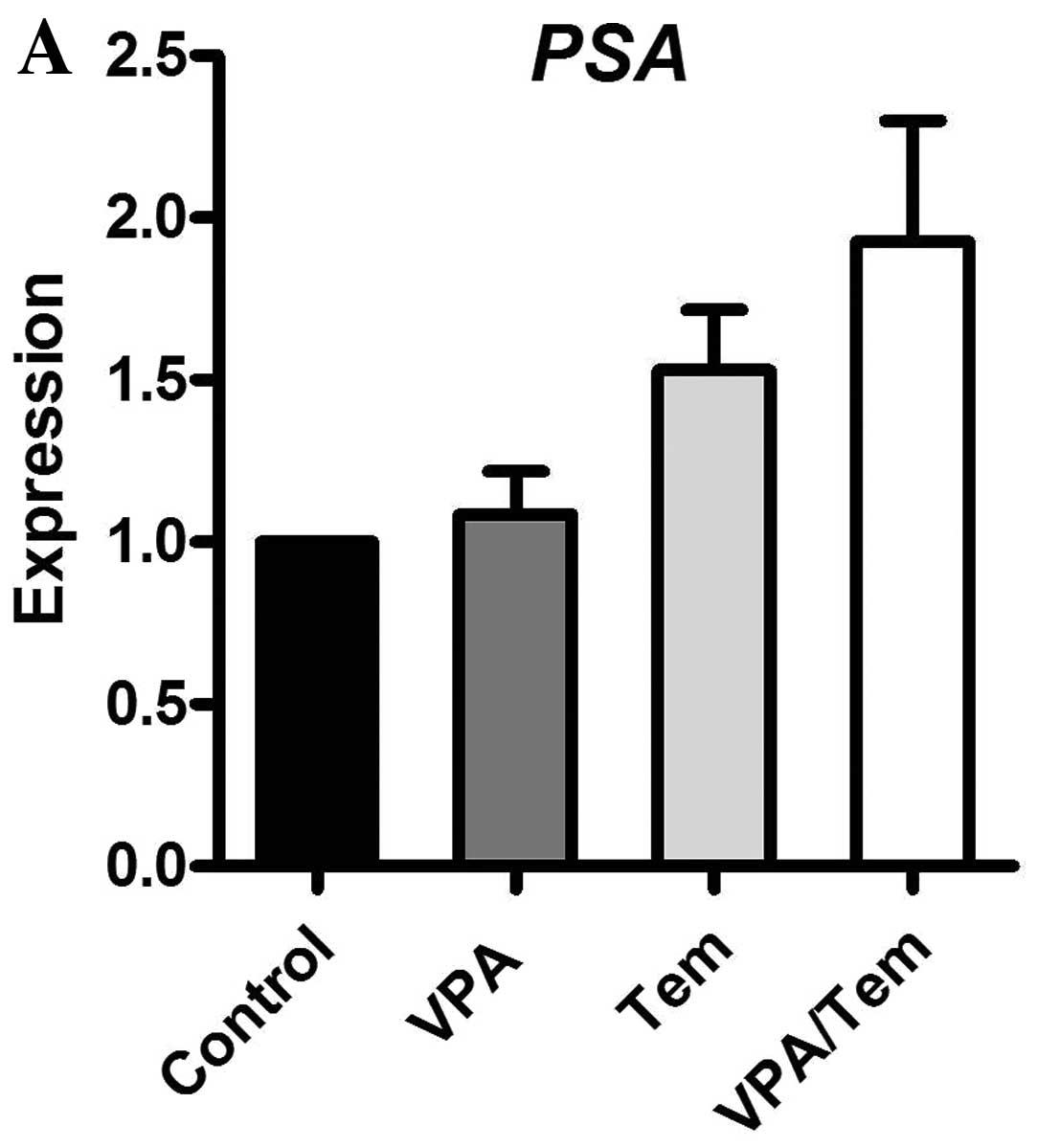
Although no difference in tumor growth was observed,
we investigated the mRNA expression of crucial regulators of cell
proliferation in prostate carcinoma. Therefore, RNA was extracted
from tumor samples and analyzed similarly to the cell culture
experiments for the mRNA expression levels of PSA, IGF1, IGF1-R and
IGFBP-3 via real-time PCR (Fig.
