Introduction
β-hemoglobin disorders, including β-thalassemia and
sickle cell anemia (SCA), are the most prevalent monogenic
inherited disorders that result from the deficient or altered
synthesis of the β-globin chain of human hemoglobin (1). β-thalassemia is characterized by
diminished or absent production of β-globin chains which causes a
relative excess of α-globin chains. The imbalance in the production
of non-α/α-globin chains is the main factor responsible for the
pathophysiology of β-thalassemia (2). In SCA, the substitution of glutamate
for valine at position 6 of the β-globin chain leads to the
synthesis of abnormal hemoglobin (HbS), which forms polymers that
cause vascular occlusion under specific physiological conditions
(3).
The only potential cures for these disorders are
bone marrow transplantation, which is useful for only a minority of
patients (4), and gene or
cellular therapies, which have faced numerous challenges (5). Therefore, these treatments have
significant limitations for widespread use, particularly in
developing countries where affected patients are often unable to
sustain the high costs of clinical management. In addition, a
number of issues need to be resolved before gene therapy can be
introduced into clinical trials, including the efficiency of
transduction, transgene expression levels and safety concerns
(6). The development of
pharmacological therapeutics for β-hemoglobin disorders, including
the possible use of fetal hemoglobin (HbF) inducers, is considered
crucial (7). It has been known
for some time that increasing HbF (α2γ2) expression is clinically
beneficial for patients with β-thalassemia or SCA. The
pharmacological reactivation of γ-globin gene expression, which is
silenced in adults, can decrease the imbalance of non-α-/α-globin
chains in β-thalassemia, and elevating the HbF level can prevent
HbS polymerization in SCA, substantially ameliorating the clinical
symptoms of patients with these disorders (8). Therefore, a considerable research
effort is underway to investigate the therapeutic approaches that
involve the pharmacological induction of HbF expression (9). This effort has led to the
development of several agents, including 5-azacytidine,
hydroxyurea, butyrate (and its analogues) and histone deacetylase
(HDAC) inhibitors, as lead compounds capable of inducing γ-globin
expression and increasing HbF levels (10–15). However, a number of factors limit
the usefulness of these compounds. Butyrate and its analogues have
short half-lives and may also cause hematopoietic suppression
(16). Hydroxyurea, a drug
commonly used for the treatment of SCA, induces a relatively weak
expression of the γ-globin gene and is not effective in the
majority of patients with β-thalassemia (17). When used long-term, 5-azacytidine
and other DNA methyltransferase inhibitors may increase the risk of
cancer, due to the non-specific modification of DNA (18). Therefore, there is an urgent need
to identify novel agents that can induce HbF expression with higher
efficiency and less toxicity.
The successful introduction of numerous compounds
derived from traditional Chinese herbal remedies or the natural
world have encouraged scientists to seek additional novel drugs
from these ancient medicinal practices (19–22). In a previous study, we reported
that serum obtained from Sprague-Dawley rats fed with Plastrum
testudinis (PT) caused an increase in γ-globin gene expression and
HbF production in K562 cells (23). PT (or turtle shell), which is the
carapace and plastron of the turtle, is a well-known traditional
Chinese medicine that has been used in China for over two thousand
years, and has been documented in the oldest Materia Medica book,
‘Shen Nong’s Classic of Materia Medica’. According to the record in
‘Shen Nong’s Herbal Classic’, PT is a valuable and top-grade
Chinese medicine that has been used in clinical practice for the
treatment of various types of anemia, bleeding disorders and bone
diseases, as well as to nourish the body and enhance immunity
(24,25). Previous studies have demonstrated
that PT promotes the proliferation and differentiation of
mesenchymal stem cells (MSCs), protects the nervous system and
exerts antioxidant activity (26–28). Although PT has been recorded as
being used for the treatment of anemia, it is not clear whether PT
could be used for the treatment of β-thalassemia and SCA, and its
mechanisms of action are currently unknown.
The aim of this study was to determine whether PT
can induce the erythroid differentiation of K562 cells and increase
γ-globin gene expression and HbF synthesis in human erythroid
cells, particularly those from patients with β-thalassemia, as well
as to investigate its mechanisms of action. Our data demonstrate
that PT is a novel therapeutic candidate for the treatment of
β-thalassemia and SCA, which functions by activating the HbF gene
through a mechanism involving epigenetic histone modifications
within the γ-globin gene promoter via activation of the p38
mitogen-activated protein kinase (MAPK) signaling pathway.
Materials and methods
Reagents and antibodies
PT was purchased from Guangdong YiFang
Pharmaceutical Co. (Guangdong, China); sodium butyrate (NaB)
SB203580 (SB), benzidine, β-mercaptoethanol, human transferrin and
phenylmethylsulfonyl fluoride (PMSF) were from Sigma (St. Louis,
MO, USA); RPMI-1640 medium was obtained from Invitrogen (Carlsbad,
CA, USA); TRIzol Reagent from Invitrogen Corporation (Grand Island,
NY, USA); L-glutamine was from Mediatech (Manassas, VA, USA); fetal
bovine serum (FBS) was from Sijiqing (Hangzhou, China); Iscove’s
modified Dulbecco’s medium (IMDM) was purchased from HyClone
(Logan, UT, USA); cyclosporin A, recombinant human stem cell factor
(rh-SCF) and recombinant human erythropoietin (rh-EPO) were
obtained from ProSpec (Ness Ziona, Israel); 10% conditioned medium
was from the Chinese Academy of Sciences Cell Bank (Shanghai,
China); dexamethasone was from Mechem (Chengdu, China); trypan blue
was obtained from Gibco BRL (Grand Island, NY, USA); the real-time
PCR kit and First Strand cDNA Synthesis kit were from GeneCopoeia
(Rockville, MD, USA); sheep anti-HbF antibody was purchased from
Abcam (Cambridge, MA, USA); anti-p38 MAPK antibody was from Cell
Signaling Technology (Beverly, MA, USA); mouse anti-β-actin
antibody was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA); HRP-labeled rabbit anti-sheep IgG (H+L) was from KPL
(Gaithersburg, USA); HRP-labeled goat anti-mouse IgG (H+L) was from
Zhongshan Golden Bridge Biotechnology (Beijing, China); the ECL
Western Blotting Luminol Reagent kit was obtained from Merck (USA);
the EZ-ChIP Chromatin Immunoprecipitation (ChIP) kit,
anti-acetyl-histone H3 polyclonal antibody and
anti-phosphoacetyl-histone H3 antibody were from Upstate
Biotechnology (Lake Placid, NY, USA); and rabbit
anti-tetra-acetyl-histone H4 polyclonal antibody was from Active
Motif (Carlsbad, CA, USA). All other chemicals and reagents used
were of analytical grade.
