Introduction
Vascular endothelial dysfunction is implicated in
the initial development of atherosclerosis (1,2).
Oxidized low-density lipoprotein (ox-LDL) inhibits nitric oxide
(NO) production and impairs endothelial function (1,2).
In endothelial cells (ECs), NO is derived from L-arginine in a
reaction catalyzed by endothelial NO synthase (eNOS) (3). Previous studies have demonstrated
that decreased eNOS activity is an important feature in endothelial
dysfunction (4). The lectin-like
ox-LDL receptor-1 (LOX-1) with a type II membrane protein structure
has been identified as a major ox-LDL receptor that is expressed in
ECs (5). Relatively long
incubation periods with ox-LDL (12–24 h) have been determined to
induce endothelial dysfunction by upregulating the expression of
LOX-1 and inhibiting the function of eNOS (6). Thus, an ox-LDL-LOX-1-eNOS pathway
appears to play a crucial role in the pathogenesis of endothelial
dysfunction. Akt has been indicated to phosphorylate eNOS at
Ser1179, and the activation of the PI3K-Akt-eNOS signaling pathway
appears to be an important mechanism for maintaining the integrity
of the endothelium of the arteries. In previous studies, ox-LDL has
been reported to inactivate Akt through the activity of LOX-1
(7) and subsequently, to
dephosphorylate the Ser1179/1177 (bovine/human) residue of eNOS.
The Ser1179/1177 dephosphorylation of eNOS decreases eNOS activity
(8,9). However, the mechanisms through which
LOX-1 interrupts the AKT signaling cascade, leading to the
dephosphorylation of eNOS at Ser1179/1177, have not yet been fully
elucidated.
Previous studies have demonstrated that endoplasmic
reticulum (ER) stress induced by ox-LDL in human vascular cells
modulates the balance between survival and apoptosis induced by
ox-LDL (10–12). Other stimuli that can trigger ER
stress include high glucose levels, oxidative stress,
Ca2+ overload, ischemia and hypoxia. Evidence of ER
stress has been detected in atherosclerotic lesions in
hypercholesterolemic mice (13)
and ER stress has been suggested to occur in acute coronary
syndrome (14). ER stress can
lead to the accumulation of unfolded and misfolded proteins,
causing an ‘unfolded protein response’ (UPR). The UPR in turn
prompts adaptive responses to ER dysfunction and emerges as a new
adaptive system that determines the fate of cells (survival or
death) (15) and thus, the UPR
may play a role in endothelial dysfunction (16). A previous study indicated that the
inactivation of Akt is involved in ER stress-mediated signaling,
which can occur upon exposure to ER stress (17). This result led us to investigate
whether ER stress initiated by LOX-1 is associated with
ox-LDL-induced eNOS dysfunction in ECs.
In the present study, we found that ER stress is an
important trigger for the Akt and eNOS downregulation induced by
ox-LDL in ECs. Short-term treatment with ox-LDL immediately induced
ER stress and caused the dephosphorylation of Akt and eNOS in
bovine aortic ECs (BAECs) prior to changes in the expression of the
eNOS and LOX-1 proteins. Of note, treatment with JTX20, a LOX-1
blocking antibody, or 4-phenylbutyric acid (PBA), a chemical
chaperone facilitating the correct folding of proteins, partly
rescued the dephosphorylation of AKT/eNOS during a short-term (0–4
h) ox-LDL treatment. Our findings demonstrated that ER stress
preferentially regulated Akt/eNOS dephosphorylation prior to the
alteration of eNOS and LOX-1 expression during the early phase of
ox-LDL treatment. The present study provides new data regarding the
inhibition of eNOS induced by ox-LDL through LOX-1-mediated ER
stress, which may be implicated in endothelial dysfunction reported
in coronary artery disease.
Materials and methods
Materials
The cell culture materials used in the present study
were obtained from Invitrogen (Carlsbad, CA, USA). BAECs and growth
medium were purchased from Cell Systems Corp. (Kirkland, WA, USA).
2′,5′-ADP-Sepharose 4B was produced by Amersham Biosciences
(Piscataway, NJ, USA). The antibody against eNOS was purchased from
the BD Transduction Laboratories/BD Biosciences (San Jose, CA,
USA). Antibodies against phosphorylated eNOS (p-eNOS; at serine
1179), Akt, phosphorylated Akt (p-Akt; at serine 473) and p-Akt (at
threonine 308) were obtained from Cell Signaling Technology
(Beverly, MA, USA). The antibody against p-eNOS (at threonine 497)
was purchased from Upstate Biotechnology (Lake Placid, NY, USA).
Antibodies against protein phosphatase (PP)2A, glucose-regulated
protein (GRP)78, p-proline-rich extensin-like receptor kinase
(PERK) and β-actin were purchased from Santa Cruz Biotechnology
(Santa Cruz, CA, USA). 3H-arginine was purchased from
PerkinElmer Life Sciences (Waltham, MA, USA). The protease
inhibitor tablet was purchased from Roche Applied Science
(Indianapolis, IN, USA). Calmodulin, NADPH, tetrahydrobiopterin,
L-arginine, N-nitro-L-arginine methyl ester, LY294002 and
recombinant human insulin-like growth factor-1 (IGF-1) were
purchased from Sigma (St. Louis, MO, USA). PBA was obtained from
Sigma-Aldrich (Strasbourg, France). ox-LDL was obtained from
Guangzhou Yiyuan Biotech Co. Ltd. (Guangzhou, China). The JTX20
antibody was obtained from Wuhan Sanying Biotechnology, Inc.
(Wuhan, China) and all the other reagents used were purchased from
Sigma, unless otherwise indicated.
Cell culture
The BAECs were cultured in endothelial growth medium
supplemented with 10% fetal bovine serum, 10 μg/ml human
recombinant epidermal growth factor, 1 mg/l hydrocortisone, 50
μg/ml gentamicin, 50 ng/ml amphotericin B and 12 μg/ml bovine brain
extracts. The BAECs between passages 4 and 10 were employed for the
experiments.
Western blot analysis
The cells were harvested and lysed on ice for 30 min
in a modified radioimmunoprecipitation assay buffer (50 mM
Tris-HCl, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 0.25% sodium
deoxycholate, 50 mM NaF, 1 mM Na3VO4, 5 mM
sodium pyrophosphate and a protease inhibitor tablet). The cell
lysates were centrifuged at 14,000 × g for 15 min, and the
supernatant was recovered. The total protein concentration was
determined using BCA protein assay reagent (Pierce Biotechnology,
Inc., Rockford, IL, USA). The lysates were denatured by boiling in
SDS sample buffer. The proteins were separated through gradient
SDS/PAGE on a 4–20% SDS-polyacrylamide gel and transferred onto
polyvinylidene difluoride membranes (Amersham Pharmacia Biotech,
Piscataway, NJ, USA) with a semi-dry transfer cell (Bio-Rad,
Hercules, CA, USA). After being blocked with 3% BSA in TBS for 1 h,
the membranes were probed with the appropriate primary antibodies.
Membrane-bound primary antibodies were detected using secondary
antibodies conjugated with horseradish peroxidase. The western
blots were then visualized through enhanced chemiluminescence
detection reagents (Sigma) according to the instructions provided
by the manufacturer. The protein bands were quantified by scanning
with the Bio-Rad GelDoc™ XR and ChemiDoc™ XRS systems and analyzed
with Quantity One 1-D analysis software version 4.6.3.