9). Valproate and the combination with temsirolimus enhanced
the expression of PSA. IGF1 was reduced upon treatment with
valproate and temsirolimus. The expression of IGF1-R was only
increased by temsirolimus. In contrast to our cell culture
experiments, the combination of valproate and temsirolimus reduced
the expression of IGFBP-3.
Discussion
In the present study, we showed that valproate and
temsirolimus mediate a decrease in the proliferation potential of a
cultured prostate carcinoma cell line. Furthermore, the enhanced
effect of a combined treatment suggests that both drugs act
synergistically. Valproate has been described to inhibit
tumorigenesis by reduction of tumor growth and induction of cell
differentiation in different cell lines, such as neuroblastoma
cells (SH-SY5Y) and a breast cancer cell line (MT-450) (9,25,26). In a previous study, we showed a
fundamental mechanism underlying this phenomenon: valproate
inhibits the activity of HDACs in LNCaP cells, leading to an
increased expression of ERβ (10,11). ERβ itself exhibits
antiproliferative, anti-invasive and pro-apoptopic capacities and
is downregulated in prostate carcinoma (27–31). In addition, valproate is a
negative regulator of the cell cycle: treatment with valproate
increases the amount of G0/G1 phase prostate carcinoma cells and
reduces the expression of Cdk1 (32). Decreased Cdk1 levels in turn
result in a reduction of androgen receptor transcription (33). On the other hand, an
antiproliferative influence has been reported for various mTOR
inhibitors in vitro and in vivo as well (34). For instance, the application of
rapamycin significantly reduces the growth rate of induced prostate
carcinomas in a mouse model (35). Furthermore, Fung et al
(36) showed a decreased
proliferation of LNCaP cells upon treatment with temsirolimus,
connected with a reduction in S-phase cells and an increase in
G1-phase cells. Similar results have also been achieved with
another mTOR inhibitor, RAD001 (32). The reduction of proliferation of
LNCaP cells treated with a combination of valproate and
temsirolimus observed in this study is in line with data from Wedel
et al (32), who reported
a synergistic effect of valproate with RAD001 on the inhibition of
the cell cycle.
We further showed that the application of valproate
on LNCaP cells results in a decrease of PSA expression, which is
most likely due to an increase in ERβ expression (12). PSA is frequently used to asses the
activity of prostate carcinoma and therefore a valid marker for
tumorigenesis and diagnosis (37). A key regulator in tumorigenesis is
the IGF-signaling pathway, which influences crucial processes such
as cell proliferation, differentiation, transformation and
apoptosis (38). Two regulatory
mechanisms are essential for triggering the IGF activity: IGFBP-3
binds IGF1 and thereby competes with the IGF-receptor whereas PSA
cleaves and inactivates IGFBP-3, thus increasing the pool of
functional IGF (39).
IGF1/IGF1-R-signaling enhances the metastatic capacity of prostate
carcinoma cells (40,41) and facilitates the nuclear import
of the androgen-receptor in the absence of androgens (42,43). In a previous study, IGF1 and
IGFBP-3 were found to be correlated with advanced prostate cancer
(44). Thus, IGF1 is a key
mediator for the development of a castration-resistant prostate
carcinoma, as it is not directly targeted by the currently used
anti-androgen therapy. We found that treatment with valproate leads
to a decreased expression of IGF1 and IGF1-R, whereas the
expression of IGFBP-3 is enhanced. Furthermore, the combined
application of valproate and temsirolimus further potentiates this
effect. By contrast, temsirolimus alone does not affect IGFBP-3
expression. In addition to its inhibitory effects on IGF-signaling,
IGFBP-3 also exhibits IGF-independent antiproliferative and
pro-apoptotic abilities (19,20).
We showed that the simultaneous treatment of a
prostate carcinoma cell line with the HDAC-inhibitor valproate and
the mTOR inhibitor temsirolimus leads to synergistic
anti-proliferative and pro-apoptotic effects by modulating the
IGF1-signaling pathway. However, why the application of
temsirolimus alone has no effect and the underlying mechanisms for
the synergistic effects of the combined therapy, remain
unclear.
