Introduction
Elevated plasma cholesterol, such as native
low-density lipoprotein (LDL) and oxidized LDL (oxLDL), is a
hallmark of numerous cardiovascular diseases including
hypercholesterolemia, atherosclerosis, hypertension, heart failure,
and diabetes. These diseases are closely linked with endothelial
dysfunction indicating decreased nitric oxide (NO) production in
the endothelium. In the vasculature, NO is a vasoprotective
molecule and plays a central role in vascular homeostasis by
regulating vasoreactivity, platelet activation, leukocyte adhesion
and smooth muscle cell migration and proliferation (1). It is well established that
endothelial arginase constrains the activity of endothelial nitric
oxide synthase (eNOS) by substrate depletion, thereby reducing NO
bioavailability and contributing to vascular diseases. oxLDL, the
primary pathogenic lipid in atherogenesis, activates human
endothelial cell arginase II by stimulating the dissociation of
arginase II from microtubules and also by inducing arginase II mRNA
transcription (2). Furthermore,
atherogenic-prone apolipoprotein E-null (ApoE−/−) mice
treated with an arginase inhibitor exhibit restored NO
bioavailability and endothelial function, reactive oxygen species
(ROS) production, and an arterial compliance similar to that
observed in wild-type (WT) mice (3). Therefore, endothelial arginase may
be a novel target for therapeutic drug design for vascular diseases
such as atherosclerosis (3).
Rhubarb is the rhizome of Rheum undulatum and
is commonly distributed in Asia. Many components of the rhizome
possess diverse biological activities and have been reported as
being able to counter allergic (4) and diabetic states (5), as having anti-oxidant properties
(4), and as functioning as a
vasorelaxant (6). Piceatannol,
one of the active components of rhubarb, was recently found to
inhibit lipooxygenase activity (7) and VSMC proliferation and migration
(8). Recently, we reported that
piceatannol-3′-O-β-D-glucopyranoside (PG) is a potent inhibitor of
arginase isoforms. PG inhibited the arginase isoforms in a
dose-dependent manner, resulting in augmented NO production by
enhancing eNOS dimer stabilization (9).
Based on these data, we hypothesized that PG
regulates vascular function. Therefore, we examined whether PG
improves NO/ROS production and endothelial dysfunction in
ApoE−/− mice fed a high-cholesterol diet (HCD). We also
investigated whether arginase inhibition by PG restores L-arginine
bioavailability and attenuates fatty streak formation in this
atherogenic mouse model.
Materials and methods
Animals
Twenty 10-week-old male wild-type (WT) (C57BL/6J)
and ApoE−/− mice (Dae Han Biolink Co.) were studied. The
study was approved in accordance with the Guide for the Care and
Use of Laboratory Animals (Institutional Review Board, Kangwon
National University).
Protocol
To determine the effect of PG on vascular
reactivity, we studied aortic rings isolated from 20 male C57BL/6J
WT mice fed a normal diet (ND) and 20 male ApoE−/− mice
fed an HCD (D12108C; Research Diet Inc., USA) for 6 weeks. Aortic
rings were incubated with or without PG (50 μmol/l) for 18 h as
previously described (9). For the
pathological assay, PG was administered in the drinking water to
ApoE−/− mice for 6 weeks when the mice were started on
the HCD. Given that each mouse consumed ~10 ml water/day this
represented a daily dose of ~500 μg/mouse/day of PG.
Western blot analysis
Aortic vessel lysates were subjected to SDS-PAGE,
and densitometry of the bands was conducted using NIH ImageJ
(9). To analyze the ratio of eNOS
dimer to monomer, proteins were separated using low-temperature
SDS-PAGE followed by western blot analysis (9).
Aortic vascular tension assay
Male C57BL/6J mice fed an HCD were anesthetized
using isoflurane, and the thoracic aorta was rapidly removed. The
aortas were placed in ice-cold oxygenated Krebs-Ringer bicarbonate
solution (in mM: NaCl 118.3, KCl 4.7, MgSO4 1.2,
KH2PO4 1.2, CaCl2 1.6,
NaHCO3 25, glucose 11.1), and cleared of adherent
connective tissues. The mouse aortas were cut into 1.5-mm rings and
suspended between two wire stirrups (150 μm) in a myograph (Multi
Myograph System DMT-620) in 10 ml Krebs-Ringer solution (95%
O2-5% CO2, pH 7.4, 37°C). One stirrup was
connected to a three-dimensional micromanipulator, and the other
was connected to a force transducer. The rings were passively
stretched at 10-min intervals in increments of 100 mg to reach
optimal tone (600 mg). After the arterial rings had been stretched
to their optimal resting tone, the contractile response to 100 mM
KCl was determined. The response to a maximal dose of KCl was used
to normalize the responses to agonist across vessel rings. The dose
response to the vasoconstrictors, PE
(10−9–10−4 M) and U46619
(10−9–10−5 M), was performed first. This was
followed by the dose response to the vasodilators, acetylcholine
(Ach, 10−9–10−5 M) and SNP
(10−9–10−5 M) after pre-constriction with PE
(10−6 M). At the end of the experiments, the
NO-dependency of vasorelaxation was confirmed by adding the
inhibitor of guanylate cyclase
[1H-[1,2,4]oxadizolo[4,3-a]quinoxalin-1-one (ODQ), 10−6
M)].
Arginase activity
Tissue lysates were prepared using lysis buffer (50
mM Tris-HCl, pH 7.5, 0.1 mM EDTA and protease inhibitors) by
homogenization at 4°C followed by centrifugation for 20 min at
14,000 × g at 4°C. The supernatants were used to assay for arginase
activity as previously described (10).
Estimation of NO or ROS generation using
DAF-FM or DHE in isolated mice aorta
Mice aortic rings were isolated and incubated
overnight at 37°C in 5% CO2 in Dulbecco’s modified
Eagle’s medium containing 2% FBS and antibiotics (1X) in the
presence of PG (50 μmol/l) (10).
The fluorescence from the aortic endothelium was measured at
different time intervals under microscopy (9).
Determination of intracellular L-arginine
concentrations
The intracellular concentration of L-arginine was
determined by high-performance liquid chromatography (HPLC) using
pre-column derivatization with o-phthalaldehyde (OPA) by
modification of a previously published method (11). L-arginine (100 μmol/l) was added
to the cell lysate (0.5 ml) as an internal standard. The samples
were extracted on solid-phase extraction cartridges (CBA Bond
Elute; Varian, Inc.). Recovery rates were 87.5±3.9%. Eluates were
dried over nitrogen and resuspended in double-distilled water for
HPLC analysis. HPLC was performed on a computer-controlled Waters
chromatography system (M600E) consisting of an automatic injector
(M7725i; Waters Co.) and a fluorescence detector (FP-1520; Jasco
Co.) located in the Central Laboratory of Kangwon National
University. Samples were incubated for exactly 1 min with OPA
reagent (5.4 mg/ml OPA in borate buffer, pH 8.4, containing 0.4%
2-mercaptoethanol) before automatic injection for the HPLC. The OPA
derivative of L-arginine was separated on a 150×4.6 mm, 3.5-μm
Zorbax Eclipse XDB-C18 column with the fluorescence detector set at
Ex 340 nm and Em 450 nm. Samples were eluted from the column with
0.96% citric acid/methanol (70:30), pH 6.8, at a flow rate of 1.5
ml/min.