For drug induction experiments, PT or NaB were
suspended as stock solutions in sterile H2O, and diluted
to the indicated concentrations in culture medium prior to
experimentation. NaB was used at a concentration of 0.5 mmol/l as a
positive control for the induction of HbF expression as previously
described (29).
K562 cell culture
The human chronic myelogenous leukemia cell line,
K562, was purchased from the Chinese Academy of Sciences Cell Bank
(Shanghai, China). K562 cells were cultured in RPMI-1640 medium
containing 2 mmol/l L-glutamine and supplemented with 10% (v/v)
heat-inactivated FBS, 100 U/mL penicillin and 100 μg/ml
streptomycin, in a humidified 5% CO2 atmosphere at 37°C,
as previously described (30).
For the experiments, K562 cells were seeded in culture flasks at a
density of 1×104 cells/ml. Cells were treated with 4
different concentrations of PT (0.5, 2.5, 5 or 10 mg/ml) for
various periods of time (24, 48, 72, 96, 120 or 144 h), and the
percentage of benzidine-positive cells was determined; 0.5 mmol/l
NaB acted as the positive control; the same volume of sterile
H2O was used in the negative control experiments.
Culture of human erythroid progenitor
cells
The experiments were approved by the Ethics
Committee of Nangfan Hospital, Guangzhou, China (no.
NFEC-201206-k2). Human erythroid progenitor cells were obtained
from the umbilical cord blood from 8 healthy donors and the
peripheral blood from 6 patients with β-thalassemia. All subjects
provided written informed consent prior to enrollment in the
study.
A two-phase liquid culture procedure was used to
culture the human erythroid progenitor cells, as previously
described (31–33). Briefly, mononuclear cells were
isolated from cord blood or peripheral blood by centrifugation
through a Ficoll-Hypaque density gradient (GE Healthcare, Uppsala,
Sweden) prior to culture. Mononuclear cells were initially cultured
in IMDM containing 10% FBS, 1 μg/ml cyclosporin A and 10%
conditioned medium, and grown in a humidified 5% CO2
incubator at 37°C. After 5–7 days in this phase 1 culture,
non-adherent cells were harvested, washed and seeded at a density
of 1×106/ml in IMDM containing 30% FBS, 1% BSA, 10
μmol/l β-mercaptoethanol, 1 μmol/l dexamethasone, 0.3
mg/ml human transferrin, 10 ng/ml rh-SCF and 1 U/ml rh-EPO; this
corresponded to phase 2 of the culture. The resulting human
progenitor cells were maintained at 37°C in a 5% CO2
incubator for 14 days and, if indicated, the compounds were added
on days 4 to 5 of phase 2; cell samples were analyzed on day 12 or
13 of phase 2.
Benzidine staining
Erythroid differentiation of K562 cells was assessed
using the benzidine/H2O2 reaction to measure
the proportion of benzidine-positive (heme-producing) cells, using
a minor modification of a previously described technique (34). K562 cells were incubated with the
indicated concentrations of PT or NaB. Following treatment for the
indicated periods of time, 0.5 ml aliquots of cell suspension were
removed, washed in PBS and added to 14 μl benzidine reaction
solution, which contained 0.4% benzidine, 12% acetic acid, 5% (w/v)
sodium nitroprusside and 1 μl 30%
H2O2. The reaction was carried out in the
dark at room temperature for 10 min, after which 500 cells were
counted under a microscope (TE2000-U, Nikon, Tokyo, Japan) and the
percentage of benzidine-positive cells (BZ%) was determined. The
experiment was repeated 3 times.
Trypan blue dye exclusion assay
The proliferation of viable cells was determined by
the trypan blue dye exclusion assay. K562 cells were grown with
inducing agents (PT or NaB) at the indicated concentrations for
periods of up to 144 h (for K562 cells). Human erythroid progenitor
cells in phase 2 culture were cultured with inducing agents from
day 6 to 14, and aliquots were removed daily or every 2 days to
determine the number of viable cells. Cell viability was assessed
by mixing aliquots of the cell suspensions with equal volumes of
0.5% trypan blue. Cells that accumulated the dye were consi dered
dead. The cell inhibition rate was calculated as: cell inhibition
rate = 1 − cell viability rate = [(Cn − C0) − (Tn − T0)]/(Cn − C0)
where C and T represent the number of cells per ml in the control
and PT group, respectively, and n and 0 represent the day on which
the count was made.
Quantitative real-time
reverse-transcription polymerase chain reaction (qRT-PCR)
Analysis of globin gene expression was performed by
qRT-PCR. Total RNA was isolated using TRIzol reagent, according to
the manufacturer’s instructions. RNA purity was determined using
absorbance at 260 and 280 nm (A260/280). cDNA synthesis was
performed using the First strand cDNA synthesis kit with
oligo(dT)18 and M-MLV reverse transcriptase, according
to the directions provided by the manufacturer. For qRT-PCR, the
reaction mixture contained: 2X All-in-One q-PCR Mix, 10 μl;
sample, 5 μl; forward primer (4 μmol/l), 2 μl;
reverse primer (4 μmol/l), 2 μl; ROX Reference dye,
0.5 μl; nuclease-free water was added to a final volume of
20 μl. The reaction parameters used were: 95°C for 10 min;
95°C for 30 sec; 60°C for 30 sec, 72°C for 1 min; this was repeated
for 40 cycles on an 8-tube ABI7500 PCR System (Applied Biosystems,
Foster City, CA, USA). The housekeeping gene, human
glyceraldehyde-3-phosphate dehydrogenase (GAPDH), was used for
qRT-PCR normalization. The relative gene expression levels were
expressed as the relative fold/percentage compared with the
corresponding control, calculated using the 2−ΔΔCt
method (35). The primers used
were designed according to NCBI sequences using Prime 5.0 software,
and were synthesized by GeneCopoeia. The primer sequences are
presented in Table I.
 | Table IPrimer sequences and sizes of the
real-time PCR products. |
Table I
Primer sequences and sizes of the
real-time PCR products.