Measurement of eNOS activity
The effect of ox-LDL on the eNOS-mediated metabolism
of 3H-arginine to 3H-citrulline was
determined, and the assay was performed under apparent
Vmax conditions, as previously described
(18–20). Briefly, BAEC lysates were
suspended in cold lysis buffer (0.3 M sucrose, 10 mM HEPES, 1%
Nonidet P-40, 0.1 mM EDTA, 1 mM dithiothreitol, 10 μg/ml leupeptin,
2 μg/ml aprotinin, 10 μg/ml soybean trypsin inhibitor and 50 μM
phenylmethylsulfonyl fluoride, pH 7.4) through vigorous vortexing.
The cell lysates (150–250 μg of protein) were combined with NADPH
(2 mM), CaCl2 (230 μM), tetrahydrobiopterin (3 μM) and
3H-arginine (0.2 μCi, 10 μM) for 20 min at 37°C in an
assay volume of 100 μl. The reaction products were measured using a
liquid scintillation counter. To determine whether ox-LDL altered
the activity of eNOS, calcium was replaced with EDTA (1.7 mM) in
the assay buffers.
Pull-down assay
Cells were harvested and lysed on ice for 30 min in
lysis buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 0.5% Nonidet
P-40, 50 mM NaF, 1 mM Na3VO4, 5 mM sodium
pyrophosphate and a protease inhibitor tablet). The cell lysates
were centrifuged at 14,000 × g for 15 min, and the supernatants
were recovered. The supernatants, which contained equal quantities
of protein, were incubated with 2′,5′-ADP-Sepharose 4B resins (50
μl in a 50% slurry) overnight at 4°C as previously described
(21). The resins were washed
twice with a standard washing buffer (50 mM Tris-HCl, 100 mM NaCl,
1 mM EDTA and 0.5% Nonidet P-40), 3 times with high-salt washing
buffer (50 mM Tris-HCl, 500 mM NaCl, 1 mM EDTA and 0.5% Nonidet
P-40) and once with a standard washing buffer. The proteins pulled
down were then eluted by boiling the beads for 5 min in SDS sample
buffer and the proteins were analyzed by western blot analysis.
Statistical analysis
Data are expressed as the means ± SD and were
analyzed using SPSS 10.0 statistical software (SPSS Inc., Chicago,
IL, USA). A one-way ANOVA, followed by LSD post hoc tests were
employed to determine the significance of differences among the
groups (P-values <0.01 and <0.05, respectively, were
considered to indicate statistically significant differences).
Results
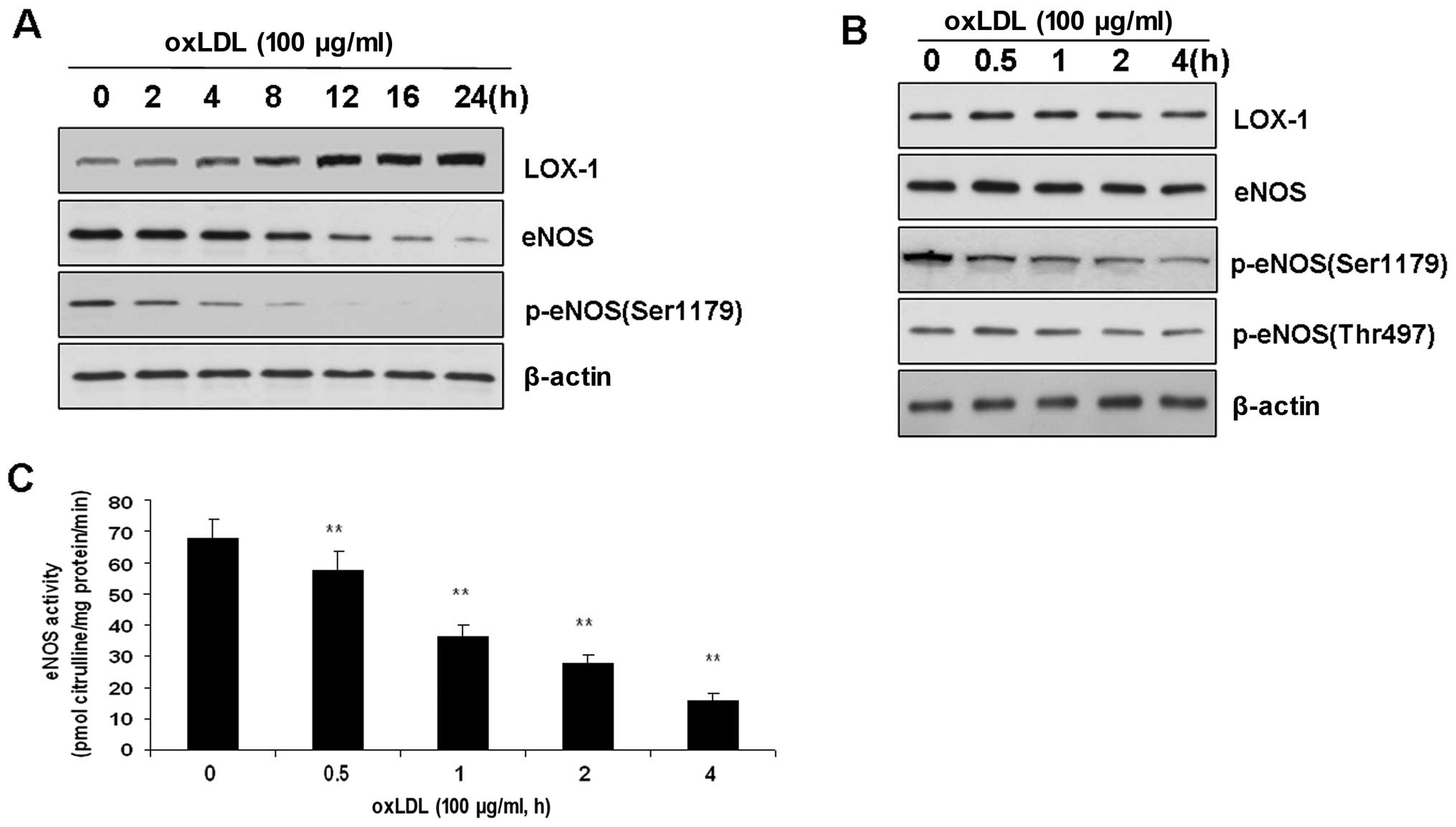
Effects of ox-LDL on LOX-1, eNOS and ER
stress sensors in BAECs
LOX-1 has been demonstrated to be a major
endothelial receptor for ox-LDL in ECs. Therefore, we investigated
whether LOX-1 expression is altered following treatment with
ox-LDL. The incubation of BAECs with ox-LDL (100 μg/ml) for 8 to 24
h increased the expression of LOX-1 compared with that of the
control (P<0.01). These results are consistent with those of
previous studies (6). However,
the expression of LOX-1 was not altered during short-term treatment
with ox-LDL prior to a 4-h incubation.
Previous studies have indicated that long-term
treatment with ox-LDL (from 12 to 24 h) induces endothelial
dysfunction by upregulating the expression of LOX-1 and inhibiting
the activity of eNOS. In this study, eNOS expression was also found
to be decreased following the treatment of BAECs with ox-LDL for 12
h (Fig. 1). Therefore, we
investigated whether the changes in LOX-1 expression accounted for
the alterations in eNOS phosphorylation and function. To determine
the effects of ox-LDL on eNOS phosphorylation, we added ox-LDL (100
μg/ml) to the medium, which exerted a time-dependent effect on the
dephosphorylation of eNOS at Ser1179 (Fig. 1). In other words, ox-LDL (100
μg/ml) reduced the level of eNOS phosphorylation. This
time-dependent effect of ox-LDL was specific to Ser1179, as neither
eNOS at Thr497 phosphorylation nor the level of total eNOS in the
BAECs was affected (Fig. 1).
Accordingly, eNOS activity was also reduced in a time-dependent
manner (Fig. 1).