Gross pathologic assessment of
plaque
The extent of atherosclerosis in the aortas was
quantified in an en face preparation. Digital color images
of the aortas after staining with Oil Red O were analyzed using
MetaMorph image analysis software (Molecular Devices, Sunnyvale,
CA, USA).
Data analysis and statistics
Aortic vasoconstrictor responses are presented as
percent change in the maximum response induced by KCl. Vasodilator
responses are expressed as a percentage of pre-constricted tension.
The EC50 and the maximal response (Emax) were
calculated using nonlinear logistic regression analysis with Prism
(GraphPad) software. All data are represented as means ± standard
error of at least four independent experiments. An unpaired
Student’s t-test or 2-way ANOVA was used to assess statistical
significance. A value of P<0.05 was accepted as significant.
Results
Vascular responses of PG-treated aortic
rings from WT mice fed an ND
It was previously demonstrated that PG inhibits
arginase activity and reciprocally increases NO production in the
endothelium by enhancing eNOS dimerization. Therefore, we tested
the vascular response to the vasoconstrictors, PE and U46619, and
the vasorelaxants, Ach and SNP, with and without preincubation with
PG.
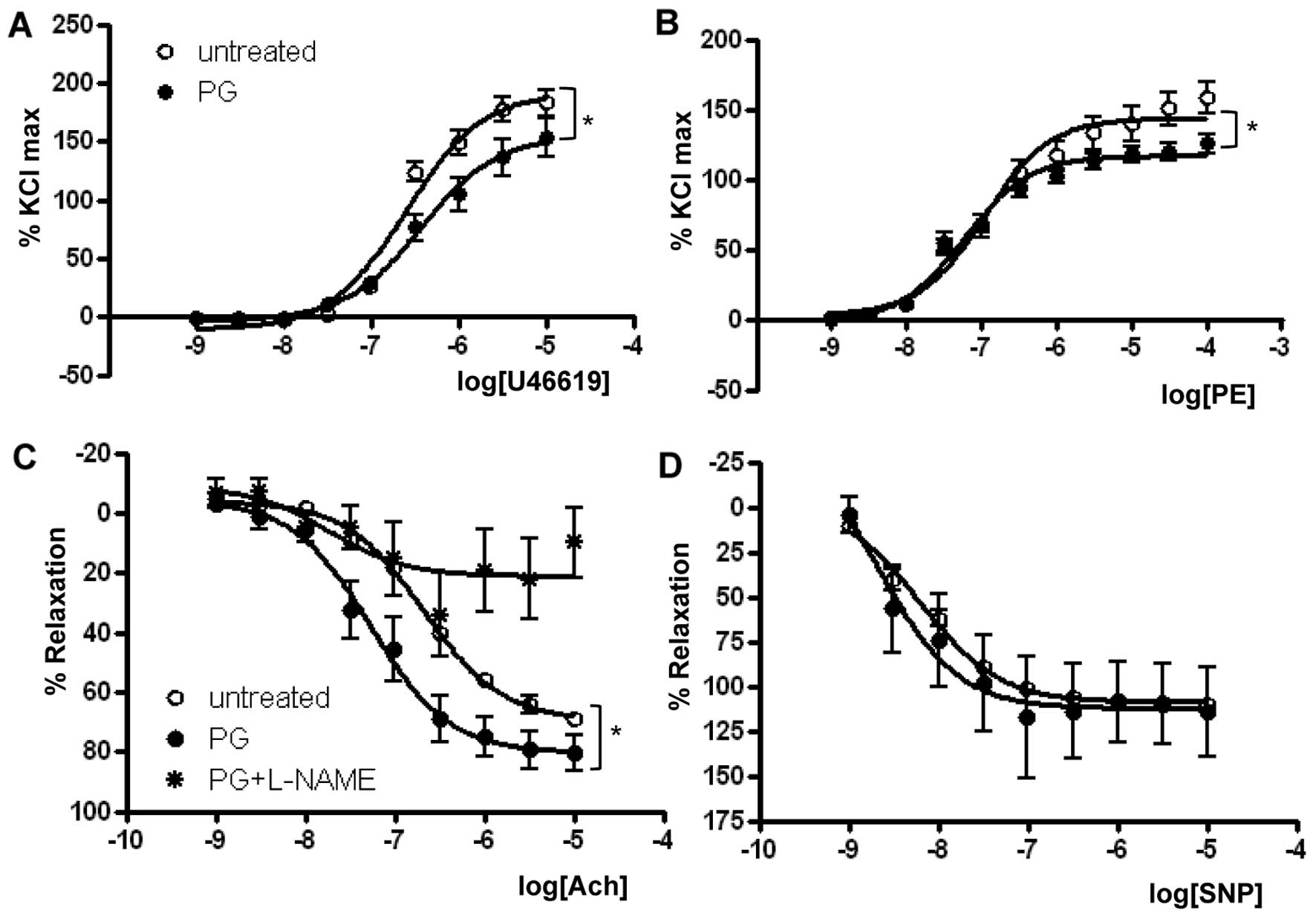
As presented in Fig.
1 and Table I, the
Emax in response to U46619 in the PG-treated group
(155.2±8.6%) was significantly reduced when compared with that in
the untreated group (192.0±5.9%, P<0.01) (Fig. 1A). Similar to the U46619 response,
PG exposure significantly reduced the Emax of
PE-dependent vasoconstriction (Fig.
1B) (untreated vs. PG, 143.9±4.4 vs. 116.9±2.3%, P<0.01,
n=7). The U46619 EC50 was significantly lower in the
PG-treated group (untreated vs. PG, −6.59±0.06 vs. −6.42±0.11 M
[log (PE)], P<0.05), and the EC50 in response to PE
was more significantly reduced in the PG-treated group (untreated
vs. PG, −6.91±0.10 vs. −7.23±0.07 M [log (PE)], P<0.05).
Therefore, we used PE to pre-constrict vessels in the experiments
to test the vasorelaxant response.
 | Table IVasoconstrictor responses in
PG-treated and -untreated aortic rings. |
Table I
Vasoconstrictor responses in
PG-treated and -untreated aortic rings.
|
LogEC50 | Emax
(%KCl) | |
|---|
|
|
| |
|---|
| Untreated | PG | Untreated | PG | n |
|---|
| U46619 | −6.59±0.06 | −6.42±0.11 | 192.0±5.94 | 155.2±8.61a | 6 |
| PE | −6.91±0.10 | −7.23±0.07a | 143.9±4.45 | 116.9±2.31a | 7 |
To determine the effect of PG on
endothelial-dependent vasorelaxation, mouse aortas were
preincubated with or without PG and were preconstricted with PE
(10−6 M). A dose-response curve to the
endothelial-dependent vasodilator Ach was then constructed. Ach
resulted in significant dose-dependent relaxation in mouse aortas
preincubated with PG. The Emax values were 69.5±1.6 vs.