| Gene | Primer | Sequences
(5′→3′) | Size (bp) |
|---|
| α-globin | Forward |
tccccaccaccaagacctac | 256 |
| Reverse |
ccttaacctgggcagagcc | |
| β-globin | Forward |
ctcatggcaagaaagtgctcg | 242 |
| Reverse |
aattctttgccaaagtgatggg | |
| γ-globin | Forward |
ggcaacctgtcctctgcctc | 250 |
| Reverse |
gaaatggattgccaaaacgg | |
| GAPDH | Forward |
gcaccgtcaaggctgagaac | 221 |
| Reverse |
tggtgaagacgccagtgga | |
| Gγ-globin | Forward |
gctgcatgtggatcctgagaac | 251 |
| Reverse |
tctgcatcatgggcagtgag | |
| Aγ-globin | Forward |
aagctttacacaggatcatgaagg | 249 |
| Reverse |
cagggtaggaagtatttatggtgg | |
| Necdin | Forward |
gtcctctgcctctgccatca | 217 |
| Reverse |
atacagggcactggccactc | |
Western blot analysis
K562 cells or human erythroid progenitor cells were
lysed on ice for 20 min in 50 mmol/l Tris-HCl (pH 8), 150 mmol/l
NaCl, 2% Nonidet P-40 (NP40), 0.5% sodium deoxycholate, 0.02%
sodium azide and 0.1% SDS supplemented with 10 mmol/l PMSF. This
was followed by centrifugation at 12,000 × g for 15 min at 4°C. The
protein concentration was determined using a Bradford assay. For
immunoblotting, 75 μg of total cellular proteins per lane
were separated by 15% SDS-PAGE, and then transferred onto
polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA,
USA). The membranes were subsequently blocked in 1X TBST (1X
TBS/0.1% Tween-20) containing 5% non-fat dry milk for 2 h at room
temperature. Following a brief wash in 1X TBST, the membrane was
incubated with primary antibodies (sheep anti-HbF antibody,
anti-p38 MAPK antibody or mouse anti-β-actin antibody) for 1–2 h at
room temperature, washed with 1X TBST, and then incubated with
secondary antibodies [HRP-labeled rabbit anti-sheep IgG (H+L) or
HRP-labeled goat anti-mouse IgG (H+L)] for 1–2 h at room
temperature. β-actin was used as an endogenous control to normalize
the differences in the amount of total protein in each sample.
Proteins on the membrane were detected by the electrogenerated
chemiluminescence method (ECL Western Blotting Luminol Reagent
kit), according to the manufacturer’s instructions. Band
intensities were measured using Bandscan 5.0 software in the
Bio-BEST-140E gel image system (SIM International, Newark, DE,
USA).
ChIP assay
Following induction with PT or NaB, aliquots of K562
cells or human erythroid progenitor cells were used for γ-globin
quantifications and analysis of histone modification, carried out
with a qRT-PCR-based ChIP assay (36). Briefly, protein-DNA crosslinking
was achieved by incubating 1×107 cells in 1%
formaldehyde for 10 min at 37°C. After cell lysis, the lysate was
sonicated to reduce DNA fragments to a size of 0.1 to 1.0 Kbp. DNA
was co-immunoprecipitated in duplicate with histones using either a
polyclonal antibody against diacetyl-histone H3, an antibody
against tetra-acetyl-histone H4 or an antibody against
phosphoacetyl-histone H3. IgG was analyzed along with a negative
control with no antibody. The immunoprecipitated DNA was purified
by phenol/chloroform extraction and dissolved in 50 μl
distilled water. In addition, 50 μl aliquots of the
sonicated lysate (total DNA) were directly subjected to DNA
extraction without immunoprecipitation, and were dissolved in 100
μl distilled water. Real-time PCR was used to determine the
results. The primer sequences are presented in Table I.
Statistical analysis
All statistical analyses were conducted using SPSS
software version 12.0 (SPSS Inc., Chicago, Il, USA). All
experiments in this study were independently repeated at least 3
times. Data are presented as the means ± SD. Statistical analysis
was performed by one-way ANOVA, or repeated measures analysis of
variance, where appropriate. The least significant difference post
hoc test was used for ANOVA statistics. P-values <0.05 were
considered to indicate statistically significant differences.
Results
PT promotes erythroid differentiation and
proliferation of K562 cells
To assess the effect of PT on the erythroid
differentiation and proliferation of K562 cells,
5×104/ml K562 cells were cultured in the absence or
presence of PT at concentrations of 0.5, 5 and 10 mg/l, or in the
presence of 0.5 mmol/l NaB as the positive control, for periods of
up to 144 h. Every 24 h, 0.5 ml of cell suspension was removed for
benzidine staining to determine the proportion and the absolute
number of benzidine-positive cells/ml of culture. Fig. 1 illustrates the effects of PT on
K562 cell differentiation and proliferation.
Fig. 1A presents
microphotographs taken after 3 days of K562 cell culture, in the
(a) absence or (b) presence of PT or (c) NaB, indicating
differentiated benzidine-positive cells. Fig. 1B and C illustrate the kinetic
characteristics of K562 erythroid differentiation with various
concentrations of PT, revealing that the induction of erythroid
differentiation by PT was both time-and dose-dependent. Maximal
stimulation of K562 erythroid differentiation was observed at a PT
concentration of 5 mg/l after 96 h of culture: the proportion of
benzidine-positive cells induced by PT (at 96 h) was 11.72±0.68%,
similar to the maximal effect of NaB (10.99±0.79% at 72 h). Of
note, the absolute number of benzidine-positive cells induced by PT
was 24084.3±3276.1 (at 96 h), a value significantly higher than
that for obtained NaB (8242.0±423.0, at 72 h).
Fig. 1D and E
illustrate the effect of PT on the proliferation of K562 cells.
Fig. 1D presents representative
results regarding cell growth. Fig.
1E shows that the inhibitory effects of PT on K562 cell
proliferation were dose-dependent: after 96 h of culture, the
inhibitory rates for 0.5, 5 and 10 mg/l PT were 3.9±0.23,
22.47±0.70 and 44.16±2.84%, respectively. The inhibitory rate for
PT was significantly lower than that for 0.5 mmol/l NaB
(75.87±3.51%). At a concentration similar to that used
therapeutically for the treatment of sickle cell disease (0.5
mmol/l), NaB caused a marked inhibition of cell proliferation.
These results suggest that a PT dose of 5 mg/l may
be suitable for erythroid induction without affecting cell
proliferation. In our opinion, it is very important that the
induction of differentiation is not associated with the inhibition
of cell growth. For this reason, PT has a significant advantage
over other known inducers, such as 5-azacytidine, hydroxyurea and
butyrate, which exert greater inhibitory effects on cell
proliferation.
PT selectively induces γ-globin gene
expression in human erythroid cells
In order to determine whether the induction of the
erythroid differentiation of K562 cells by PT was associated with a
selective upregulation of γ-globin gene expression, total RNA was
extracted from the K562 cells that were either untreated, or
treated with either PT (5 mg/ml) or NaB (0.5 mmol/l).
Representative results from the qRT-PCR analysis are shown in
Fig. 2A, and these indicate that
the erythroid differentiation induced by PT after 96 h of treatment
was associated with a sharp increase in γ-globin mRNA accumulation.