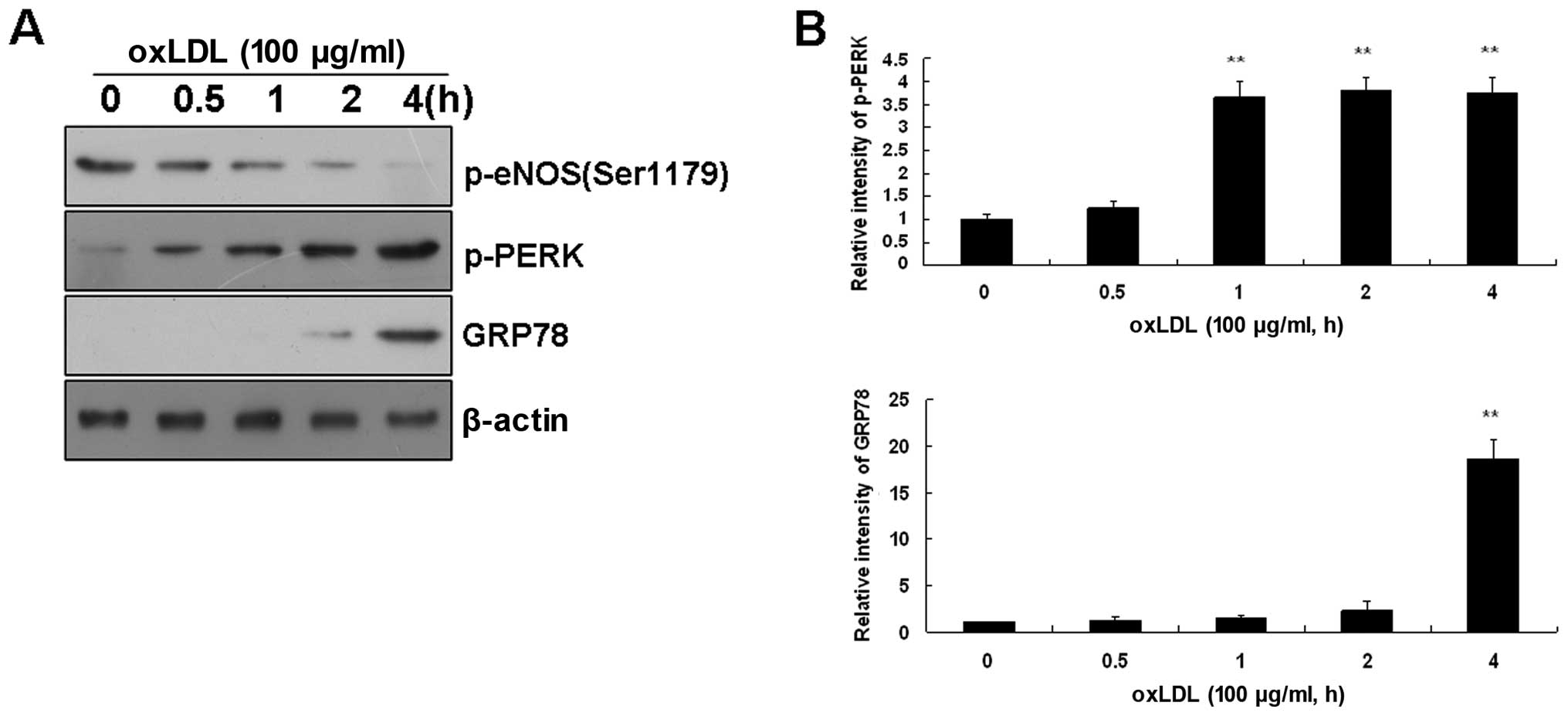
Effects of ox-LDL on ER stress sensors in
BAECs
As ox-LDL can trigger ER stress in ECs, and ER
stress is characterized by the activation of ER stress sensors,
which leads to UPR induction, we also investigated alterations in
ER stress sensors in BAECs. The incubation of BAECs with ox-LDL
(100 μg/ml) induced the time-dependent activation of ER stress
sensors, as assessed by the phosphorylation of PERK. This
activation was particularly noticeable immediately after 2 h
following treatment with ox-LDL (Fig.
2). As expected, ox-LDL-activated PERK induced GRP78 expression
(Fig. 2) and GRP78 has been
reported to regulate protein biosynthesis (22).
Taken together, these data indicate that short-term
treatment with ox-LDL immediately activates ER stress and induces
eNOS dephosphorylation in BAECs prior to the alteration of total
eNOS and LOX-1 expression.
Changes in Akt and protein phosphatase
activation in ox-LDL-treated BAECs
Akt has been described as a kinase responsible for
the phosphorylation of eNOS at Ser1179 under various conditions.
Therefore, we investigated whether Akt activity is altered by a
short-term treatment with ox-LDL and if so, whether this change is
associated with the changes in the phosphorylation of eNOS at
Ser1179. Our results revealed that Akt was activated, as
demonstrated by the phosphorylation of its Thr308 and Ser473
residues. As depicted in Fig. 3A,
the dephosphorylation of Akt also occurred in the BAECs subjected
to short-term treatment with ox-LDL. The time course of p-Akt
levels was largely parallel with that of p-eNOS Ser1179 levels.
These data suggested that the failure of Akt phosphorylation at
Thr308 and Ser473 contributed to the dephosphorylation of eNOS
Ser1179 during the short-term treatment of BAECs with ox-LDL.