80.3±4.0%, and the EC50 was −6.66±0.05 vs. −7.26±0.14 M
[log (Ach)] for untreated and PG-incubated aortas, respectively
(n=8, P<0.05) (Fig. 1C and
Table II). Interestingly, the
enhanced relaxation response to Ach by PG incubation was completely
prevented by treatment with L-NAME, a general NOS inhibitor.
| Table IIVasodilator responses in PG-treated
and untreated aortic rings. |
Table II
Vasodilator responses in PG-treated
and untreated aortic rings.
|
LogEC50 | Emax
(%KCl) | |
|---|
|
|
| |
|---|
| Untreated | PG | Untreated | PG | n |
|---|
| Ach | −6.66±0.05 | −7.26±0.14a | 69.5±1.61 | 80.3±4.02a | 8 |
| SNP | −8.19±0.07 | −8.64±0.57 | 109.0±1.62 | 112.5±10.7 | 5 |
The percentage of relaxation to SNP was similar
between the untreated and PG-treated mice (Fig. 1D). Emax values for
untreated and PG-treated groups were 109.0±1.6 and 112.5±10.7%,
respectively. EC50 values for untreated and PG-treated
groups were −8.19±0.07 and −8.64±0.57 M [log (SNP)], respectively.
Preincubation with PG and the presence of L-NAME as an eNOS
inhibitor did not alter vascular responses to SNP in any groups
(data not shown).
PG inhibits arginase activity in mice fed
an HCD and is associated with increased NO generation and decreased
ROS production
We analyzed the lipid profiles from sera (n=10)
isolated from WT mice fed an ND and ApoE−/− mice fed an
HCD. The total cholesterol (100±2.3 vs. 989.2±12.9 mg/dl,
P<0.01), LDL content (3.0±1.2 vs. 572.7±0.9 mg/dl, P<0.01),
and the triglyceride level (31.0±1.9 vs. 86.0±3.8 mg/dl, P<0.01)
of the ApoE−/− mice fed an HCD were all significantly
higher than levels of the WT mice fed an ND. HDL concentration was
also significantly higher in ApoE−/− mice fed an HCD
(91.9±2.8 vs. 688.6±9.6 mg/dl, P<0.01).
We wished to determine the effect of PG on arginase
activity in aortic vessels of ApoE−/− mice fed an HCD.
The HCD induced an increase in arginase activity, which was
dominantly presented in the endothelium (ApoE−/− + HCD
vs. WT + ND, 119±1 vs. 100±1%, P<0.01; ApoE−/− + HCD
vs. ApoE−/− + HCD + Endoth, 96±4 vs. 119±1%, P<0.01)
(Fig. 2A). Preincubation of
aortic rings with PG significantly decreased arginase activity in
ApoE−/− mice fed an HCD (ApoE−/− + HCD +
Endoth + PG vs. ApoE−/− + HCD + Endoth, 92±2 vs. 119±1%,
P<0.01) and in WT fed an ND. We next tested whether PG-dependent
arginase inhibition resulted in increased NO production using an
NO-sensitive fluorescence dye, DAF-FM
[3-amino-4-(N-methylamino)-2′,7′-difluorofluorescein] diacetate,
over the indicated time intervals. HCD induced a decrease in the
slope of DAF fluorescence (Fig.
2B) (ApoE−/− + HCD vs. WT + ND, 0.13±0.02 vs.
0.32±0.05 change in DAF fluorescence/sec, P<0.01). This was
improved by incubation with PG (ApoE−/− + HCD + PG vs.
ApoE−/− + HCD, 0.44±0.11 vs. 0.13±0.02 change in DAF
fluorescence/sec, P<0.01). On the other hand, incubation with
NG-nitro-L-arginine methyl ester (L-NAME) acutely
decreased the slope of DAF fluorescence (0.06±0.01 change in DAF
fluorescence/sec).
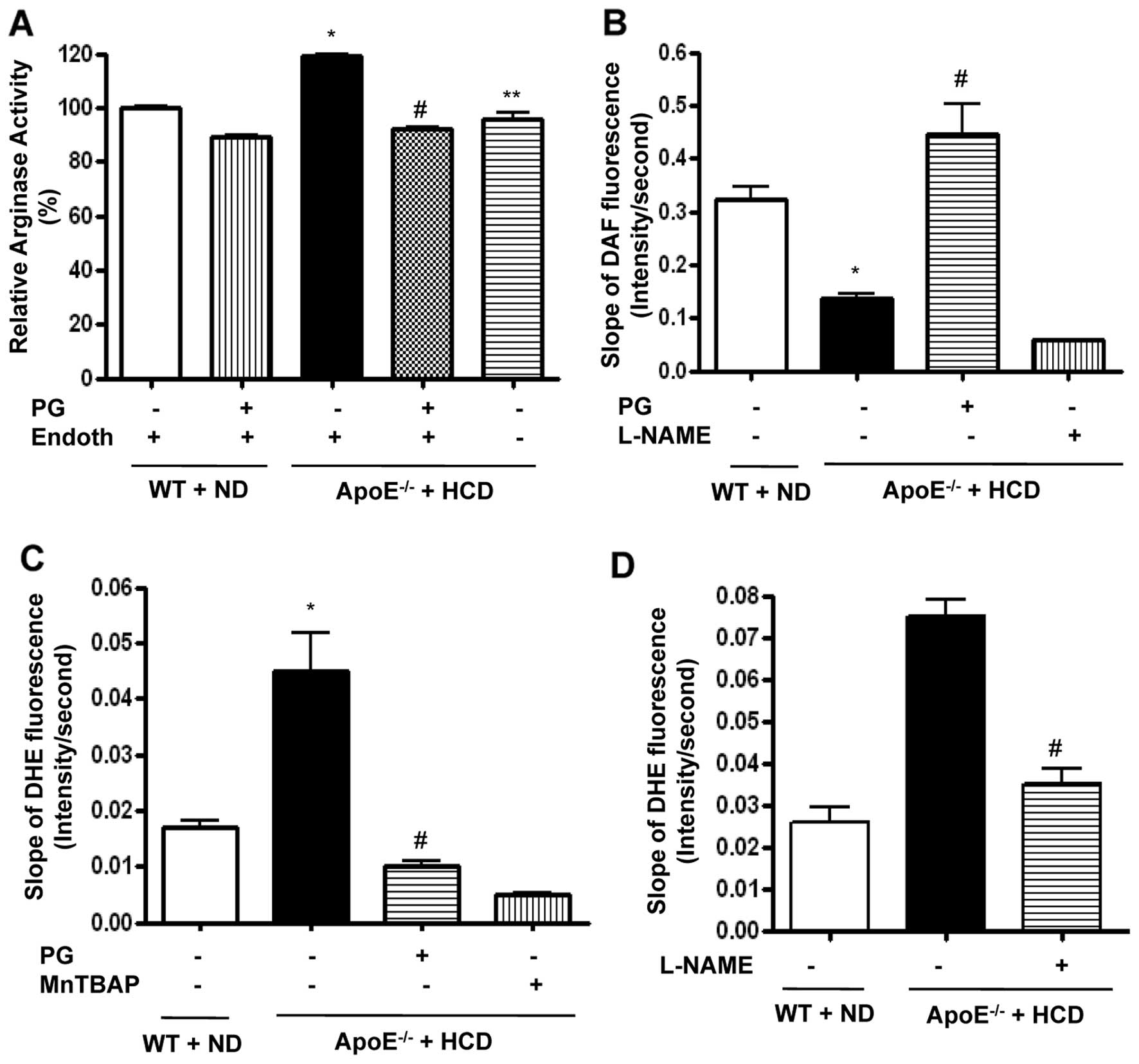 | Figure 2PG-dependent arginase inhibition
improves endothelial NO production in ApoE−/− mice fed
an HCD. Arginase activity was significantly increased in the
endothelium of vessels from ApoE−/− mice fed an HCD for
six weeks. This was blocked following incubation with PG (50 μg/ml,
*P<0.01, #P<0.01,
**P<0.01, n=4). (B) Vascular NO was measured in mouse
aortas (en face, endothelial side up) by monitoring the
change in the slope of DAF fluorescence (*P<0.01,
#P<0.01). L-NAME (10 μmol/l) was used as a control,
n=6. (C) ROS production was measured in mouse aortas using
O2•−-specific fluorescent dye (DHE), and the slope of
DHE fluorescence over time was determined (*P<0.01,
#P<0.01). MnTBAP (1 μmol/l) was used as a negative
control, n=6. (D) Isolated vessels were treated with L-NAME (10
μmol/l), and DHE signals were measured (#P<0.01). |
To determine whether increased NO production by
arginase inhibition contributes to ROS reduction, we measured
O2•− generation using the O2•−-sensitive dye,
dihydroethidium (DHE), in the endothelia of WT and
ApoE−/− mice. The time-dependent intensity of DHE
fluorescence was increased in ApoE−/− mice fed an HCD
compared to WT mice fed an ND (Fig.