Relative to the untreated cells, the maximal increases in γ-globin
mRNA levels induced by PT and NaB were 8.17±1.68-fold (96 h) and
7.20±0.80-fold (72 h), respectively (P<0.01). The n-fold
increase in γ-globin mRNA accumulation was measured using the GAPDH
sequence as an internal control. Of note, no major increases in α-
or β-globin mRNA accumulation were observed (0.6±0.02-fold,
P<0.01 and 0.96±0.02-fold, P>0.05, respectively) in the
erythroid cells treated with PT, compared with the untreated cells.
These data suggest that this compound merits further study for its
possible effects on the expression of the γ-globin gene in
erythroid cells isolated from patients with β-thalassemia.
To evaluate the selective effects of PT on γ-globin
mRNA accumulation in human erythroid progenitor cells, we employed
a two-phase liquid culture system as previously described (31–33). In phase 2 of this procedure (in
the presence of EPO), the erythroid progenitor cells continue their
proliferation and mature into Hb-containing orthochromatic
normoblasts. PT (5 mg/ml) or NaB (0.5 mmol/l) were added on day 6
of phase 2. Cells were harvested on day 12 and total mRNA was
isolated. The expression of globin genes in the erythroid
progenitor cells from 6 patients with β-thalassemia were then
analyzed by qRT-PCR using GAPDH mRNA expression as an internal
control. The results shown in Fig.
2B indicate that PT caused a selective increase in γ-globin
mRNA accumulation (mean, 4.94±1.17-fold; range, 3.674- to
5.997-fold; P<0.05), with no effect on the accumulation of
α-globin mRNA (mean, 0.72±0.06-fold; range, 0.682- to 0.787-fold)
or β-globin mRNA (mean, 0.97±0.28-fold; range, 0.669- to
1.23-fold). The effects of NaB on γ-, α- and β-globin genes were
similar to those of PT; however, a moderate inhibition of cell
growth was observed in the PT-treated erythroid progenitor cells
compared to those treated with NaB.
PT enhances HbF synthesis in human
erythroid cells
First, the effect of PT on HbF induction in K562
cells was examined. K562 cells were cultured for 96 h in the
absence or presence of PT at 0.5, 2.5, 5 or 10 mg/l, and the levels
of total protein were measured. The relative concentration of HbF
was determined by western blot analysis, with β-actin used as an
internal control for quantification. Representative results are
shown in Fig. 3A, illustrating
that treatment with PT increased HbF levels compared with the
untreated control. The group data from 3 independent experiments
revealed that PT significantly increased HbF levels in a
concentration-dependent manner, from 1.17±0.04-fold at 0.5 mg/ml to
2.23±0.17-fold at 5 mg/ml, compared with the untreated control.
Thus, PT at 5 mg/l caused the optimal increase in HbF levels,
similar to the observations made in the erythroid induction
experiments (Fig. 1B and C). PT
at 5 mg/l exerted a moderate inhibitory effect.
Second, the time dependence of the HbF induction in
response to 5 mg/l PT was investigated (Fig. 3B). The K562 cells were treated for
0, 24, 48, 72, 96 or 120 h, and the relative HbF levels were
assessed by western blot analysis. As shown in Fig. 1B and C, the expression level of
HbF increased until it peaked at 96 h, and then slowly declined at
120 h. The group data from 3 independent experiments (Fig. 3B) indicated that the HbF level
significantly increased 2.95±0.09-fold over the basal levels (0 h),
and then gradually declined. These data also demonstrated that 0.5
mmol/l NaB was a powerful inducer of HbF expression: the optimal
effect of NaB was a 1.90±0.03-fold increase following 72 h of
culture (Fig. 3B).
Third, we examined the effects of PT on HbF
induction in cells derived from 6 patients with β-thalassemia. The
HbF levels induced by PT in these patients increased 1.98±0.11-fold
(ranging from 1.97±0.15 to 2.04±0.09) compared with the untreated
controls (P<0.01), and this change was significantly higher
compared to the 1.62±0.23-fold increase induced by NaB (P<0.01)
(Fig. 3C).
PT activates p38 MAPK signaling
In this series of experiments, we determined whether
PT can activate the p38 MAPK pathway. The levels of phosphorylated
p38 MAPK (p-p38) induced by PT were evaluated by western blot
analysis (37). K562 cells
treated with PT at various concentrations (0.5, 2.5, 5 or 10 mg/ml)
displayed a steady increase in p-p38 expression, as shown in
Fig. 4A. The increase in p38 MAPK
phosphorylation by PT occurred in a dose-dependent manner, with the
levels of induced p-p38 ranging from 1.14±0.15-fold (0.5 mg/ml PT)
to 5.02±0.52-fold (5 mg/ml PT) (P<0.05). Time-course studies
were then performed using the K562 cells treated with 5 mg/ml PT
for 6 to 96 h (Fig. 4B). The
levels of p-p38 were normalized to those of total p38 (t-p38). As
shown in Fig. 4B, we observed a
1.28±0.15-fold induction of p-p38 expression at 6 h (P>0.05), a
1.7±0.22-fold increase at 12 h (P<0.01), a 6.52±0.47-fold
increase at 72 h (P<0.01), and then a decline to 4.62±0.36-fold
at 96 h (P<0.01). In addition, SB203580 (SB), a p38 inhibitor,
was analyzed for its ability to block PT-activated p-p38 in K562
cells. As shown in Fig. 4C, in
the absence of SB, PT caused a 5.10±0.35-fold increase in p-p38
levels (P<0.05), consistent with our previous data. By contrast,
pre-treatment with 10 μmol/l SB for 1 h significantly
reduced the effect of the PT-mediated p-p38 induction (a
2.17±0.05-fold increase of p-p38, corresponding to an inhibitory
effect of 57.27 %; P<0.01). As a positive control, the effects
of SB were also determined on the effects of NaB, a known p38
activator. Pre-treatment with 10 μmol/l SB inhibited
NaB-mediated p-p38 induction by 71.38% (NaB-induced changes in
p-p38 levels of 5.12±0.15-fold in the absence of SB and
1.46±0.03-fold in its presence; P<0.01). These data provide
evidence that PT activates the p38 MAPK signaling pathway.
 | Figure 4Effects of Plastrum testudinis (PT)
on γ-globin gene expression and fetal hemoglobin (HbF) synthesis
via the activation of the p38 MAPK pathway, analyzed by western
blot analysis and qRT-PCR. Total proteins were isolated from K562
cells treated with the indicated concentrations of PT or NaB for
the indicated periods of time, to determine (A) the dose-dependence
(B) and time-course of p38 MAPK activation. (C) The experiment was
performed in the absence or presence of SB203580 (SB). Levels of
phosphorylated p38 (p-p38) were normalized to the total p38
(t-p38). The untreated K562 cell p-p38 levels were normalized to 1,
so that the values of each group are presented as the fold-increase
relative to the negative control group. (D) p38 MAPK is required
for γ-globin induction by PT. The qRT-PCR assay was performed in
K562 cells treated with PT or NaB, alone or in combination with SB
pre-treatment. (E) p38 MAPK is required for HbF synthesis induced
by PT. Western blot analyses were performed in K562 cells treated
with PT and NaB, alone or in combination with SB pre-treatment. The
amount of γ-globin mRNA and HbF synthesis was calculated as the
ratio to GAPDH mRNA and β-actin protein, respectively. The amounts
of γ-globin mRNA and HbF in the negative control group were
normalized to 1, so that the values of each group are presented as
the fold-increase relative to the negative control group. The data
represent the means ± SD of 3 independent experiments.
