Introduction
Retinoic acid (RA), a metabolite of vitamin A,
influences cell growth, differentiation and apoptosis in adult
tissues, as well as during embryonic development (1,2).
RA signaling is mediated through binding to members of the RA
receptor (RAR) family, including RAR-α and retinoid X receptor-α
(RXR-α) (1). These are nuclear
receptors and as such, RAR-RXR heterodimers control the expression
of target genes through interactions with RA response elements
(RARE) and by recruiting coactivators to the promoter region of
target genes (3).
In addition to altering cell cycle progression and
inducing apoptosis, retinoids have also been shown to inhibit
activator protein-1 (AP-1) signaling (4,5).
AP-1 transcription factors, heterodimers that include members of
the c-Fos and c-Jun families, are required for the development and
progression of malignant tumors (6). Although it has been shown that the
transactivation of RARE and the subsequent upregulation of RAR-α is
involved in the inhibition of AP-1 activity (5), another study reported that retinoids
inhibit AP-1 activity through transrepression (4).
Altered RA signaling, as a result of a chromosomal
translocation resulting in a RAR-α-promyelocytic leukemia (PML)
gene fusion or abnormal RAR-α transcription, has been reported in a
number of inflammatory and neoplastic diseases, including breast
cancer and promyelocytic leukemia (7). RA analogs, including
all-trans retinoic acid (ATRA) and its metabolite,
9-cis-RA (8), are thought
influence cell cycle progression in a time-dependent manner
(9); however, the involvement of
p53 and p21waf may be cell type-dependent (9,10).
ATRA has been shown to inhibit the growth of gastric cancer cells
by upregulating RAR-α expression (5).
RA analogs have been used in the treatment of a
variety of cancers; however, a high rate of early mortality
(11–13), serious therapy-related sequelae,
including myelodysplastic syndrome and acute myeloid leukemia
(14,15), as well as a high incidence of
side-effects (16) have been
observed with this treatment. In addition, the tight metabolic
control of ATRA (17) and of
9-cis RA (18) levels
under normal physiological conditions may limit their use in
anticancer therapeutics. Thus, the identification and
characterization of novel RA analogs that inhibit cancer cell
growth without the associated side-effects would be important for
the clinical treatment of cancer. The aim of this study was to
compare the effects of a novel RA analog,
4-amino-2-trifluoromethyl-phenyl retinate (ATPR) (19), with those of ATRA (20,21) on the growth and distribution of
gastric cancer cells in different cell cycle phases, using the AGS,
MKN-74 and SC-M1 cell lines. Cyclin E, Bcl-2 and Bax expression, as
well as AP-1 activity were also measured following treatment with
ATPR and ATRA. ATPR inhibited cancer cell proliferation to a
greater extent compared with ATRA, possibly through the
RXR-mediated inhibition of AP-1 activity.
Materials and methods
Cell cultures
Three gastric cancer cell lines, AGS, MKN-74 and
SC-M1, were purchased from the Institute of Cell Biology, Shanghai,
China. Cells were maintained in RPMI-1640 medium (Invitrogen Life
Technologies, Carlsbad, CA, USA) containing 10% fetal bovine serum
(FBS; Invitrogen Life Technologies), streptomycin (500 μg/ml) and
penicillin (500 μg/ml), in a 5% CO2 atmosphere at
37°C.
Receptor ligand binding assay
Binding affinity of ATPR, ATRA and 9-cis-RA
(purchased from Sigma-Aldrich, St. Louis, MO, USA) to RAR-α and
RXR-α was assessed by time-resolved fluorescence resonance energy
transfer (TR-FRET) analysis using LanthaScreen™ TR-FRET RXR-α and
RAR-α Coactivator Assay kits (catalog nos. PV4797 and PV4409,
respectively; Invitrogen Life Technologies), following the
manufacturer’s instructions. In a cell-free system, the FRET signal
reflects the binding of the ligand to the nuclear receptor and
subsequent recruitment of coactivator peptides. Briefly, the
following 11 serial dilutions of ATPR, ATRA and 9-cis-RA
were prepared in RPMI-1640 medium from corresponding starting
solutions (10−3 M) in dimethyl sulfoxide (DMSO):
1×10−6, 2×10−7, 4×10−8,
8×10−9, 1.6×10−9, 3.2×10−10,
6.4×10−11, 1.28×10−11, 2.56×10−12,
5.12×10−13 and 1.02×10−13 M.