2C) (ApoE−/− + HCD vs. WT + ND, 0.045±0.015 vs.
0.017±0.003 change in DHE fluorescence/sec, P<0.01).
Preincubation with PG reduced the slope of DHE fluorescence in
ApoE−/− fed an HCD (ApoE−/− + HCD + PG vs.
ApoE−/− + HCD, 0.01±0.002 vs. 0.045±0.015 change in DHE
fluorescence/sec, P<0.01). MnTBAP, a ROS scavenger, completely
quenched DHE signal. We next measured ROS production in the
presence of NOS inhibitor, L-NAME. Interestingly, L-NAME prevented
ROS production in ApoE−/− mice fed an HCD (Fig. 2D) (ApoE−/− + HCD +
L-NAME vs. ApoE−/− + HCD, 0.035±0.01 vs. 0.075±0.01
change in DHE fluorescence/sec, P<0.01).
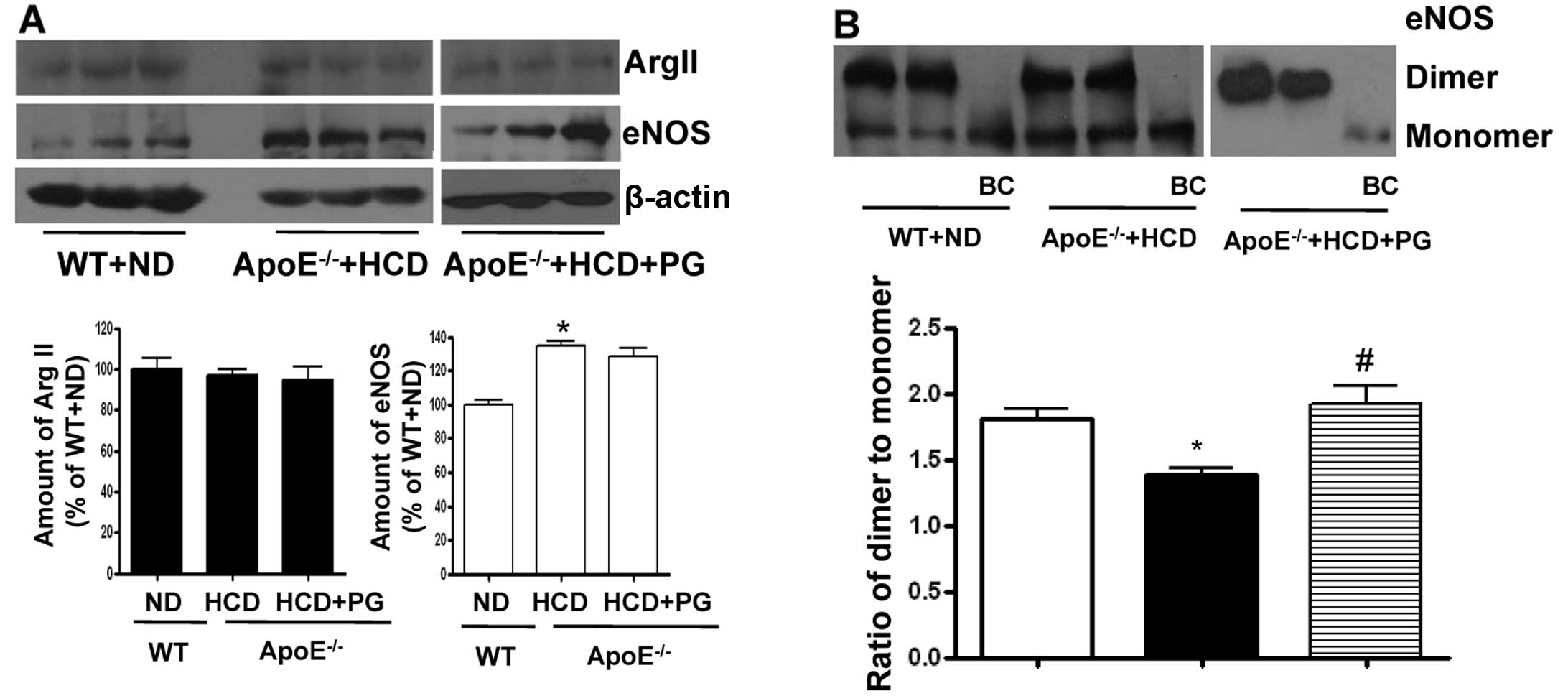
PG enhances the stability of the eNOS
dimer in ApoE−/− mice fed an HCD without affecting
protein expression levels
To understand the underlying mechanism of how PG
increases NO production and reduces ROS generation in aorta from
ApoE−/− mice fed an HCD, we performed a western blot
analysis of arginase II and eNOS in the endothelium. The expression
level of arginase II was not significantly different between
untreated- and PG-treated aortas isolated from ApoE−/−
mice fed an HCD (ApoE−/− + HCD vs. ApoE−/− +
HCD+PG, 96.9±5.1 vs. 94.8±10.0%, ns) compared to that of WT fed an
ND (100±9.1%) (Fig. 3A). However,
eNOS protein levels were significantly increased in
ApoE−/− mice fed an HCD (ApoE−/− + HCD vs. WT
+ ND, 134.2±5.9 vs. 100±4.7%, P<0.01). PG incubation had no
effect on the protein levels of eNOS (ApoE−/− + HCD vs.
ApoE−/− + HCD + PG, 134.2±5.9 vs. 128.5±8.3%, ns). On
the other hand, the ratio of eNOS dimer/monomer was significantly
decreased in the aortas of ApoE−/− mice fed an HCD from
1.83±0.27 to 1.18±0.08 (P<0.01). When incubated with PG, this
value in ApoE−/− mouse aortas was restored to 1.92±0.25
(Fig. 3B) (P<0.01).
Effect of PG on aortic vascular
reactivity in ApoE−/− mice fed an HCD
PG-dependent arginase inhibition enhanced vascular
function in WT mice fed an ND (Fig.