**P<0.01, vs. the negative control;
#P<0.05 and ##P<0.01, PT group vs. the
PT + SB group; ΔΔP<0.01, the NaB group vs. the NaB +
SB group. |
In the following set of experiments, the correlation
between PT-induced p38 MAPK phosphorylation and PT-induced
increases in γ-globin gene expression and HbF production was
further determined. The data presented in Fig. 4D and E illustrate that in the
absence of SB, PT and NaB significantly increased γ-globin mRNA
expression by 5.28±0.67- and 5.48±0.80-fold, respectively, and HbF
production by 2.31±0.09 and 2.36±0.03-fold, respectively
(P<0.001). By contrast, following pre-treatment with 10
μmol/l SB for 1 h, exposure to PT or NaB resulted in a
2.70±0.37- and 2.26±0.36-fold reduction in γ-globin gene
expression, and a 1.67±0.11- and 1.29±0.13-fold reduction in HbF
production, respectively (P<0.001). These results indicate that
the p38 MAPK pathway contributes to the induction of γ-globin gene
expression and the production HbF by PT.
PT regulates epigenetic histone
modifications within the γ-globin gene promoter regions through the
p38 MAPK signaling pathway
To investigate whether the PT-induced γ-globin
expression involves epigenetic histone modifications within the
γ-globin gene promoter region via the p38 MAPK signaling pathway,
we used a qRT-PCR-based ChIP assay as previously described
(38,39). The acetylation levels of histone
H3 (acH3) and H4 (acH4), and the acetylation and phosphorylation
levels of histone H3 (ph/acH3), were determined within the Gγ- and
Aγ-globin gene promoters and the necdin gene promoter (as a
negative control) in human erythroid cells (K562 cells and
erythroid progenitor cells from normal cord blood). We found high
levels of acH3, acH4 and ph/acH3 within the promoters of the Gγ-
and Aγ-globin genes in human erythroid cells, in which the Gγ- and
Aγ-globin genes are expressed. By contrast, there were low levels
of acH3, acH4 and ph/acH3 within the promoter of the necdin gene,
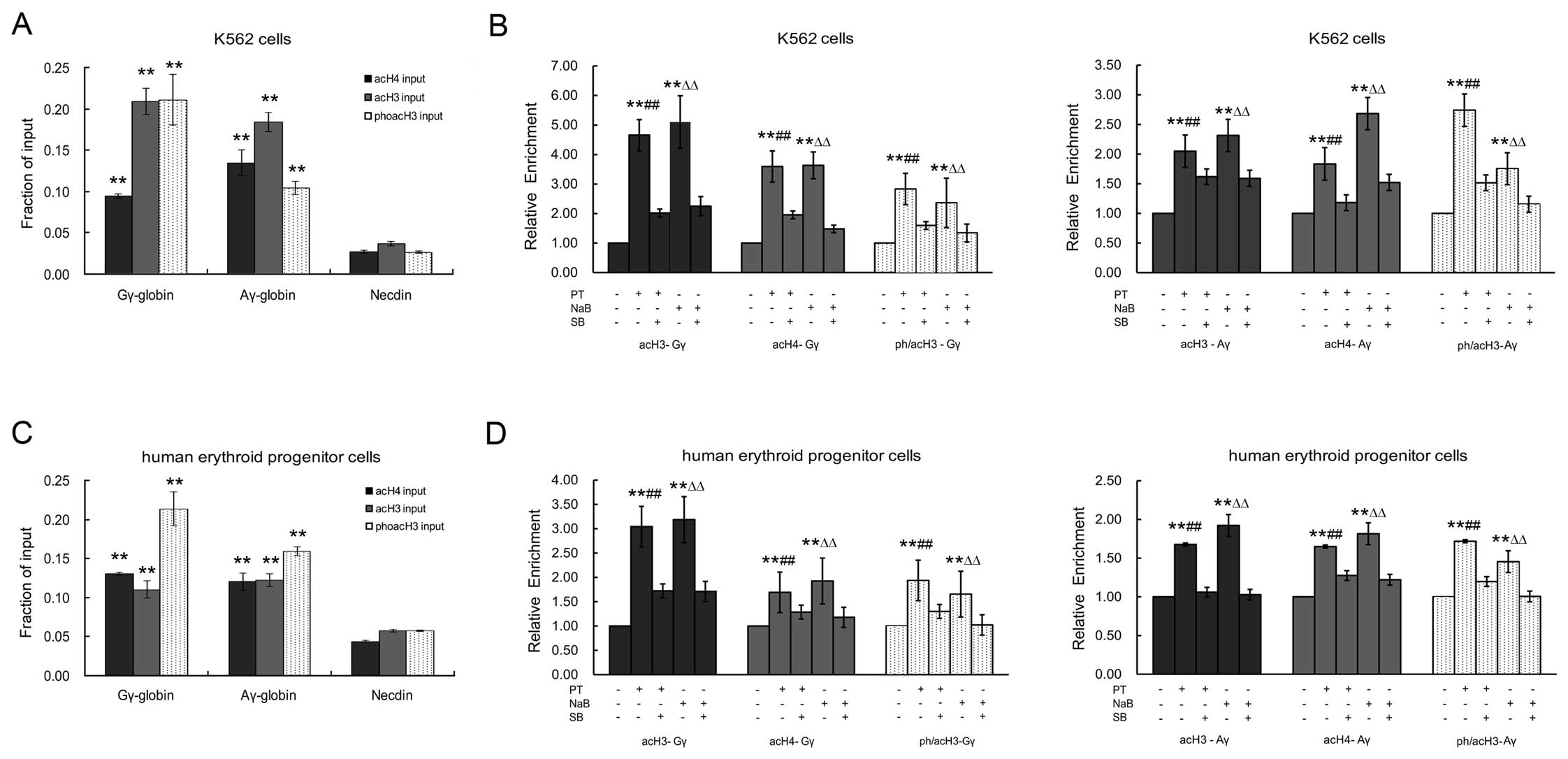
which is not expressed in erythroid cells (P<0.05; Fig. 5A and C).