To investigate whether the observed effect of
valproate and temsirolimus on cultured prostate carcinoma cells is
sufficient to inhibit tumor growth in vivo, we implanted
LNCaP cells into nude mice. However, we observed no significant
difference in tumor volume over the time of drug administration
between the treated and untreated groups. Which concentration of
the drug actually reaches the transformed cells due to its specific
bioavailability has yet to be clarified. For valproate it has been
shown that the dose rate we used in this study is sufficient to
obtain a blood concentration, which is used in patients (45); however, different vascularization
of the tumor may also influence the concentration of the drug at
its destination. To our knowledge, for temsirolimus, no data are
available about its bioavailability and its final concentration in
targeted tissues.
In contrast to our finding, valproate has been shown
in other studies to inhibit tumor growth in human stomach and liver
carcinomas (46,47).
Contradictory reports have been published regarding
the in vivo effect of mTOR inhibitors. The mTOR inhibitor
RAD001 was able to reduce the growth of prostate carcinoma in mice
as well as prostate carcinoma in mice tibiae (35,48). By contrast, the treatment of
LNCaP-derived tumors in SCID mice with RAD001 did not affect tumor
growth, which is in line with our results (49).
Consistent with unchanged tumor volumes, levels of
human PSA in the blood of treated versus untreated mice also showed
no differences. Therefore, we concluded that the activity of the
androgen receptor is not permanently altered upon drug treatment in
our experiment. This is in contrast to our findings from the cell
culture experiment, where valproate reduced the expression of PSA
2.9-fold, whereas temsirolimus and the combined application of both
drugs did not show any effects.
Whether the potential of valproate on PSA expression
is not sufficient to reduce PSA levels in the blood or whether
other mechanisms, which are not present in vitro, interfere,
remain to be clarified.
In addition, the results from the expression
analysis of PSA, IGF1, IGF1-receptor and IGFBP-3 differ from our
cell culture experiments.
While PSA mRNA is decreased in cultured prostate
carcinoma cells, its expression in vivo is increased by 53%
upon treatment with temsirolimus and combined therapy with
valproate and temsirolimus even potentiates this phenomenon.
However, this elevated expression is not reflected by an increase
in serum-PSA. Clearly, expression alterations induced by short-term
stimulation of cultured cells do not persist in long-term in
vivo treatments. In addition, the mRNA expression of IGF-axis
genes does not reflect tumor growth features in vivo.
Although we did not observe any significant growth
inhibition of prostate carcinoma cell-derived tumors in our nude
mouse model upon treatment with valproate, temsirolimus or a
combination of both, staining against the cell cycle marker Ki-67
in histological sections of the tumors showed a remarkable
reduction in the proliferation potential when the mice were treated
with a combination of valproate and temsirolimus. By contrast,
tumors from untreated animals or mice treated with valproate or
temsirolimus alone showed a significant higher proliferation
potential. This is in line with our finding from the cell culture
experiments, where the combined therapy exhibits synergistic
effects. A similar phenomenon was observed for the combination of
valproate with another mTOR inhibitor, RAD001, in cultured prostate
carcinoma cells (32). However,
we can only speculate why the reduced proliferation potential does
not result in a reduced tumor growth. One possibility is that the
period of treatment was too short, transformed cells proliferate in
the beginning, without interference of a drug, and reach a steady
state situation upon treatment with the valproate-temsirolimus
combination. This is reflected by a reduced Ki-67-staining in tumor
sections. Prolonged and extended studies are needed to clarify
whether these tumors would finally arrest growth.