1 and Table I). This also
reciprocally increased NO production and decreased ROS generation
by enhancing the stability of the eNOS dimer in ApoE−/−
mice fed an HCD. Therefore, we tested whether PG restores impaired
vascular function in ApoE−/− mice fed an HCD.
The vasoconstriction response induced by a 60 mM
K+ solution was significantly higher in thoracic aortas
isolated from ApoE−/− mice fed an HCD compared to those
isolated from WT mice fed an ND (Fig.
4A) (ApoE−/− + HCD vs. WT + ND, 150.2±9.6 vs.
100±11.4%, P<0.01). PG incubation had no effect on
K+-induced vasoconstriction (ApoE−/− + HCD
vs. ApoE−/− + HCD + PG, 150.2±9.6 vs. 147.3±13.4%, ns).
The vasoconstriction response to PE was markedly reduced in
ApoE−/− mice fed an HCD in a dose-response manner. As
presented in Fig. 4B, the
Emax value in aortas from ApoE−/− mice fed an
HCD compared with WT mice fed an ND was significantly decreased (WT
+ ND vs. ApoE−/− + HCD, 150.5±4.16 vs. 119.9±3.15%,
P<0.01). LogEC50 values were also significantly
decreased (WT + ND vs. ApoE−/− + HCD, −8.2±0.07 vs.
−7.9±0.06 M [log (PE)], P<0.01). Interestingly, preincubation of
vessels from ApoE−/− mice fed an HCD with PG resulted in
a marked decrease in the maximal vasoconstrictor response to PE
(ApoE−/− + HCD vs. ApoE−/− + HCD + PG,
119.9±3.15 vs. 79.6±5.2%, P<0.01).
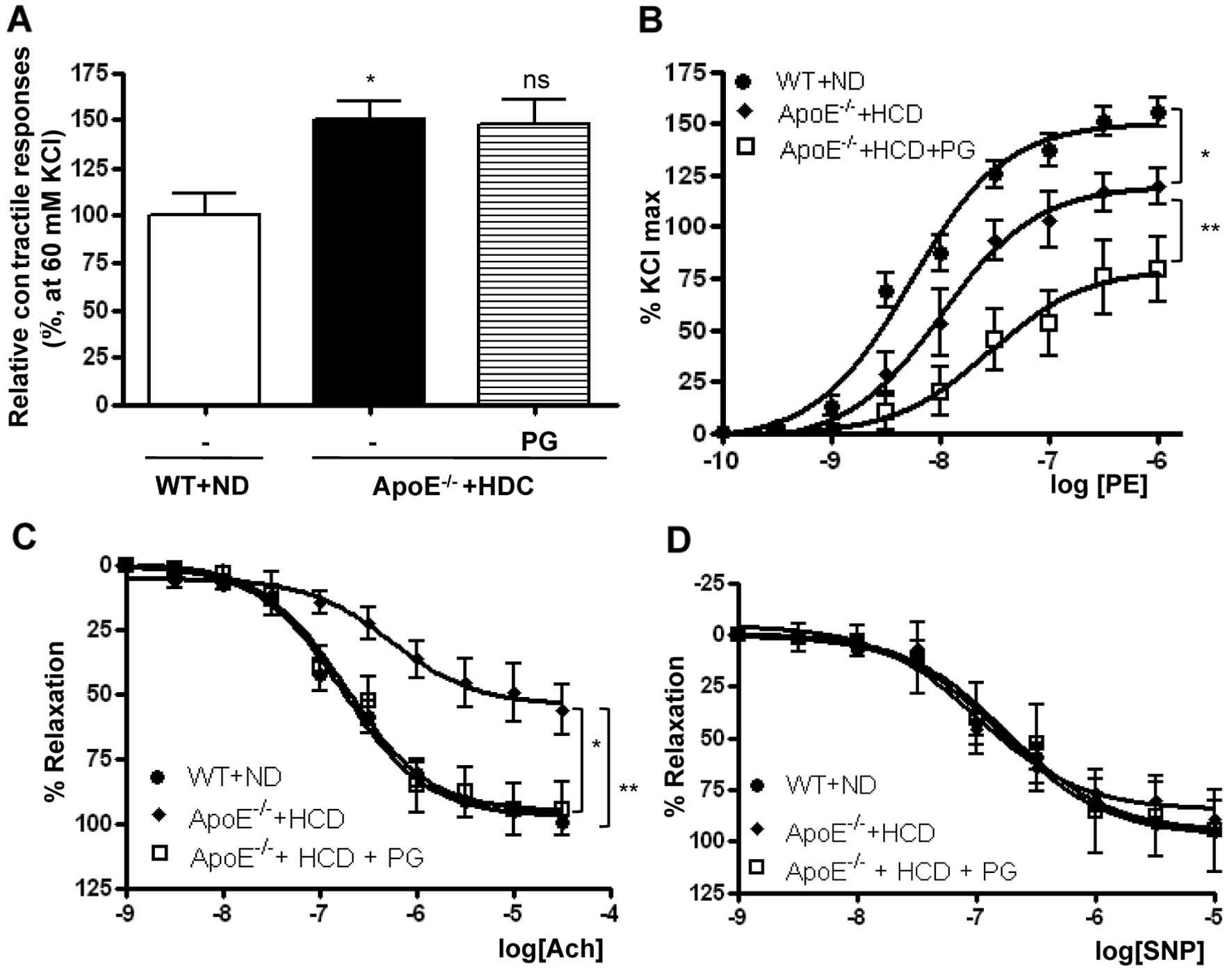 | Figure 4Preincuabtion with PG improves
endothelial dysfunction in aortas from ApoE−/− mice fed
an HCD. (A) Maximal tension to a K+ solution (60 mM KCl)
in ApoE−/− mice + HCD was significantly higher compared
with that in WT mice fed an ND (*P<0.01, n=8). (B)
The maximal vascular tension (Emax) and the
EC50 response to PE (10−9–10−4
mol/l) were significantly reduced in aortas of ApoE−/−
mice fed an HCD compared with WT mice fed an ND, which was
attenuated by preincubation with PG (50 μg/ml,
*P<0.01, **P<0.01, n=8). (C) Vessels
were preconstricted to 50–75% of the contractile Emax
with PE (10−6 mol/l), and a cumulative dose-response to
Ach was performed. The vasorelaxant Emax to Ach was
markedly attenuated in rings isolated from ApoE−/− mice
fed an HCD compared with WT mice fed an ND, whereas PG
preincubation with aortas from ApoE−/− mice fed an HCD
enhanced the vasorelaxant response (*P<0.01,
**P<0.01, n=8). (D) In contrast, the efficacy of
vasorelaxant responses to SNP was not changed in all rings. |
To determine the effect of PG on the
endothelium-dependent vasorelaxation in ApoE−/− mice fed
an HCD, mouse aortas were preconstricted with PE (10−6
M), and dose-response curves were constructed to the
endothelium-dependent vasorelaxant, Ach, and the
endothelium-independent NO donor, SNP. Ach resulted in significant
dose-dependent relaxation in mouse aortas. The vasorelaxant
responses in aortic rings from ApoE−/− mice fed an HCD
were significantly attenuated compared with those from WT mice fed
an ND. The Emax was 54.3±4.5 vs. 97.1±2.1% (Fig. 4C) (ApoE−/− + HCD vs. WT
+ ND, P<0.01). The reduced response of the aortic rings from
HCD-fed mice was markedly improved by incubation with PG
(ApoE−/− + HCD vs. ApoE−/− + HCD + PG,
54.3±4.5 vs. 95.3±4.5%, P<0.01). However, the logEC50
values were not significantly changed (WT + ND, −6.77±0.06;
ApoE−/− + HCD, −6.94±0.12; ApoE−/− + HCD +
PG, −6.74±0.13 M [log (Ach)]). SNP induced a maximal relaxant
response in aortas from the HCD-fed mice that was similar to
responses in the aortas from the ND-fed WT mice and from aortas
from the HCD-fed mice following PG incubation (Fig. 4D).