In the K562 cells treated with PT or NaB, the levels
of acH3, acH4 and ph/acH3 within the Gγ- and Aγ-globin gene
promoters were significantly higher than those in the untreated
cells (Fig. 5B). The increases in
acH3, acH4 and ph/acH3 expression within the Gγ-globin gene
promoter were 4.56-, 3.57- and 2.81-fold for PT, and 5.03-, 3.61-
and 2.30-fold for NaB (P<0.05), respectively. The increases in
acH3, acH4 and ph/acH3 expression within the Aγ-globin gene
promoter were 2.04-, 1.81- and 2.74-fold for PT, and 2.31-, 2.67-
and 1.75-fold for NaB (P<0.05), respectively.
Similarly, the levels of acH3, acH4 and ph/acH3
within the Gγ- and Aγ-globin gene promoters were also significantly
higher in the erythroid progenitor cells from cord blood treated
with PT or NaB compared to the untreated cells (Fig. 5D). The increases in acH3, acH4 and
ph/acH3 expression within the Gγ-globin gene promoter were 3.02-,
1.68- and 1.91-fold for PT, and 3.18-, 1.92- and 1.72-fold for NaB
(P<0.05), respectively. The increases in acH3, acH4 and ph/acH3
expression within the Aγ-globin gene promoter were 1.66-, 1.62- and
1.72-fold for PT, and 1.9-, 1.78- and 1.45-fold for NaB
(P<0.05), respectively.
Of note, pre-treatment with SB reduced the effects
of PT and NaB on acH3, acH4 and ph/acH3 expression within the Gγ-
and Aγ-globin gene promoters, in the K562 cells and erythroid
progenitor cells. As shown in Fig.
5B, the effects of PT on acH3, acH4 and ph/acH3 expression
within the Gγ-globin gene promoter in the K562 cells were inhibited
by 48.87, 54.82 and 57.23%, respectively, while those of NaB were
inhibited by 44.27, 41.05 and 57.43%, respectively. For acH3, acH4
and ph/acH3 expression within the Aγ-globin gene promoter, the
inhibitory effects of SB were 79.10, 64.83 and 54.93% for PT, and
68.42, 56.56 and 65.78% for NaB, respectively. In the erythroid
progenitor cells (Fig. 5D),
pre-treatment with SB decreased the effects of PT on the levels of
acH3, acH4 and ph/acH3 expression within the Gγ-globin gene
promoter by 56.94, 76.18 and 67.67, respectively, and the
corresponding values for NaB were 53.62, 60.81 and 61.33%,
respectively. For the Aγ-globin gene promoter, the values were
63.28, 77.43 and 69.65% for PT, and 53.78, 68.17 and 62.74% for
NaB, respectively. These effects of SB were consistent with those
on the transcriptional expression of γ-globin. Taken together, our
results suggest that the p38 MAPK pathway plays an important role
in the mechanism by which PT and NaB enhance the acetylation of
histone H3 and H4, and the phosphorylation of histone H3.
Discussion
In this study, to our knowledge, we demonstrate for
the first time that PT induces γ-globin mRNA accumulation and
promotes HbF production in K562 cells and human erythroid
progenitor cells from patients with β-thalassemia. First, we
assessed the effects of PT on erythroid differentiation, cell
proliferation, α-, β- and γ-globin gene expression and HbF
synthesis in K562 cells. K562 cells represent a useful in
vitro model which may be used to screen for novel drugs capable
of inducing differentiation and to elucidate the molecular
mechanisms that regulate the expression of human globin genes
(40,41). Our results demonstrated that PT
significantly increased the number of benzidine-positive K562
cells, suggesting that PT induced the erythroid differentiation of
these cells. Furthermore, this effect on erythroid differentiation
was associated with an increase in γ-globin mRNA accumulation and
HbF production. Of note, the optimal doses of PT which induced
γ-globin expression, had no apparent cell growth-inhibitory effect.
By contrast, NaB, which caused similar effects on differentiation,
HbF synthesis and γ-globin mRNA accumulation, significantly
inhibited K562 cell proliferation (Fig. 1). Another interesting result was
that the effects of PT on γ-globin mRNA accumulation were clearly
selective, since there were no detectable effects on the
accumulation of α- and β-globin mRNA (Fig. 2). The selective effect of PT is
crucial since it can decrease the imbalance of non-α-/α-globin
chains in β-thalassemia (2).
Another significant aspect of this study is that we also evaluated
the effects of PT on the induction of γ-globin mRNA expression and
HbF production in human erythroid progenitor cells from patients
with β-thalassemia, and found that PT was superior at inducing HbF
expression compared to NaB. More importantly, we found that PT may
be a safer drug since it had no apparent cytotoxic effects and
caused a significantly lower inhibition of human erythroid
progenitor cell proliferation compared with NaB. Therefore, our
study has identified a novel HbF inducer from a traditional Chinese
medicine that shows great promise for the treatment of
β-thalassemia and SCA.
However, the mechanism by which PT induces γ-globin
gene expression is unknown. Research efforts to elucidate the
mechanisms for reactivating γ-globin gene expression have been
ongoing in order to aid the development of specific gene-based
therapeutics for the treatment of hemoglobin-opathies. Significant
progress has been made over the past decade (42–45). In particular, evidence has emerged
demonstrating an important role of cell signaling pathways,
including cyclic guanosine monophosphate (cGMP), p38 MAPK, ROS and
cytokine signaling pahtways, in mediating the effects of
drug-induced γ-globin gene expression (7). Among these, the p38 MAPK signaling
pathway has attracted particular attention since it has been
demonstrated to play a role in the induction of γ-globin gene
expression by various drugs (37,46). MAPKs are key regulatory enzymes
that transduce external signals into a complexity of intracellular
responses. The direct activation of p38 MAPK in K562 stable lines
with MAPK kinase kinase (MKK)3 and MKK6, the immediate upstream
activators of p38 MAPK, has been shown to increase γ-globin mRNA
expression in the absence of HbF inducers (37). Of note, we found that the enforced
p38 MAPK expression in K562 stable lines directly stimulated
γ-globin gene expression and HbF production in the absence of
inducers, suggesting a critical role for p38 MAPK in regulating
γ-globin gene expression. In this study, we therefore first
determined whether PT can activate the p38 MAPK signaling pathway.
Our results demonstrated that PT enhanced p38 MAPK phosphorylation,
and that this was accompanied by an increase in γ-globin mRNA
expression and HbF production. However, pre-treatment with the p38
MAPK inhibitor, SB203580, abolished the effects of PT on p38 MAPK
phosphorylation and γ-globin gene expression and HbF production
(Fig. 4). This evidence supports
the hypothesis that the p38 MAPK signaling pathway plays an
important role in PT-induced γ-globin gene expression.