We hypothesize that the combined therapy of prostate
carcinoma with the HDAC inhibitor valproate and the mTOR inhibitor
temsirolimus exhibits synergistic effects on cell proliferation and
apoptosis and is thus potentially capable of inhibiting tumor
growth. The mTOR kinase is known to be the main signal integrating
point receiving inputs via the PTEN/PI3K pathway through the action
of Akt. In castration-resistant prostate cancer, mutated PTEN is
expressed opening the PTEN/PI3K/Akt pathway for constitutive
activation of anti-apoptotic mechanisms and activation of the mTOR
pathway in the absence of androgens. Clinical trials with mTOR
inhibitors in advanced prostate cancer led to the conclusion that
it is likely that such agents will need to be combined with other
therapies (50). Here, we
combined the mTOR inhibitor temsirolimus with the HDAC inhibitor
valproate with anti-androgen capacities, as we previously
demonstrated (12). However, our
in vivo data show that further studies are warranted to
evaluate the optimal conditions (dose rate, application form,
formulation) for the application of these two drugs in the therapy
of prostate carcinoma.
Abbreviations:
|
LNCaP
|
lymph node metastasis of prostate
cancer;
|
|
mTOR
|
mammalian target of rapamycin;
|
|
IGFBP-3
|
insulin-like growth factor-binding
protein-3;
|
|
IGF1-R
|
insulin-like growth factor 1
receptor;
|
|
ARP
|
acidic ribosomal protein;
|
|
PSA
|
prostate-specific antigen;
|
|
HDAC
|
histone-deacetylase;
|
|
PTEN
|
phosphatase and tensin homolog;
|
|
BrdU
|
bromodeoxyuridine;
|
|
ELISA
|
enzyme-linked immunosorbent assay;
|
|
PBS
|
phosphate-buffered saline;
|
|
mRNA
|
messenger RNA;
|
|
RT-PCR
|
reverse transcription-polymerase chain
reaction
|
References
|
1.
|
Börgermann C, Loertzer H, Luboldt HJ, et
al: PSA - Quo vadis? Urologe A. 48:1012–1014. 2009.(In German).
|
|
2.
|
Tannock IF, de Wit R, Berry WR, et al:
Docetaxel plus prednisone or mitoxantrone plus prednisone for
advanced prostate cancer. N Engl J Med. 351:1502–1512. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Centenera MM, Harris JM, Tilley WD and
Butler LM: The contribution of different androgen receptor domains
to receptor dimerization and signaling. Mol Endocrinol.
22:2373–2382. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Knudsen KE and Penning TM: Partners in
crime: deregulation of AR activity and androgen synthesis in
prostate cancer. Trends Endocrinol Metab. 21:315–324. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Wang L, Zou X, Berger AD, et al: Increased
expression of histone deacetylaces (HDACs) and inhibition of
prostate cancer growth and invasion by HDAC inhibitor SAHA. Am J
Transl Res. 1:62–71. 2009.PubMed/NCBI
|
|
6.
|
Weichert W, Röske A, Gekeler V, et al:
Histone deacetylases 1, 2 and 3 are highly expressed in prostate
cancer and HDAC2 expression is associated with shorter PSA relapse
time after radical prostatectomy. Br J Cancer. 98:604–610. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Halkidou K, Gaughan L, Cook S, et al:
Upregulation and nuclear recruitment of HDAC1 in hormone refractory
prostate cancer. Prostate. 59:177–189. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Xu WS, Parmigiani RB and Marks PA: Histone
deacetylase inhibitors: molecular mechanisms of action. Oncogene.
26:5541–5552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Göttlicher M, Minucci S, Zhu P, et al:
Valproic acid defines a novel class of HDAC inhibitors inducing
differentiation of transformed cells. EMBO J. 20:6969–6978.
2001.PubMed/NCBI
|
|
10.
|
Xia Q, Sung J, Chowdhury W, et al: Chronic
administration of valproic acid inhibits prostate cancer cell
growth in vitro and in vivo. Cancer Res. 66:7237–7244. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Thelen P, Schweyer S, Hemmerlein B, et al:
Expressional changes after histone deacetylase inhibition by
valproic acid in LNCaP human prostate cancer cells. Int J Oncol.