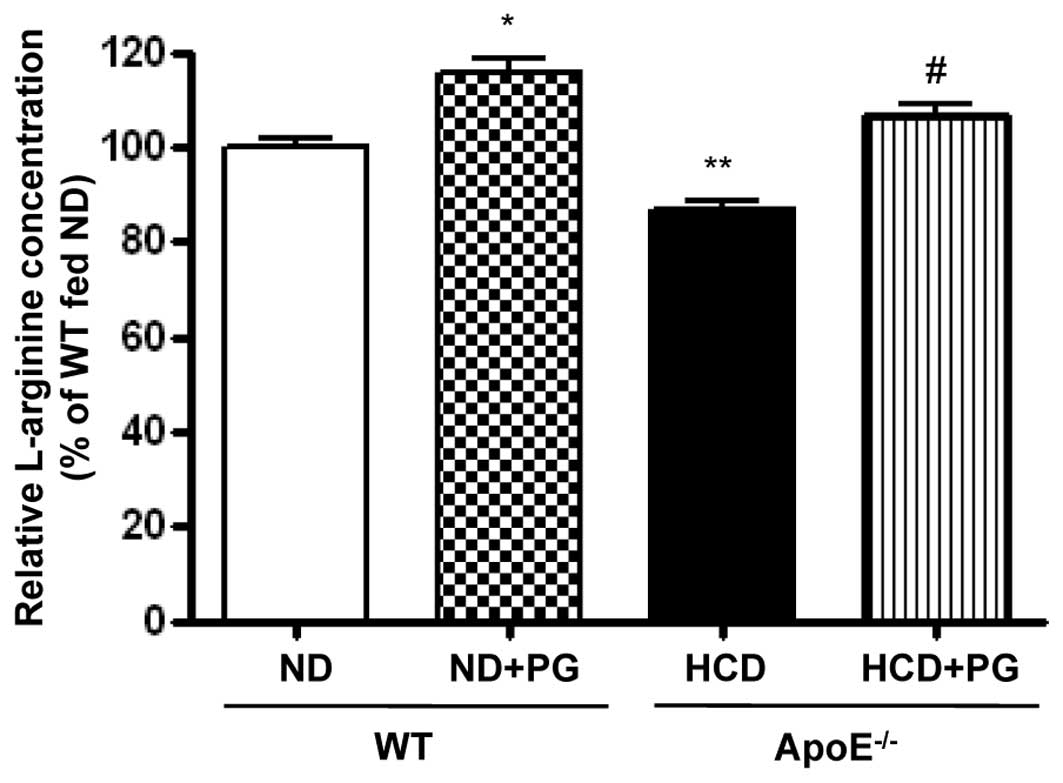
PG incubation restores intracellular
L-arginine content
Given that upregulation of arginase activity
regulates NOS activity by limiting L-arginine bioavailability, we
measured the intracellular L-arginine concentration by OPA
derivatization. Arginase inhibition by PG resulted in an increase
in L-arginine levels in WT aorta (WT + ND + PG vs. WT + ND,
115.5±6.5 vs. 100±3.8%, P<0.05) (Fig. 5). On the other hand, L-arginine
content was significantly decreased in the aortas of
ApoE−/− mice fed an HCD (ApoE−/− + HCD vs. WT
+ ND, 86.7±4.0 vs. 100±3.8%, P<0.01). This decreased content was
attenuated by incubation with PG (ApoE−/− + HCD + PG vs.
ApoE−/− + HCD, 106.4±4.6 vs. 86.7±4.0%, P<0.01).
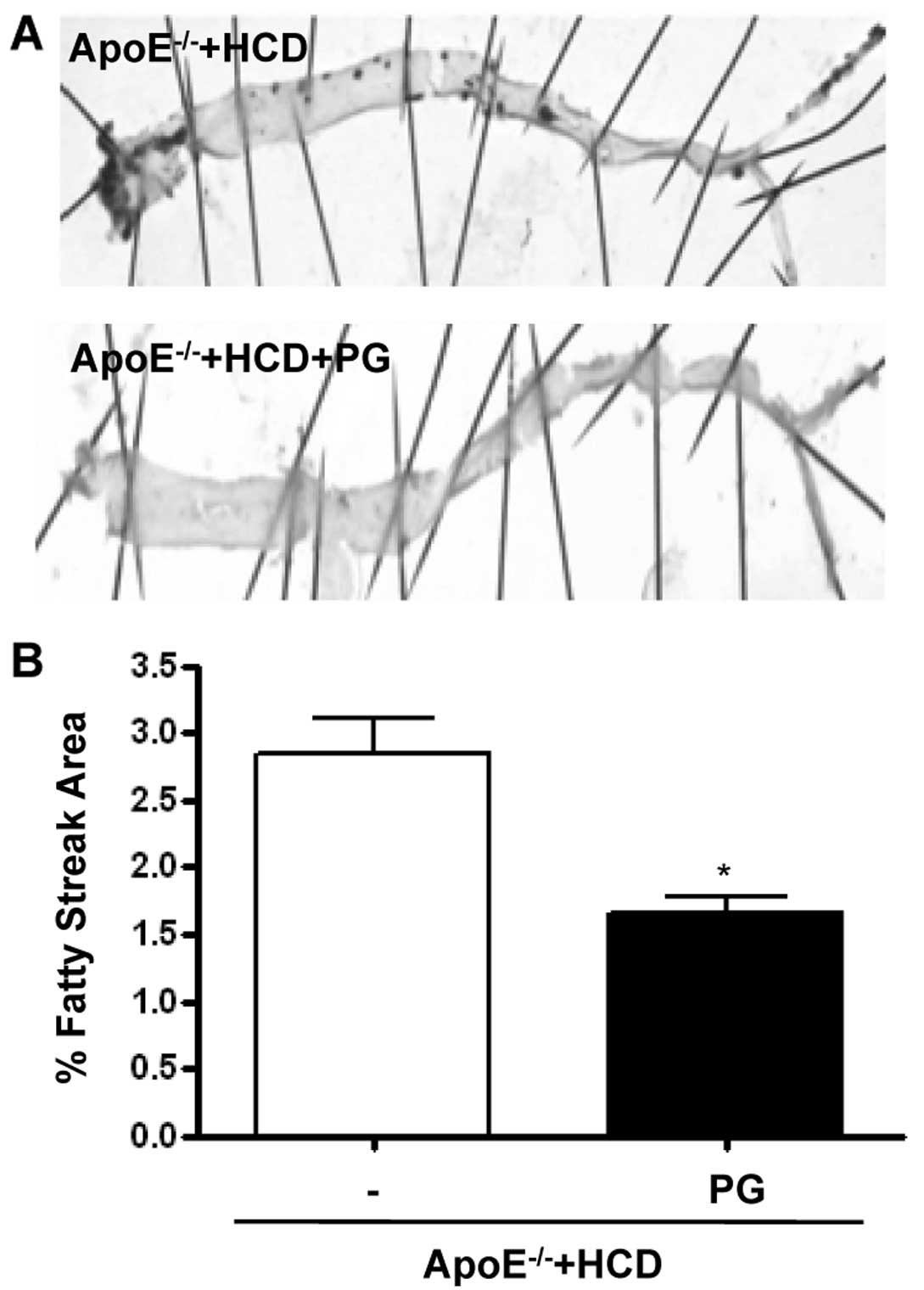
PG decreases fatty streak formation in
ApoE−/− mice fed an HCD
PG reduced the area of the aorta with fatty streaks
(Fig. 6A), as quantified by
staining with the lipid dye Oil Red O (Fig. 6B) (ApoE−/− + HCD + PG
vs. ApoE−/− + HCD, 2.8±0.4 vs. 1.6±0.2%, P<0.01).