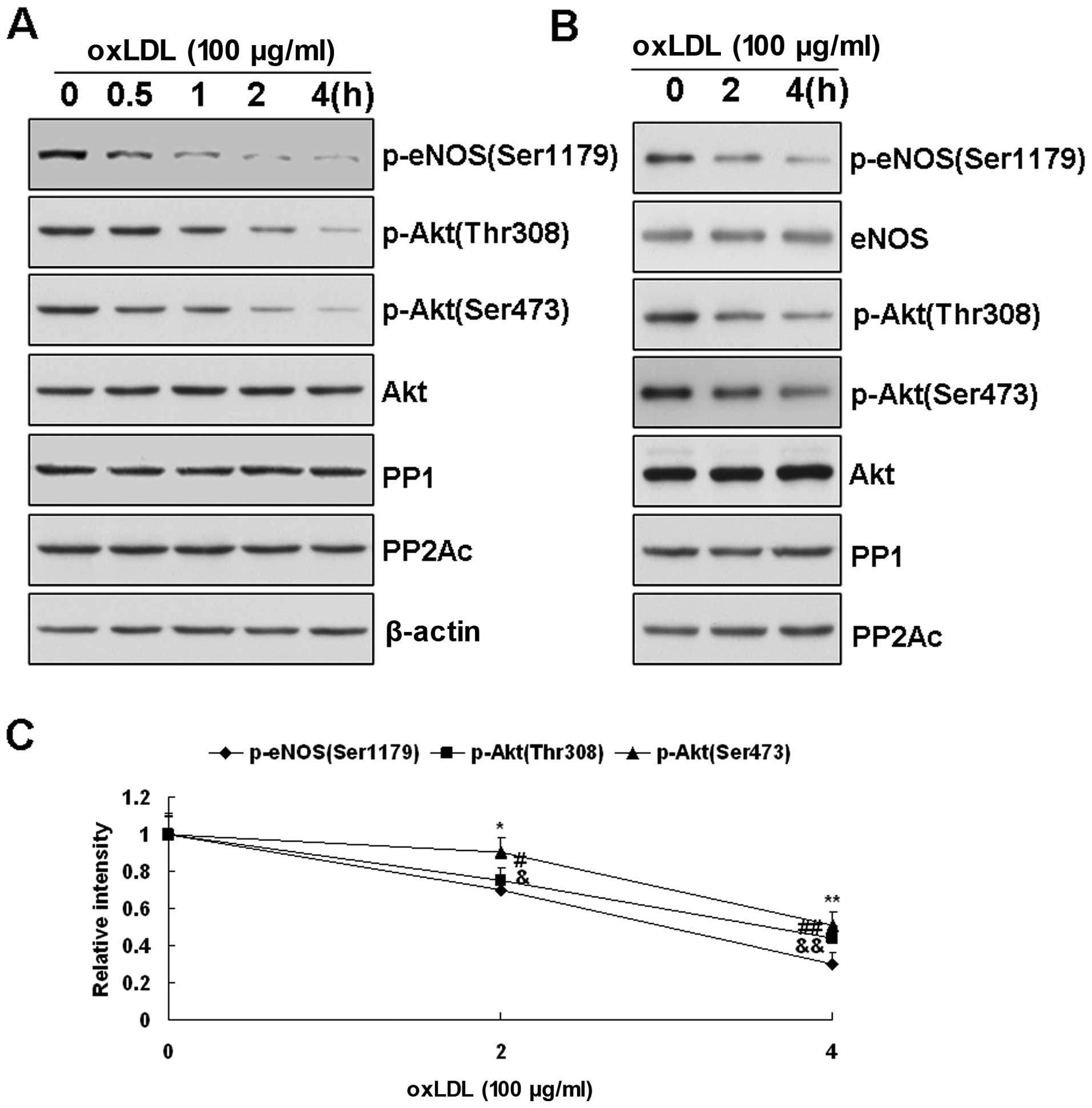 | Figure 3(A) Changes in Akt, protein
phosphatase (PP)2A and PP1 in oxidized low-density lipoprotein
(ox-LDL)-treated bovine aortic endothelial cells (BAECs) measured
by western blot analysis. Effects of ox-LDL (100 μg/ml) on Akt
phosphorylation at Thr308 and Ser473 in BAECs. As the
phosphorylation of endothelial nitric oxide synthase (eNOS) at
Ser1179 decreased upon ox-LDL treatment, the phosphorylated form of
Akt at Thr308 and Ser473 was also reduced in a time-dependant
manner. However, the Akt protein levels depicted did not change
according to the results presented here. (B) Effects of ox-LDL (100
μg/ml) on the expression of PP2A and PP1 in BAECs after treatment
for 0, 2 and 4 h. No significant change was observed in the levels
of PP2A and PP1 in the BAECs before and after ox-LDL treatment. (B
and C) Representative blots are presented from 3 independent
experiments. (B) Effects of ox-LDL on the association of PP1, PP2A
and Akt with eNOS. After 2 or 4 h of treatment of ox-LDL (200
μg/ml), eNOS in the BAECs was pulled down with the
2′,5′-ADP-Sepharose beads, and eNOS-associated proteins were
measured by western blot analysis with the corresponding
antibodies. Consistent with the results obtained with the cell
lysates, ox-LDL treatment decreased the phosphorylation of Akt (at
Ser473 and Thr308) and eNOS (at Ser1179). However, in the samples
containing equal amounts of eNOS input, the levels of PP1, PP2A and
Akt associated with eNOS were not significantly altered by ox-LDL
treatment. All the data presented are representative of 3
independent experiments. (C) Quantitative densitometry analyses of
Akt phosphorylation at Thr308 and Ser473 associated with eNOS. Data
are presented as the means ± SE (*,&,#P<0.05;
**,&&,##P<0.01 vs. the levels observed prior
to treatment with ox-LDL, n=3). |
Both Akt and eNOS at Ser1179 have been reported to
be dephosphorylated by PP2A (23,24). To determine whether the loss of
Akt and eNOS phosphorylation at Ser1179 was due to the increased
PP2A and PP1 activity, we measured the effects of ox-LDL on the
expression levels of PP2A and PP1. PP2A is a heterotrimeric enzyme
consisting of a catalytic subunit C, a structural subunit A and a
regulatory subunit B (24). As
depicted in Fig. 3A, none of the
PP2A subunits exhibited changes in expression levels in the
ox-LDL-treated BAECs. These data indicated that the inactivation of
Akt, as well as the loss of eNOS phosphorylation at Ser1179 in the
short-term ox-LDL-treated BAECs, was not due to the elevated
expression of PP2A and PP1.
Effects of ox-LDL on the association of
PP1, PP2A and Akt with eNOS
Akt and PP2A have been reported to be associated
with eNOS (21), raising the
possibility that the increased association with protein
phosphatases induced the dephosphorylation of Akt and eNOS at
Ser1179. To investigate this hypothesis, we pulled down eNOS from
BAECs after 2 and 4 h of ox-LDL treatment and measured the levels
of eNOS-associated Akt, PP2A and PP1. As depicted in Fig. 3B, whereas the levels of p-Akt and
p-eNOS (Ser1179) were both reduced, the quantities of Akt, PP1 and
PP2A associated with eNOS were not significantly affected by ox-LDL
treatment. These results indicated that the loss of Akt and eNOS
phosphorylation at Ser1179 following short-term treatment with
ox-LDL was not due to the increased association of PP1 and PP2A
with eNOS.
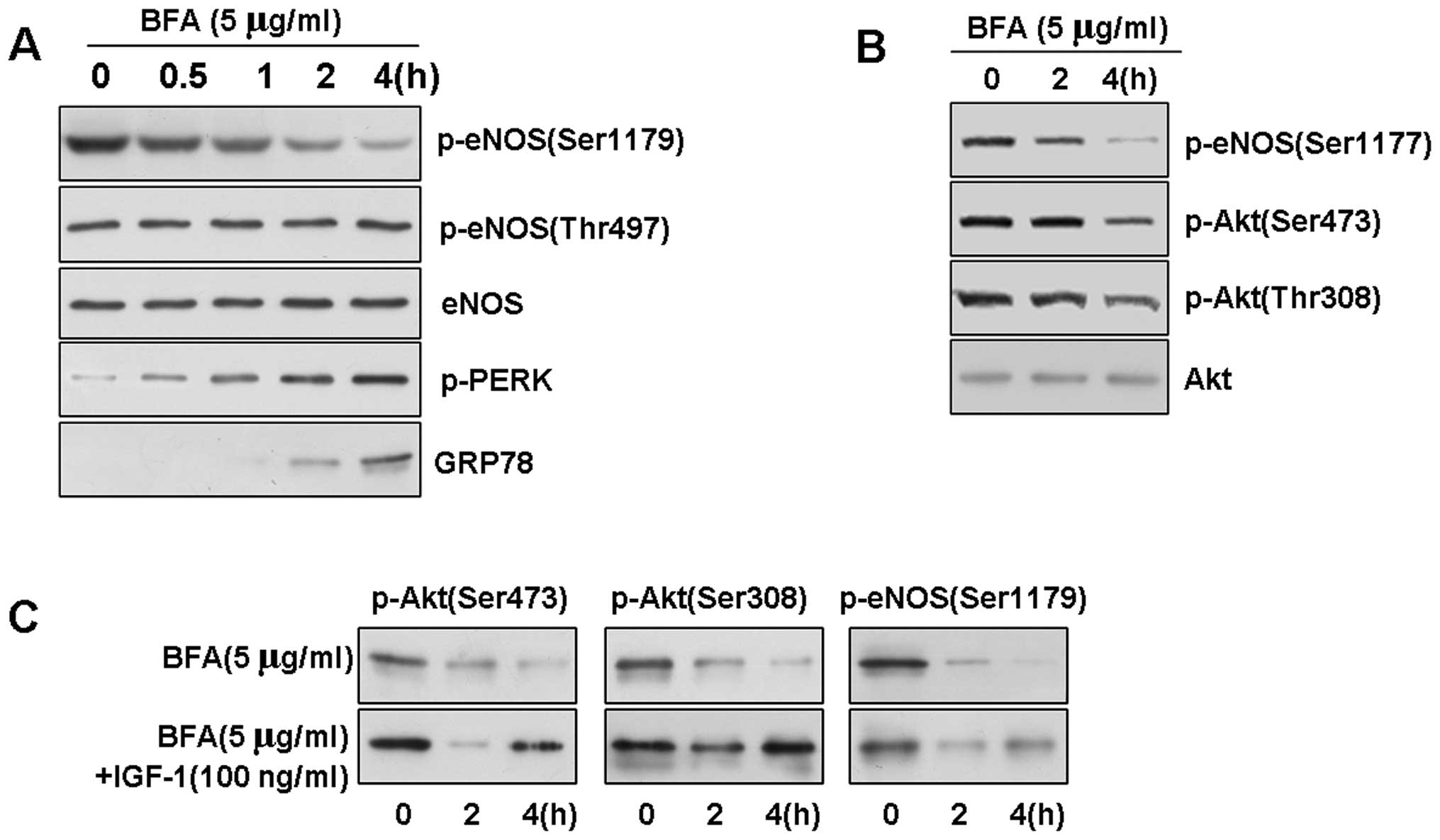
ER stress downregulates eNOS and Akt
phosphorylation in BAECs
To further understand the mechanisms through which
treatment with ox-LDL leads to the loss of Akt phosphorylation and
eNOS phosphorylation at active sites, we investigated whether ER
stress affects eNOS phosphorylation in BAECs. We treated the BAECs
with an ER stress-inducing reagent, Brefeldin A (BFA; which
inhibits ER-Golgi transport, at a concentration of 5 μg/ml), to
induce UPR signaling. As assessed by western blot analysis, further
evaluation of the phosphorylation of eNOS indicated that BFA
induced the rapid dephosphorylation of eNOS at Ser1179 (Fig. 4A and B). Surprisingly, this
time-dependent result of ER stress was also specific, as neither
eNOS phosphorylation at Thr497 nor the level of total eNOS protein
in the BAECs was affected.