It is not known how the activation of the p38 MAPK
signaling pathway results in γ-globin gene expression. During the
past few years, evidence has accumulated that gene expression is
controlled by alterations in chromatin structure produced by
epigenetic modifications, including the methylation of DNA, and the
acetylation, methylation, phosphorylation and ubiquitination of
histones (47–49). The pattern of acetylation of
histone H3 and H4 within the promoters of the α- and β-globin genes
has been characterized, and suggests that histone acetylation may
play a pivotal role in the regulation of development-stage and
tissue-specific expression of the genes of α- and β-globin clusters
(29,50,51). However, the correlation between
γ-globin gene expression and histone phosphorylation within its
promoter is not clear. It has been reported that the MAPK-mediated
phosphoacetylation of histone H3 can induce gene regulation
(52). In this study, we further
investigated whether PT can induce not only the hyperacetylation
but also the hyperphosphorylation of histones within the γ-globin
gene promoter regions in human erythroid cells; we also examined
the correlation between the p38 MAPK signaling pathway and histone
modifications within the γ-globin gene promoter regions. The
experimental approach of ChIP (38,39), used routinely to measure the
levels and distribution of epigenetic markers, allows one to obtain
molecular snapshots of a specific chromosomal region in living
cells. Using ChIP, we provide evidence that PT induces the
hyperacetylation of histone H3 and H4, as well as the
hyperphosphorylation of histone H3 within the γ-globin gene
promoter regions, and that this is accompanied by a significant
increase in γ-globin gene expression and HbF synthesis. These
results indicate that the PT-induced γ-globin expression involves
histone modifications within the γ-globin gene promoter regions. Of
note, the PT-induced γ-globin gene expression and histone
modifications within its promoter regions were blocked by
pre-treatment with the p38 MAPK inhibitor, SB203580, consistent
with the hypothesis that the p38 MAPK signaling pathway plays an
important role in the induction of γ-globin expression by PT
through epigenetic histone modifications within the γ-globin gene
promoter regions.
In conclusion, we identified a novel effect of PT, a
well-known traditional Chinese medicine, to selectively induce
γ-globin gene expression in human erythroid progenitor cells,
suggesting that PT may have potential for development as a novel
therapeutic agent for the treatment of β-thalassemia and SCA. To
our knowledge, we are the first to clarify that the mechanism
underlying the PT-induced γ-globin gene expression may involve the
hyperacetylation of histone H3 and H4 and the hyperphosphorylation
of histone H3 within the γ-globin gene promoter region, via
activation of the p38 MAPK signaling pathway. Future studies are
required to identify which components of PT are effective at
inducing γ-globin gene expression, and in vivo studies are
also warranted to further explore the benefits of PT.
Acknowledgements
This study was supported by a grant
(no. 7005146) from the Natural Science Foundation of Guangdong
Province and a special co-fund (no. 2011B032200002) from the
Department of Science and Technology and Academy of Tradition
Chinese Medicine of Guangdong Province, P.R. China.
References
|
1
|
Sankaran VG: Targeted therapeutic
strategies for fetal hemoglobin induction. Hematology Am Soc
Hematol Educ Program. 2011:459–465. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Higgs DR, Engel JD and Stamatoyannopoulos
G: Thalassaemia. Lancet. 379:373–383. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Frenette PS and Atweh GF: Sickle cell
disease: old discoveries, new concepts, and future promise. J Clin
Invest. 117:850–858. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lawson SE, Roberts IA, Amrolia P, Dokal I,
Szydlo R and Darbyshire PJ: Bone marrow transplantation for
beta-thalassaemia major: the UK experience in two paediatric
centres. Br J Haematol. 120:289–295. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sadelain M, Lisowski L, Samakoglu S,
Rivella S, May C and Riviere I: Progress toward the genetic
treatment of the beta-thalassemias. Ann N Y Acad Sci. 1054:78–91.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sadelain M: Recent advances in globin gene
transfer for the treatment of beta-thalassemia and sickle cell
anemia. Curr Opin Hematol. 13:142–148. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pace BS and Zein S: Understanding
mechanisms of gamma-globin gene regulation to develop strategies
for pharmacological fetal hemoglobin induction. Dev Dyn.
235:1727–1737. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weatherall DJ and Clegg JB: The
Thalassaemia Syndromes. 4th edition. Blackwell Science; Oxford:
2001, View Article : Google Scholar
|
|
9
|
Gambari R and Fibach E: Medicinal
chemistry of fetal hemoglobin inducers for treatment of
beta-thalassemia. Curr Med Chem. 14:199–212. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
DeSimone J, Koshy M, Dorn L, et al:
Maintenance of elevated fetal hemoglobin levels by decitabine
during dose interval treatment of sickle cell anemia. Blood.
99:3905–3908. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bradai M, Abad MT, Pissard S, Lamraoui F,
Skopinski L and de Montalembert M: Hydroxyurea can eliminate
transfusion requirements in children with severe beta-thalassemia.
Blood. 102:1529–1530. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mankidy R, Faller DV, Mabaera R, et al:
Short-chain fatty acids induce gamma-globin gene expression by
displacement of a HDAC3-NCoR repressor complex. Blood.
108:3179–3186. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Johnson J, Hunter R, McElveen R, Qian XH,
Baliga BS and Pace BS: Fetal hemoglobin induction by the histone
deacetylase inhibitor, scriptaid. Cell Mol Biol (Noisy-le-grand).
51:229–238. 2005.PubMed/NCBI
|
|
14
|
Witt O, Monkemeyer S, Ronndahl G, et al:
Induction of fetal hemoglobin expression by the histone deacetylase
inhibitor apicidin. Blood. 101:2001–2007. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Perrine SP, Castaneda SA, Boosalis MS,
White GL, Jones BM and Bohacek R: Induction of fetal globin in
beta-thalassemia: Cellular obstacles and molecular progress. Ann N
Y Acad Sci. 1054:257–265. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Quek L and Thein SL: Molecular therapies
in beta-thalassaemia. Br J Haematol. 136:353–365. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fathallah H, Sutton M and Atweh GF:
Pharmacological induction of fetal hemoglobin: Why haven’t we been
more successful in thalassemia? Ann N Y Acad Sci. 1054:228–237.