Discussion
We previously established that PG is an active
component of rhubarb that inhibits arginase I and II activity and
reciprocally regulates NO/ROS generation by enhancing the stability
of the eNOS dimer. Here, we demonstrated that in aortic rings
isolated from WT mice fed an ND, PG attenuated the response to
vasoconstrictors U46619 and PE and enhanced the response to the
vasorelaxant Ach in an endothelium-dependent manner. PG did not
alter the response to the NO donor, SNP. Furthermore, in
atherogenic model mice (ApoE−/−) fed an HCD, arginase
activity was increased, NO production was decreased, ROS production
was increased. All of these were significantly reversed by
treatment with PG. Furthermore, PG treatment enhanced eNOS coupling
by increasing L-arginine bioavailability and reduced fatty streak
formation. PG treatment attenuated the impaired vascular responses
to both, PE and Ach, in ApoE−/− mice fed an HCD.
Arginase negatively regulates eNOS-dependent NO
production in endothelial cells. Thus, increased arginase activity
has been shown to contribute to reduced NO bioavailability in
several pathologies including vascular dysfunction (10,12,13), asthma (14), erectile dysfunction (15), aging (12), and atherosclerosis (3,16).
oxLDL, a prime atherogenic agent, increases arginase activity by
two distinct mechanisms: transcriptional upregulation and a
posttranslational mechanism that dissociates the enzyme from the
microtubule, resulting in activation (10). This oxLDL-dependent effect on
arginase activity is one possible mechanism by which high
cholesterol may lead to endothelial dysfunction dysregulating NO
production.
NO has multiple vasoprotective characteristics
(1). NO-based therapeutics are
under investigation and include dietary L-arginine (17–19), drug-eluting stents (20), inhalational NO gas (21,22), and NOS gene therapy (23,24). Here we investigated the potential
of PG, an inhibitor of arginase, as a novel NO-based therapeutic.
PG improved HCD-induced endothelial dysfunction by attenuating the
vasoconstriction response to PE and U46619, and it also augmented
the vasorelaxation response to Ach by increasing NO
bioavailability.
In aortic rings isolated from ApoE−/−
mice fed an HCD, vasoconstriction induced by a high K+
solution was increased when compared to those isolated from WT mice
fed an ND. This increase observed in ApoE−/− mouse
tissues may be due to changes in the properties of the smooth
muscle itself, rather than from injury to the endothelial cells.
Alterations in smooth muscle cell function would be expected to
occur when considering the morphological and biochemical changes
observed in vascular tissues during cholesterol-induced
atherogenesis; i.e., an increase in foam cells or in cell
proliferation or a decrease in Na+/K+-ATPase
activity (25).
The expression of inducible nitric oxide synthase
(iNOS) has also been reported in atherosclerotic lesions (26). Therefore, increased iNOS
expression and/or activity may be an additional possible mechanism
to explain the decreased contraction in the aortic rings of these
mice in response to PE. This enhanced iNOS activity does not
require an increase in cytosolic Ca2+, and this may help
explain how the observed decreased contraction was
Ca2+-independent. Indeed, elevated cGMP content was
found in atherosclerotic aortas from atherogenic rabbits (27). This observation indicates that an
absence of NO or relative deficiency of NO resulted in compensatory
upregulation of a downstream pathway.
Paradoxically, eNOS expression/abundance is actually
increased in most animal models of atherosclerosis (28). This is consistent with
observations in eNOS-deficient and eNOS-overexpressing mice in
which an HCD resulted in decreased and increased measures of
atherosclerosis, respectively (29,30). Interestingly, NO production in
ApoE−/− mice fed an HCD was decreased despite an
increase in eNOS expression, suggesting that coupling rather than
protein abundance is critical. Furthermore, eNOS inhibition with
L-NAME in ApoE−/− mice fed an HCD significantly
decreased ROS production, which suggests that uncoupled eNOS is an
important ROS-producing enzyme in atherogenesis (Fig. 2D). Several mechanisms could
explain eNOS uncoupling under pathophysiological conditions,
including: i) substrate (L-arginine) depletion; ii) cofactor (BH4)
depletion; iii) loss of dimerization; and iv) altered eNOS
phosphorylation. These are interrelated and depend on the spatial
confinement of NO signaling and the nitroso-redox milieu. NOS
uncoupling in the setting of cofactor (BH4) or substrate
(L-arginine) limitations could be amplified by an overabundance of
the enzyme itself. This is consistent with our results (Figs. 2 and 3). Our data suggest that the
upregulation of arginase results in NOS uncoupling, and arginase
inhibition results in recoupling (Fig. 3B), with restoration of the
nitroso-redox balance of endothelium function (Fig. 4).
In fact, we found that the L-arginine concentration
was 66.6 μmol/mg protein in WT mice fed an ND and 57.7 μmol/mg
protein in ApoE−/− mice fed an HCD. Previous studies
(31–33) have demonstrated that endothelial
cells contain two pools of L-arginine: i) pool I, regulated by the
cationic transporter and can be depleted by cationic amino acid
L-lysine, ii) pool II (pool IIA and IIB), accessible to eNOS but is
not freely exchangeable with extracellular L-lysine (or
L-arginine). Arg II specifically in mitochondria utilizes pool IIB.
Pool IIB may be influenced by arginase and thus modulates the local
concentration of L-arginine available to eNOS. Although our study
did not distinguish the L-arginine pools, we demonstrated that
arginase activity is involved in the regulation of the
intracellular L-arginine concentration (Fig. 5). The relationship between
arginase activity and L-arginine concentration was also shown in
the plasma of mice (32).
Atherosclerosis is defined as a chronic inflammatory
disease that is the result of activation and inhibition of multiple
complex interacting mechanisms. Overall atherosclerotic process and
fatty streak formation are inhibited by NO and enhanced by ROS.
Despite advanced fatty streak development in the ApoE−/−
mice, arginase inhibition with PG, thereby increasing NO
bioavailability and decreasing ROS production, significantly
decreased fatty streak formation (Fig. 6).