To explore the possible role of Akt in ER stress, we
then examined Akt phosphorylation in the stressed cells by western
blot analysis. We determined that the basal level of p-Akt was
gradually dephosphorylated upon exposure to ER stress in the BAECs.
We observed a slight decrease in the level of phosphorylation at 2
h, and Akt at Thr308 and Ser473 was significantly dephosphorylated
at 4 h in response to BFA (5 μg/ml). Conversely, the expression
level of total Akt protein was not affected by ER stress (data not
shown), indicating that the downregulation of Akt phosphorylation
was not induced by the downregulation total Akt protein.
Previous studies have indicated that phosphorylated
PERK (p-PERK) and GRP78 are induced in response to ER stress
(25). Since we observed that ER
stress downregulated Akt activation, we investigated whether BFA
affects the PI3K/Akt pathway in BAECs. BFA, at a concentration of 5
μg/ml, rapidly reduced the level of Akt and eNOS phosphorylation,
but had no effect on the quantity of Akt (data not shown), and
treatment with 100 ng/ml of IGF-1, an activator of PI3K, reversed
the BFA-induced decrease in Akt and eNOS phosphorylation (Fig. 4C).
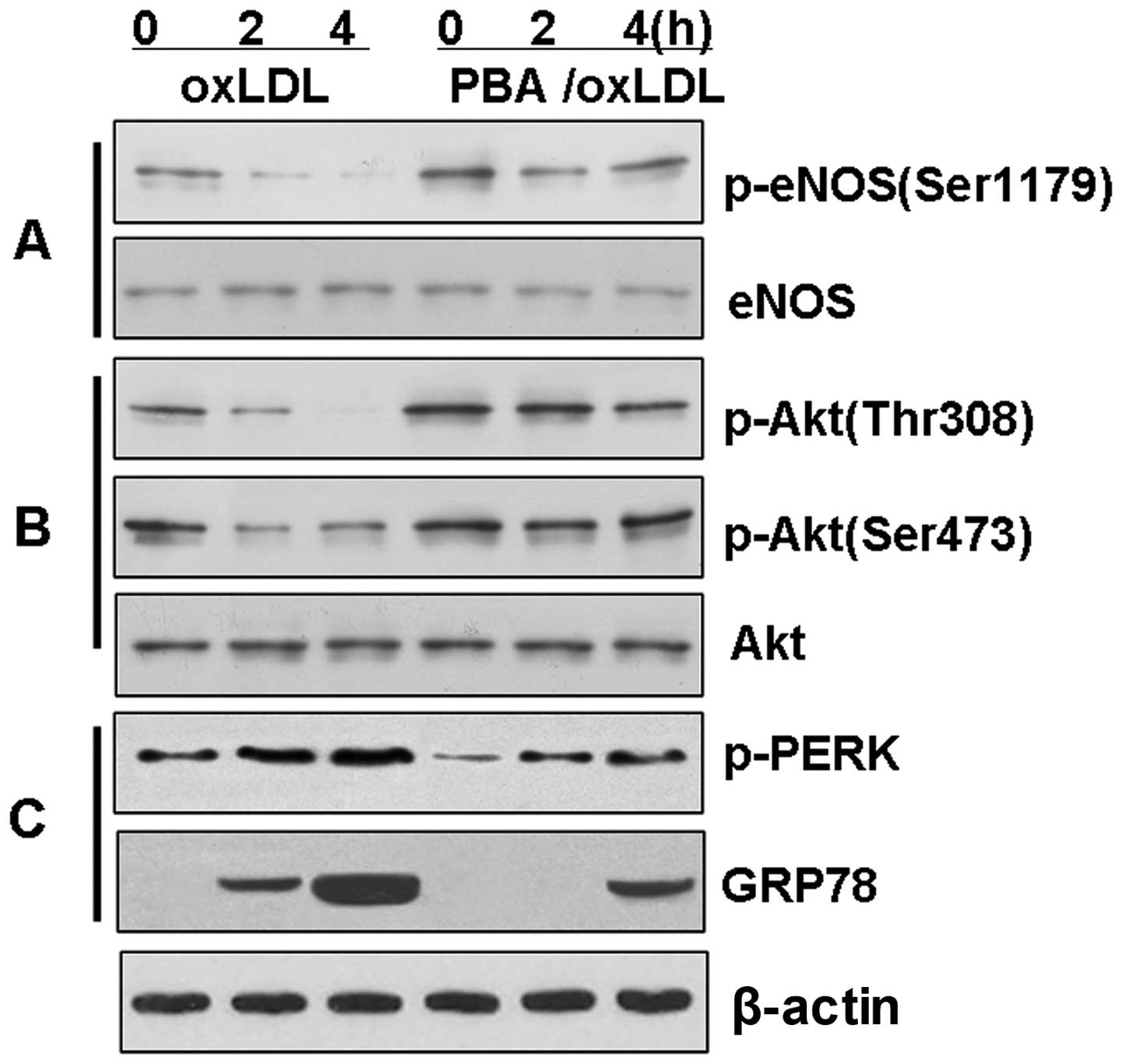
The chemical chaperone, PBA, reduces ER
stress and prevents the loss of Akt and eNOS phosphorylation in
BAECs
To investigate the hypothesis that the
downregulation of eNOS phosphorylation is associated with the Akt
signaling pathway through the induction of ER stress, the BAECs
were treated with ox-LDL in the presence or absence of PBA in order
to inhibit ER stress. The induction of p-PERK and GRP78 by ox-LDL
was significantly reduced (P<0.01) in the BAECs pre-treated with
PBA (10 mM) for 4 h (Fig. 5).