24:25–31. 2004.PubMed/NCBI
|
|
12.
|
Stettner M, Kaulfuss S, Burfeind P, et al:
The relevance of estrogen receptor-beta expression to the
antiproliferative effects observed with histone deacetylase
inhibitors and phytoestrogens in prostate cancer treatment. Mol
Cancer Ther. 6:2626–2633. 2007. View Article : Google Scholar
|
|
13.
|
Yuan R, Kay A, Berg WJ and Lebwohl D:
Targeting tumorigenesis: development and use of mTOR inhibitors in
cancer therapy. J Hematol Oncol. 2:452009. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Li J, Yen C, Liaw D, et al: PTEN, a
putative protein tyrosine phosphatase gene mutated in human brain,
breast, and prostate cancer. Science. 275:1943–1947. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Steck PA, Pershouse MA, Jasser SA, et al:
Identification of a candidate tumour suppressor gene, MMAC1, at
chromosome 10q23.3 that is mutated in multiple advanced cancers.
Nat Genet. 15:356–362. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Jiang BH and Liu LZ: PI3K/PTEN signaling
in angiogenesis and tumorigenesis. Adv Cancer Res. 102:19–65. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Kremer CL, Klein RR, Mendelson J, et al:
Expression of mTOR signaling pathway markers in prostate cancer
progression. Prostate. 66:1203–1212. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Pollak M: Insulin and insulin-like growth
factor signalling in neoplasia. Nat Rev Cancer. 8:915–928. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Han J, Jogie-Brahim S, Harada A and Oh Y:
Insulin-like growth factor-binding protein-3 suppresses tumor
growth via activation of caspase-dependent apoptosis and cross-talk
with NF-κB signaling. Cancer Lett. 307:200–210. 2011.PubMed/NCBI
|
|
20.
|
Franklin SL, Ferry RJ Jr and Cohen P:
Rapid insulin-like growth factor (IGF)-independent effects of IGF
binding protein-3 on endothelial cell survival. J Clin Endocrinol
Metab. 88:900–907. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Igawa T, Lin FF, Lee MS, et al:
Establishment and characterization of androgen-independent human
prostate cancer LNCaP cell model. Prostate. 50:222–235. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
|
|
23.
|
Gerdes J, Lemke H, Baisch H, et al: Cell
cycle analysis of a cell proliferation-associated human nuclear
antigen defined by the monoclonal antibody Ki-67. J Immunol.
133:1710–1715. 1984.PubMed/NCBI
|
|
24.
|
Scholzen T and Gerdes J: The Ki-67
protein: from the known and the unknown. J Cell Physiol.
182:311–322. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Yuan PX, Huang LD, Jiang YM, et al: The
mood stabilizer valproic acid activates mitogen-activated protein
kinases and promotes neurite growth. J Biol Chem. 276:31674–31683.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Blaheta RA and Cinatl J Jr: Anti-tumor
mechanisms of valproate: a novel role for an old drug. Med Res Rev.
22:492–511. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Cheng J, Lee EJ, Madison LD and Lazennec
G: Expression of estrogen receptor β in prostate carcinoma cells
inhibits invasion and proliferation and triggers apoptosis. FEBS
Lett. 566:169–172. 2004.
|
|
28.
|
Horvath LG, Henshall SM, Lee CS, et al:
Frequent loss of estrogen receptor-beta expression in prostate
cancer. Cancer Res. 61:5331–5335. 2001.PubMed/NCBI
|
|
29.
|
Latil A, Bièche I, Vidaud D, et al:
Evaluation of androgen, estrogen (ER alpha and ER beta), and
progesterone receptor expression in human prostate cancer by
real-time quantitative reverse transcription-polymerase chain
reaction assays. Cancer Res. 61:1919–1926. 2001.
|
|
30.
|
Pasquali D, Rossi V, Esposito D, et al:
Loss of estrogen receptor beta expression in malignant human
prostate cells in primary cultures and in prostate cancer tissues.
J Clin Endocrinol Metab. 86:2051–2055. 2001.PubMed/NCBI
|
|
31.
|
Pasquali D, Staibano S, Prezioso D, et al:
Estrogen receptor beta expression in human prostate tissue. Mol
Cell Endocrinol. 178:47–50. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Wedel S, Hudak L, Seibel JM, et al:
Inhibitory effects of the HDAC inhibitor valproic acid on prostate
cancer growth are enhanced by simultaneous application of the mTOR
inhibitor RAD001. Life Sci. 88:418–424. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Chen S, Xu Y, Yuan X, et al: Androgen
receptor phosphorylation and stabilization in prostate cancer by
cyclin-dependent kinase 1. Proc Natl Acad Sci USA. 103:15969–15974.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Azim H, Azim HA Jr and Escudier B:
Targeting mTOR in cancer: renal cell is just a beginning. Target
Oncol. 5:269–280. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Zhang W, Zhu J, Efferson CL, et al:
Inhibition of tumor growth progression by antiandrogens and mTOR
inhibitor in a Pten-deficient mouse model of prostate cancer.