2005.
|
|
18
|
Saunthararajah Y, Lavelle D and DeSimone
J: DNA hypo-methylating agents and sickle cell disease. Br J
Haematol. 126:629–636. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Normile D: Asian medicine. The new face of
traditional Chinese medicine. Science. 299:188–190. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bianchi N, Zuccato C, Lampronti I,
Borgatti M and Gambari R: Fetal hemoglobin inducers from the
natural world: A novel approach for identification of drugs for the
treatment of {beta}-thalassemia and sickle-cell anemia. Evid Based
Complement Alternat Med. 6:141–151. 2009.PubMed/NCBI
|
|
21
|
Fang S, Wu Z, Zhang X, et al: Clinical
observation on YiSuiShengXueGranule on treating 156 patients with
β-thalassemia major and the molecular mechanism study. Biol Pharm
Bull. 30:2084–2087. 2007.PubMed/NCBI
|
|
22
|
Yang M, Qian XH, Zhao DH and Fu SZ:
Effects of Astragalus polysaccharide on the erythroid lineage and
microarray analysis in K562 cells. J Ethnopharmacol. 127:242–250.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guo ZM, Li HJ and Qian XH: γ-globin
synthesis in K562 cells induced with Tortois plastron,
Astragali, Salviae miltiorrhizae and Codonopsis
pilosulae. J Exp Hematology. 16:520–524. 2008.(In Chinese).
|
|
24
|
Miao XY: Shennong Grass after Drainage
(Shen Nong’s Herbal Classic). China Press of Traditional Chinese
Medicine; Beijing: 1997
|
|
25
|
State Pharmacopoeia Commission of the
People’s Republic of China. Carapax et Plastrum Testudinis.
Pharmacopoeia of The People’s Republic China. 1:15–16. 2000.
|
|
26
|
Chen DF, Zeng HP, Du SH, et al: Extracts
from Plastrum testudinis promote proliferation of rat
bone-marrow-derived mesenchymal stem cells. Cell Prolif.
40:196–212. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xie XM, Li XC, Zhong YS, Huang CH and Chen
DF: Study of antioxidant activities of Plastrum Testudinis in
vitro. China Pharmacy. 17:1368–1370. 2006.
|
|
28
|
Chen DF, Li H and Du SH: Effects of
plastrum testudinis on differentiation of transplanted mesenchymal
stem cells into neurons in the injured spinal cord of rat. Chinese
J of Neuroanatomy. 22:233–237. 2006.
|
|
29
|
Fathallah H, Weinberg RS, Galperin Y,
Sutton M and Atweh GF: Role of epigenetic modifications in normal
globin gene regulation and butyrate-mediated induction of fetal
hemoglobin. Blood. 110:3391–3397. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guo ZM, Li HJ and Qian XH: Accumulation of
Gγ-globin mRNA and induction of HbF in K562 cells induced by serum
contained Huangqi. Pharm J Chin PLA. 24:15–19. 2008.
|
|
31
|
Fibach E, Manor D, Oppenheim A and
Rachmilewitz EA: Proliferation and maturation of human erythroid
progenitors in liquid culture. Blood. 73:100–103. 1989.PubMed/NCBI
|
|
32
|
Fibach E: Cell culture and animal models
to screen for promising fetal hemoglobin-stimulating compounds.
Semin Hematol. 38:374–381. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu QQ, Qian XH and Xu MJ: Effect of
low-dose hydroxyurea with sodium butyrate on globin gene expression
in human erythroid progenitor cells. Nan Fang Yi Ke Da Xue Xue Bao.
29:2073–2076. 20812009.(In Chinese).
|
|
34
|
Lam LT, Ronchini C, Norton J, Capobianco
AJ and Bresnick EH: Suppression of erythroid but not megakaryocytic
differentiation of human K562 erythroleukemic cells by notch-1. J
Biol Chem. 275:19676–19684. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
|
|
36
|
Im H, Grass JA, Johnson KD, Boyer ME, Wu J
and Bresnick EH: Measurement of protein-DNA interactions in vivo by
chromatin immunoprecipitation. Methods Mol Biol. 284:129–146.
2004.PubMed/NCBI
|
|
37
|
Pace BS, Qian XH, Sangerman J, et al: p38
MAP kinase activation mediates gamma-globin gene induction in
erythroid progenitors. Exp Hematol. 31:1089–1096. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mukhopadhyay A, Deplancke B, Walhout AJ
and Tissenbaum HA: Chromatin immunoprecipitation (ChIP) coupled to
detection by quantitative real-time PCR to study transcription
factor binding to DNA in Caenorhabditis elegans. Nat Protoc.
3:698–709. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Haring M, Offermann S, Danker T, Horst I,
Peterhansel C and Stam M: Chromatin immunoprecipitation:
optimization, quantitative analysis and data normalization. Plant
Methods. 3:112007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rutherford TR, Clegg JB and Weatherall DJ:
K562 human leukaemic cells synthesise embryonic haemoglobin in
response to haemin. Nature. 280:164–165. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bianchi N, Ongaro F, Chiarabelli C, et al:
Induction of erythroid differentiation of human K562 cells by
cisplatin analogs. Biochem Pharmacol. 60:31–40. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mabaera R, West RJ, Conine SJ, et al: A
cell stress signaling model of fetal hemoglobin induction: what
doesn’t kill red blood cells may make them stronger. Exp Hematol.
36:1057–1072. 2008.PubMed/NCBI
|
|
43
|
Kiefer CM, Hou C, Little JA and Dean A:
Epigenetics of beta-globin gene regulation. Mutat Res. 647:68–76.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Thein SL, Menzel S, Lathrop M and Garner
C: Control of fetal hemoglobin: new insights emerging from genomics
and clinical implications. Hum Mol Genet. 18:R216–R223. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bauer DE and Orkin SH: Update on fetal
hemoglobin gene regulation in hemoglobinopathies. Curr Opin
Pediatr. 23:1–8. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Witt O, Sand K and Pekrun A:
Butyrate-induced erythroid differentiation of human K562 leukemia
cells involves inhibition of ERK and activation of p38 MAP kinase
pathways. Blood. 95:2391–2396. 2000.PubMed/NCBI
|
|
47
|
Kouzarides T: Chromatin modifications and
their function. Cell. 128:693–705. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bernstein BE, Meissner A and Lander ES:
The mammalian epigenome. Cell. 128:669–681. 2007. View Article : Google Scholar
|
|
49
|
Turner BM: Defining an epigenetic code.
Nat Cell Biol. 9:2–6. 2007. View Article : Google Scholar
|
|
50
|
Wozniak RJ and Bresnick EH: Epigenetic
control of complex loci during erythropoiesis. Curr Top Dev Biol.
82:55–83. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fathallah H, Portnoy G and Atweh GF:
Epigenetic analysis of the human alpha- and beta-globin gene
clusters. Blood Cells Mol Dis. 40:166–173. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Clayton AL and Mahadevan LC: MAP
kinase-mediated phosphoacetylation of histone H3 and inducible gene
regulation. FEBS Lett. 546:51–58. 2003. View Article : Google Scholar : PubMed/NCBI
|