Further evaluation of the phosphorylation of eNOS revealed that PBA
reversed the dephosphorylation of Akt and eNOS induced by ox-LDL
(P<0.01). Again, this time-dependent effect of PBA did not
affect the level of total Akt or the level of total eNOS in the
BAECs.
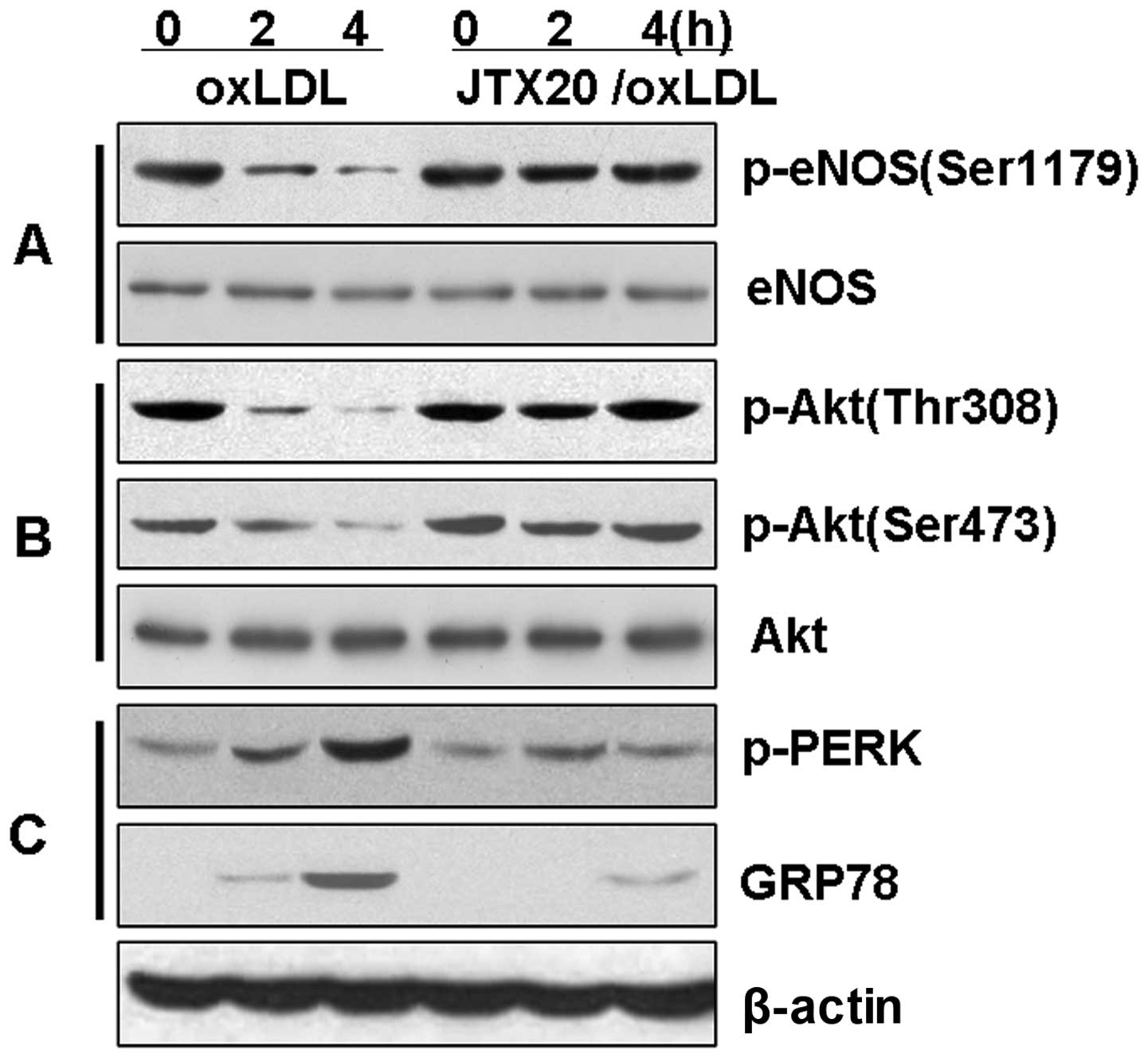
The anti-LOX-1 blocking antibody, JTX20,
reduces ER stress and inhibits the loss of Akt and eNOS
phosphorylation in BAECs
We then investigated the hypothesis that the
downregulation of eNOS phosphorylation is associated with the Akt
signaling pathway through the induction of ER stress by LOX-1
(Fig. 6). The BAECs were treated
with ox-LDL in the presence or absence of the anti-LOX-1 blocking
antibody, JTX20, to block eNOS dephosphorylation. The induction of
eNOS and Akt dephosphorylation by ox-LDL was significantly reduced
(P<0.01) in the BAECs pre-treated with JTX20 (40 μg/ml) for 1 h
(Fig. 6). Further evaluation of
ER stress indicated that JTX20 reversed the ox-LDL-induced
expression of p-PERK and GRP78 (P<0.01). These data strongly
suggest that the effects of ox-LDL on the PI3K-Akt-eNOS signaling
pathway or the induction of ER stress are mediated through the
activation by ox-LDL of the LOX-1 receptor and subsequent
intracellular signaling pathways.
Discussion
ox-LDL has been demonstrated to be an important
pathogenic factor in the formation of atherosclerotic plaque.
Endothelial dysfunction is the initial change that occurs in the
vascular wall during the course of atheroma formation. LOX-1 has
been identified as the major receptor for ox-LDL in ECs. As one of
the key intrinsic molecules, LOX-1 has been reported to induce
endothelial dysfunction after being triggered by ox-LDL (5). When bound to and activated by
ox-LDL, LOX-1 enhances NO catabolism as a result of superoxide
generation and decreases the release of NO through reduced eNOS
activity. LOX-1 has been shown to be associated with the reduced
expression of constitutive eNOS (26). As previously described, the
incubation of human coronary artery ECs (HCAECs) with ox-LDL for 24
h increased the expression of LOX-1 (mRNA and protein) compared
with the control. The incubation of HCAECs with ox-LDL markedly
reduced eNOS expression (as measured by RT-PCR and western blot
analysis) (26).
In the present study, we examined the effects of
LOX-1 in eNOS expression and activity in ox-LDL-treated BAECs. Our
results demonstrated that ox-LDL did not upregulate the expression
of LOX-1 until after 4 h (Fig.
1), and the level of LOX-1 began to increase after 4 h of
treatment with ox-LDL. At the same time, a significant reduction in
total eNOS expression was observed after 12 h of ox-LDL treatment
in the BAECs. These results are consistent with those of a previous
study (26). Of note, a
significant decrease was observed in eNOS phosphorylation at
Ser1179 following short-term (<4 h) treatment with ox-LDL, and
this change was independent of the expression of LOX-1 protein and
the level of total eNOS protein in the BAECs (Fig. 1). These findings indicated that
the changes in the phosphorylation of eNOS occurred more rapidly
than the alteration of its protein level. Thus, ox-LDL mediated the
reduction of eNOS activity through two separate mechanisms, one
being a mechanism with rapid effects without an increase in LOX-1
expression and the other being a mechanism with relatively slow
effects with increased LOX-1 expression. The former mechanism
required a decrease in eNOS phosphorylation at Ser1179, and the
latter involved a decrease in the expression of eNOS.
LOX-1 is a 50 kDa type II transmembrane glycoprotein
comprising 273 amino acids (5).
The protein contains a short N-terminal cytoplasmic domain, a
single transmembrane domain and an extracellular domain comprising
a neck domain followed by a C-terminal, C-type, lectin-like,
ligand-binding domain (27).