Cancer Res. 69:7466–7472. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Fung AS, Wu L and Tannock IF: Concurrent
and sequential administration of chemotherapy and the Mammalian
target of rapamycin inhibitor temsirolimus in human cancer cells
and xenografts. Clin Cancer Res. 15:5389–5395. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Thompson IM Jr, Leach RJ and Ankerst DP:
Prostate cancer detection: a view of the future. Eur Urol.
59:191–193. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Moschos SJ and Mantzoros CS: The role of
the IGF system in cancer: from basic to clinical studies and
clinical applications. Oncology. 63:317–332. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Jogie-Brahim S, Feldman D and Oh Y:
Unraveling insulin-like growth factor binding protein-3 actions in
human disease. Endocr Rev. 30:417–437. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Marelli MM, Moretti RM, Procacci P, et al:
Insulin-like growth factor-I promotes migration in human
androgen-independent prostate cancer cells via the ανβ3 integrin
and PI3-K/Akt signaling. Int J Oncol. 28:723–730. 2006.PubMed/NCBI
|
|
41.
|
Goya M, Miyamoto S, Nagai K, et al: Growth
inhibition of human prostate cancer cells in human adult bone
implanted into nonobese diabetic/severe combined immunodeficient
mice by a ligand-specific antibody to human insulin-like growth
factors. Cancer Res. 64:6252–6258. 2004. View Article : Google Scholar
|
|
42.
|
Wen Y, Hu MC, Makino K, et al: HER-2/neu
promotes androgen-independent survival and growth of prostate
cancer cells through the Akt pathway. Cancer Res. 60:6841–6845.
2000.PubMed/NCBI
|
|
43.
|
Manin M, Baron S, Goossens K, et al:
Androgen receptor expression is regulated by the phosphoinositide
3-kinase/Akt pathway in normal and tumoral epithelial cells.
Biochem J. 366:729–736. 2002.PubMed/NCBI
|
|
44.
|
Rowlands MA, Holly JM, Hamdy F, et al:
Serum insulin-like growth factors and mortality in localised and
advanced clinically detected prostate cancer. Cancer Causes
Control. 23:347–354. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Shabbeer S, Kortenhorst MS, Kachhap S, et
al: Multiple molecular pathways explain the anti-proliferative
effect of valproic acid on prostate cancer cells in vitro and in
vivo. Prostate. 67:1099–1110. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Yagi Y, Fushida S, Harada S, et al:
Effects of valproic acid on the cell cycle and apoptosis through
acetylation of histone and tubulin in a scirrhous gastric cancer
cell line. J Exp Clin Cancer Res. 29:1492010. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Machado MC, Bellodi-Privato M, Kubrusly
MS, et al: Valproic acid inhibits human hepatocellular cancer cells
growth in vitro and in vivo. J Exp Ther Oncol. 9:85–92.
2011.PubMed/NCBI
|
|
48.
|
Morgan TM, Pitts TE, Gross TS, et al:
RAD001 (Everolimus) inhibits growth of prostate cancer in the bone
and the inhibitory effects are increased by combination with
docetaxel and zoledronic acid. Prostate. 68:861–871. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Schayowitz A, Sabnis G, Goloubeva O, et
al: Prolonging hormone sensitivity in prostate cancer xenografts
through dual inhibition of AR and mTOR. Br J Cancer. 103:1001–1007.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Corcoran NM, Costello AJ and Hovens CM:
Interfering with cell-survival signalling as a treatment strategy
for prostate cancer. BJU Int. 97:1149–1153. 2006. View Article : Google Scholar : PubMed/NCBI
|