In summary, we present the novel molecule PG that
enhanced vascular function in WT mice, and improved impaired
vascular function and reduced fatty streak formation in an
atherosclerotic mouse (ApoE−/−) model fed an HCD. PG
inhibited arginase activity and reciprocally increased NO
production through enhanced stability of the eNOS dimer in aortic
rings isolated from ApoE−/− mice fed an HCD. These
insights suggest PG as the basis for development of safe and
effective preventative therapies for atherosclerotic disease.
Acknowledgements
This study was supported by the Basic Science
Research Program of the National Research Foundation of Korea
(NRF), funded by the Ministry of Education, Science and Technology
(2012-046921 and 2012-0006812).
References
|
1
|
Moncada S and Higgs A: The
L-arginine-nitric oxide pathway. N Engl J Med. 329:2002–2012. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ryoo S, Lemmon CA, Soucy KG, et al:
Oxidized low-density lipoprotein-dependent endothelial arginase II
activation contributes to impaired nitric oxide signaling. Circ
Res. 99:951–960. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ryoo S, Gupta G, Benjo A, et al:
Endothelial arginase II: a novel target for the treatment of
atherosclerosis. Circ Res. 102:923–932. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Matsuda H, Morikawa T, Toguchida I, Park
JY, Harima S and Yoshikawa M: Antioxidant constituents from
rhubarb: structural requirements of stilbenes for the activity and
structures of two new anthraquinone glucosides. Bioorg Med Chem.
9:41–50. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Choi SZ, Lee SO, Jang KU, et al:
Antidiabetic stilbene and anthraquinone derivatives from Rheum
undulatum. Arch Pharm Res. 28:1027–1030. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moon MK, Kang DG, Lee JK, Kim JS and Lee
HS: Vasodilatory and anti-inflammatory effects of the aqueous
extract of rhubarb via a NO-cGMP pathway. Life Sci. 78:1550–1557.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ngoc TM, Minh PT, Hung TM, et al:
Lipoxygenase inhibitory constituents from rhubarb. Arch Pharm Res.
31:598–605. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Choi KH, Kim JE, Song NR, et al:
Phosphoinositide-3-kinase is a novel target of piceatannol for
inhibiting PDGF-BB-induced proliferation and migration in human
aortic smooth muscle cells. Cardiovasc Res. 85:836–844. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Woo A, Min B and Ryoo S:
Piceatannol-3′-O-beta-D-glucopyranoside as an active component of
rhubarb activates endothelial nitric oxide synthase through
inhibition of arginase activity. Exp Mol Med. 42:524–532. 2010.
|
|
10
|
White AR, Ryoo S, Li D, et al: Knockdown
of arginase I restores NO signaling in the vasculature of old rats.
Hypertension. 47:245–251. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Boger RH, Bode-Boger SM, Mugge A, et al:
Supplementation of hypercholesterolaemic rabbits with L-arginine
reduces the vascular release of superoxide anions and restores NO
production. Atherosclerosis. 117:273–284. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Berkowitz DE, White R, Li D, et al:
Arginase reciprocally regulates nitric oxide synthase activity and
contributes to endothelial dysfunction in aging blood vessels.
Circulation. 108:2000–2006. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Santhanam L, Lim HK, Miriel V, et al:
Inducible NO synthase dependent S-nitrosylation and activation of
arginase1 contribute to age-related endothelial dysfunction. Circ
Res. 101:692–702. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Maarsingh H, Zaagsma J and Meurs H:
Arginase: a key enzyme in the pathophysiology of allergic asthma
opening novel therapeutic perspectives. Br J Pharmacol.
158:652–664. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Christianson DW: Arginase: structure,
mechanism, and physiological role in male and female sexual
arousal. Acc Chem Res. 38:191–201. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang Z and Ming XF: Endothelial arginase:
a new target in atherosclerosis. Curr Hypertens Rep. 8:54–59. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wilson AM, Harada R, Nair N,
Balasubramanian N and Cooke JP: L-arginine supplementation in
peripheral arterial disease: no benefit and possible harm.
Circulation. 116:188–195. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Walker HA, McGing E, Fisher I, et al:
Endothelium-dependent vasodilation is independent of the plasma
L-arginine/ADMA ratio in men with stable angina: lack of effect of
oral L-arginine on endothelial function, oxidative stress and
exercise performance. J Am Coll Cardiol. 38:499–505. 2001.
View Article : Google Scholar
|
|
19
|
Blum A, Hathaway L, Mincemoyer R, et al:
Oral L-arginine in patients with coronary artery disease on medical
management. Circulation. 101:2160–2164. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ansel GM and Lumsden AB: Evolving
modalities for femoropopliteal interventions. J Endovasc Ther.
16(Suppl 2): II82–II97. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ichinose F, Roberts JD Jr and Zapol WM:
Inhaled nitric oxide: a selective pulmonary vasodilator: current
uses and therapeutic potential. Circulation. 109:3106–3111. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Griffiths MJ and Evans TW: Inhaled nitric
oxide therapy in adults. N Engl J Med. 353:2683–2695. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barbato JE, Kibbe MR and Tzeng E: The
emerging role of gene therapy in the treatment of cardiovascular
diseases. Crit Rev Clin Lab Sci. 40:499–545. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kibbe MR and Tzeng E: Nitric oxide
synthase gene therapy in vascular pathology. Semin Perinatol.
24:51–54. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ibengwe JK and Suzuki H: Changes in
mechanical responses of vascular smooth muscles to acetylcholine,
noradrenaline and high-potassium solution in hypercholesterolemic
rabbits. Br J Pharmacol. 87:395–402. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Arthur JF, Yin ZL, Young HM and Dusting
GJ: Induction of nitric oxide synthase in the neointima induced by
a periarterial collar in rabbits. Arterioscler Thromb Vasc Biol.
17:737–740. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rupin A, Behr D and Verbeuren TJ:
Increased activity of guanylate cyclase in the atherosclerotic
rabbit aorta: role of non-endothelial nitric oxide synthases. Br J
Pharmacol. 119:1233–1238. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kawashima S: The two faces of endothelial
nitric oxide synthase in the pathophysiology of atherosclerosis.
Endothelium. 11:99–107. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ozaki M, Kawashima S, Yamashita T, et al:
Overexpression of endothelial nitric oxide synthase accelerates
atherosclerotic lesion formation in apoE-deficient mice. J Clin
Invest. 110:331–340. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shi W, Wang X, Shih DM, Laubach VE, Navab
M and Lusis AJ: Paradoxical reduction of fatty streak formation in
mice lacking endothelial nitric oxide synthase. Circulation.
105:2078–2082. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Closs EI, Scheld JS, Sharafi M and
Forstermann U: Substrate supply for nitric-oxide synthase in
macrophages and endothelial cells: role of cationic amino acid
transporters. Mol Pharmacol. 57:68–74. 2000.PubMed/NCBI
|
|
32
|
Simon A, Plies L, Habermeier A, Martine U,
Reining M and Closs EI: Role of neutral amino acid transport and
protein breakdown for substrate supply of nitric oxide synthase in
human endothelial cells. Circ Res. 93:813–820. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Erdely A, Kepka-Lenhart D, Salmen-Muniz R,
et al: Arginase activities and global arginine bioavailability in
wild-type and ApoE-deficient mice: responses to high fat and high
cholesterol diets. PLoS One. 5:e152532010. View Article : Google Scholar : PubMed/NCBI
|