Previous studies have indicated that the biological effects of
ox-LDL, exerted through LOX-1 binding, involve a number of
signaling pathways, including the Akt, tyrosine kinase and
mitogen-activated protein kinase (MAPK) pathways (28). The Akt cascade is a signal
transduction pathway that mediates the activation of eNOS. eNOS at
Ser1179 was first reported to be phosphorylated by Akt (29,30). Since then, Akt has remained the
key kinase that phosphorylates eNOS at Ser1179 under various
circumstances, although a number of other kinases, including 5′
adenosine monophosphate-activated protein kinase (AMPK), protein
kinase G (PKG), protein kinase A (PKA), extracellular
signal-regulated kinase (ERK)1/2 and
Ca2+/calmodulin-dependent kinase II (5,31),
have been reported to phosphorylate eNOS at Ser1179 (32).
Therefore, we investigated the hypothesis that
changes in Akt activity may account for the alteration of eNOS
phosphorylation at Ser1179 following short-term treatment with
ox-LDL. Akt has been reported to be activated following its
phosphorylation at the Thr308 and Ser473 residues. As depicted in
Fig. 3A, Akt was dephosphorylated
at the Thr308 and Ser473 residues during a short-term treatment of
BAECs with ox-LDL. The time course for Akt dephosphorylation was
largely parallel with that of eNOS dephosphorylation at Ser1179.
However, these dephosphorylation events may result from the
activity of a phosphatase. To characterize the eNOS
dephosphorylation at Ser1179 and determine whether it was the
result of the cleavage of phosphates by phosphatases, we examined
the expression of PP2A and PP1 and their association with eNOS by
pull-down assay during the short-term treatment of BAECs with
ox-LDL. Our data indicated that the inactivation of Akt, as well as
the loss of eNOS phosphorylation at Ser1179, was not accompanied by
the elevated expression of PP2A and PP1 and an increase in their
association with eNOS (Fig. 3).
LOX-1 exhibits a short 36-amino acid cytosolic tail with no
homology to known catalytic or signaling domains with the purpose
of transducing signals to activate downstream functions. One gap in
the current understanding of LOX-1 biology is the identity of the
downstream mediating signals activated by LOX-1. However, to date,
the correlation between the activation or the inactivation of the
Akt-eNOS pathway and the activation of LOX-1 has not been
determined. Our data demonstrated that the short-term treatment of
BAECs with ox-LDL activated Akt by phosphorylating its Thr308 and
Ser473 residues, thus inducing eNOS phosphorylation at Ser1179.
However, this transient event was not associated with LOX-1
expression, and did not induce a change in eNOS protein levels and
did not stimulate the activities of PP2A and PP1.
ER stress has been demonstrated to be induced by
ox-LDL in human vascular cells (10). Once stimulated, ER stress can
modulate the balance between cell survival and apoptosis. When the
ER environment is perturbed and the folding of nascent proteins is
impaired, a quality control system termed the UPR is mobilized
(33–35). Initially, the UPR is an adaptive
response in which affected cells attempt to overcome the
accumulation of misfolded proteins by increasing their
protein-folding capacity. However, when ER stress is excessive and
prolonged, cells undergo apoptotic cell death. Thus, the ER stress
response exhibits a conditional ability to protect cells against
offensive agents or to activate the cell death program. The extent
to which ER stress following stimulation with ox-LDL correlates
with the modification of eNOS activity has not yet been fully
elucidated.
The levels of ox-LDL-induced ER stress and the UPR
can be assessed by the phosphorylation of an ER stress sensor
(p-PERK) and the expression of ER-resident chaperones (GRP78)
(36). The UPR is an adaptive
response that first tends to restore ER activity and cellular
homeostasis but preferentially induces apoptosis when ER stress is
prolonged, depending on the nature of the agent and the intensity
of the stress (37). In this
study, to our knowledge, we provide the first evidence of the
induction of ER stress by ox-LDL and its potential involvement in
the dephosphorylation of eNOS during the short-term treatment of
BAECs with ox-LDL. We observed that the incubation of BAECs with
ox-LDL immediately induced the phosphorylation of PERK, which was
observed as early as 1 h after the initiation of ox-LDL treatment
(Fig. 2). This finding indicated
that the level of p-PERK began to increase as soon as ox-LDL
treatment was initiated. Furthermore, as p-PERK levels began to
increase rapidly, the level of phosphorylated eNOS at Ser1179 began
to decrease rapidly, as observed between 0.5 and 4 h. The most
significant evidence indicating the ox-LDL-dependent switch of ER
stress was provided by the persistent expression of GRP78, an
ER-localized chaperone stimulated through the ATF6 and PERK
pathways (38,39), which markedly increased at 4 h
after the initiation of treatment.
Taken together, these data demonstrated that
short-term treatment with ox-LDL immediately induced ER stress and
caused the dephosphorylation of eNOS in BAECs prior to the
alteration of total eNOS and expression of LOX-1. Based on these
data, we performed a series of experiments to investigate these
cellular pathways.
A previous study indicated that the inactivation of
Akt is involved in ER stress-mediated signaling. As demonstrated,
Akt was gradually inactivated in response to exposure to ER stress
(17). In this study, our data
demonstrated that BFA, an ER stress-inducing reagent, induced the
rapid dephosphorylation of Akt and eNOS at Ser1179 (Fig. 4). However, IGF-1 (an activator of
PI3K) partially reversed the BFA-induced increase in Akt and eNOS
dephosphorylation (Fig. 4C). In
addition, the induction of p-PERK and GRP78 by ox-LDL was observed
to be significantly reduced in the BAECs pre-treated with PBA (a
chemical chaperone that has the ability to decrease levels of ER
stress) for 14 h. PBA also reversed the ox-LDL-induced decrease in
Akt and eNOS phosphorylation (Fig.
5). Taken together, these data indicate that the ER stress
induced by ox-LDL induces the dephosphorylation of eNOS at Ser1179
in BAECs, and that this induction is associated with Akt
dephosphorylation.
This finding led us to investigate whether the
inhibition of eNOS activity induced by ER stress mediated by ox-LDL
is associated with LOX-1 in BAECs. Accordingly, we treated BAECs
with ox-LDL in the presence or absence of the anti-LOX-1 blocking
antibody, JTX20, and subsequently examined the level of eNOS
phosphorylation. The JTX20 antibody reversed the ox-LDL-induced
increase in p-PERK and GRP78 expression. JYX20 also significantly
reduced the ox-LDL-induced eNOS and Akt dephosphorylation observed
in the BAECs (Fig. 6).
In conlcusion, although long-term treatment with
ox-LDL upregulated LOX-1 expression and downregulated eNOS
expression, thereby mediating eNOS activity, our results also
indicated that eNOS was downregulated through dephosphorylation at
Ser1179 through a rapid regulatory mechanism associated with the
Akt signaling pathway. This mechanism was induced by
LOX-1-triggered ER stress during the short-term treatment of BAECs
with ox-LDL, and the mechanism involved was independent of the
stimulation of LOX-1 expression and independent of changes in eNOS
enzyme levels.
Acknowledgements
This study was partly supported by grants from the
National Natural Science Foundation of China (grant nos. 30770446,
31000471 and 31171027), the Scientific Research Foundation for
Returned Overseas Chinese Scholars, the State Education Ministry,
the Hubei Province Natural Science Foundation (grant no.
2006S2153), the Fundamental Research Funds for the Central
Universities, HUST (grant no. 2010JC055, 2010MS082 and 2011TS010)
and the Scientific and Technological Project in Hubei Province
(grant no. 2007AA301B31-2).
References
|
1
|
Ross R: The pathogenesis of
atherosclerosis: a perspective for the 1990s. Nature. 362:801–809.
1993. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stocker R and Keaney JF Jr: Role of
oxidative modifications in atherosclerosis. Physiol Rev.
84:1381–1478. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Griffith OW and Stuehr DJ: Nitric oxide
synthases: properties and catalytic mechanism. Annu Rev Physiol.
57:707–736. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cheng J, Ou JS, Singh H, et al:
20-hydroxyeicosatetraenoic acid causes endothelial dysfunction via
eNOS uncoupling. Am J Physiol Heart Circ Physiol. 294:H1018–H1026.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sawamura T, Kume N, Aoyama T, et al: An
endothelial receptor for oxidized low-density lipoprotein. Nature.
386:73–77. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sugimoto K, Ishibashi T, Sawamura T, et
al: LOX-1-MT1-MMP axis is crucial for RhoA and Rac1 activation
induced by oxidized low-density lipoprotein in endothelial cells.
Cardiovasc Res. 84:127–136. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ogura S, Kakino A, Sato Y, et al: Lox-1:
the multifunctional receptor underlying cardiovascular dysfunction.
Circ J. 73:1993–1999. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fulton D, Gratton JP, McCabe TJ, et al:
Regulation of endothelium-derived nitric oxide production by the
protein kinase Akt. Nature. 399:597–601. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dimmeler S, Fleming I, Fisslthaler B, et
al: Activation of nitric oxide synthase in endothelial cells by
Akt-dependent phosphorylation. Nature. 399:601–605. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sanson M, Auge N, Vindis C, et al:
Oxidized low-density lipoproteins trigger endoplasmic reticulum
stress in vascular cells: prevention by oxygen-regulated protein
150 expression. Circ Res. 104:328–336. 2009. View Article : Google Scholar
|
|
11
|
Tu BP and Weissman JS: Oxidative protein
folding in eukaryotes: mechanisms and consequences. J Cell Biol.
164:341–346. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Banhegyi G, Csala M, Szarka A, et al: Role
of ascorbate in oxidative protein folding. Biofactors. 17:37–46.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhou J, Werstuck GH, Lhotak S, et al:
Association of multiple cellular stress pathways with accelerated
atherosclerosis in hyperhomocysteinemic apolipoprotein E-deficient
mice. Circulation. 110:207–213. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Myoishi M, Hao H, Minamino T, et al:
Increased endoplasmic reticulum stress in atherosclerotic plaques
associated with acute coronary syndrome. Circulation.
116:1226–1233. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu C, Bailly-Maitre B and Reed JC:
Endoplasmic reticulum stress: cell life and death decisions. J Clin
Invest. 115:2656–2664. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dong Y, Zhang M, Liang B, et al: Reduction
of AMP-activated protein kinase alpha2 increases endoplasmic
reticulum stress and atherosclerosis in vivo. Circulation.
121:792–803. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hyoda K, Hosoi T, Horie N, et al: PI3K-Akt
inactivation induced CHOP expression in endoplasmic
reticulum-stressed cells. Biochem Biophys Res Commun. 340:286–290.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sessa WC, Harrison JK, Luthin DR, et al:
Genomic analysis and expression patterns reveal distinct genes for
endothelial and brain nitric oxide synthase. Hypertension.
21:934–938. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sessa WC, Barber CM and Lynch KR: Mutation
of N-myristoylation site converts endothelial cell nitric oxide
synthase from a membrane to a cytosolic protein. Circ Res.
72:921–924. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rosenkranz-Weiss P, Sessa WC, Milstien S,
et al: Regulation of nitric oxide synthesis by proinflammatory
cytokines in human umbilical vein endothelial cells. Elevations in
tetrahydrobiopterin levels enhance endothelial nitric oxide
synthase specific activity. J Clin Invest. 93:2236–2243. 1994.
View Article : Google Scholar
|
|
21
|
Wei Q and Xia Y: Proteasome inhibition
down-regulates endothelial nitric-oxide synthase phosphorylation
and function. J Biol Chem. 281:21652–21659. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Doms RW, Lamb RA, Rose JK and Helenius A:
Folding and assembly of viral membrane proteins. Virology.
193:545–562. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Greif DM, Kou R and Michel T:
Site-specific dephosphorylation of endothelial nitric oxide
synthase by protein phosphatase 2A: evidence for crosstalk between
phosphorylation sites. Biochemistry. 41:15845–15853. 2002.
View Article : Google Scholar
|
|
24
|
Millward TA, Zolnierowicz S and Hemmings
BA: Regulation of protein kinase cascades by protein phosphatase
2A. Trends Biochem Sci. 24:186–191. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kaufman RJ: Stress signaling from the
lumen of the endoplasmic reticulum: coordination of gene
transcriptional and translational controls. Genes Dev.
13:1211–1233. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mehta JL, Li DY, Chen HJ, et al:
Inhibition of LOX-1 by statins may relate to upregulation of eNOS.
Biochem Biophys Res Commun. 289:857–861. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Aoyama T, Sawamura T, Furutani Y, et al:
Structure and chromosomal assignment of the human lectin-like
oxidized low-density-lipoprotein receptor-1 (LOX-1) gene. Biochem
J. 339:177–184. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li D, Yang B and Mehta JL: Ox-LDL induces
apoptosis in human coronary artery endothelial cells: role of PKC,
PTK, bcl-2, and Fas. Am J Physiol. 275:H568–H576. 1998.PubMed/NCBI
|
|
29
|
Li D and Mehta JL: Upregulation of
endothelial receptor for oxidized LDL (LOX-1) by oxidized LDL and
implications in apoptosis of human coronary artery endothelial
cells: evidence from use of antisense LOX-1 mRNA and chemical
inhibitors. Arterioscler Thromb Vasc Biol. 20:1116–1122. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Oka K, Sawamura T, Kikuta K, et al:
Lectin-like oxidized low-density lipoprotein receptor 1 mediates
phagocytosis of aged/apoptotic cells in endothelial cells. Proc
Natl Acad Sci USA. 95:9535–9540. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mehta JL and Li DY: Identification and
autoregulation of receptor for OX-LDL in cultured human coronary
artery endothelial cells. Biochem Biophys Res Commun. 248:511–514.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shiojima I and Walsh K: Role of Akt
signaling in vascular homeostasis and angiogenesis. Circ Res.
90:1243–1250. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang X and Robbins J: Heart failure and
protein quality control. Circ Res. 99:1315–1328. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Glembotski CC: Endoplasmic reticulum
stress in the heart. Circ Res. 101:975–984. 2007. View Article : Google Scholar
|
|
35
|
Lai E, Teodoro T and Volchuk A:
Endoplasmic reticulum stress: signaling the unfolded protein
response. Physiology (Bethesda). 22:193–201. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schroder M and Kaufman RJ: The mammalian
unfolded protein response. Annu Rev Biochem. 74:739–789. 2005.
View Article : Google Scholar
|
|
37
|
Yamaguchi H and Wang HG: CHOP is involved
in endoplasmic reticulum stress-induced apoptosis by enhancing DR5
expression in human carcinoma cells. J Biol Chem. 279:45495–45502.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ma Y, Brewer JW, Diehl JA and Hendershot
LM: Two distinct stress signaling pathways converge upon the CHOP
promoter during the mammalian unfolded protein response. J Mol
Biol. 318:1351–1365. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ron D and Habener JF: CHOP, a novel
developmentally regulated nuclear protein that dimerizes with
transcription factors C/EBP and LAP and functions as a
dominant-negative inhibitor of gene transcription. Genes Dev.
6:439–453. 1992. View Article : Google Scholar : PubMed/NCBI
|