MTT assay
The SC-M1 and MKN-74 cells were seeded onto 96-well
plates (200 μl/well) at a density of 2.5×104 cells/ml
and cultured for 9–12 h. The cells were then incubated in medium
containing DMSO or 5, 10 and 500 μM ATPR or ATRA for 3 or 6 days.
In order to determine cell growth, cells were incubated with 20
μl/well MTT solution (5 mg/ml; Sigma-Aldrich) for 4 h at 5%
CO2 and 37°C. Following the removal of the medium, DMSO
was added to each well (100 μl/well) and incubated for 5 min with
continuous shaking. The optical density (OD) was detected at 540 nm
with a SpectraMax 190 microplate reader (Molecular Devices,
Sunnyvale, CA, USA). A 100% proliferation was assigned to
DMSO-treated cells, and the suppressive effects of ATRA and ATPR
(5, 10 and 500 μM) on cell growth were determined. The MTT assay
was performed in triplicate in 3 independent biological
repetitions.
Cell cycle analysis
The AGS and MKN-74 cells (3×105 cells)
were cultured in 6-well plates for 16 h, and were treated with ATRA
or ATPR (both at 10 and 50 μM). After 24 h, the medium was removed
to collect floating cells. The attached cells were trypsinized with
0.25% Trypsin/EDTA, pooled with the cells collected from the
culture medium, and fixed with cold (−20°C) ethanol overnight. The
fixed cells were washed twice in phosphate-buffered saline (PBS)
following centrifugation at 1,500 rpm for 5 min and stained with a
solution containing RNase A (100 μg/ml), 20 μg/ml propidium iodide
(PI) (both from Sigma-Aldrich) and 0.1% Triton X-100 in PBS for 30
min. The cells were washed twice in PBS prior to analysis on a
FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ,
USA). This experiment was performed at least 3 times
independently.
Western blot analysis
MKN-74 cells were treated with ATPR (10 and 50 μM)
as described above in the cell cycle analysis, and protein extracts
were harvested by incubation in cell lysis buffer (SoluLyse-M
Mammalian protein extraction reagent; Genlantis, San Diego, CA,
USA) in the presence of protease inhibitors (ProBlock Gold
Mammalian; Gold Biotechnology, Inc., St. Louis, MO, USA) on ice for
10 min. Chromosomal DNA was fragmented by ultrasonication. Protein
concentration was determined using the Bradford protein assay
(Bio-Rad, Hercules, CA, USA). Proteins (10 μg) were separated using
SDS-PAGE and transferred onto a nitrocellulose membrane. After
non-specific binding was blocked by incubation for 60 min in PBS
with Tween 20 (PBST) buffer supplemented with 5% skim milk, the
membrane was incubated with the following primary antibodies:
anti-cyclin E (Invitrogen Life Technologies), anti-Bcl-2 and
anti-Bax (both from Epitomics, Burlingame, CA, USA) in the
concentrations suggested by the corresponding manufacturers. The
expression of β-actin, serving as the loading control, was assessed
using an anti-β-actin antibody (Millipore, Billerica, MA, USA). The
membrane was developed in ECL reagent (Millipore) and visualized
using a CCD camera (Fusion FX7; Vilber Lourmat, Eberhardzell,
Germany). Following autoradiography, data from the films were
analyzed using GelQuant.NET software (available at www.biochemlabsolutions.com).
Analysis of AP-1 activity
The MKN-74 and SC-M1 cells (4×104 cells)
cultured in 96-well plates were transfected using Attractene
transfection reagent (Qiagen, Hilden, Germany) following the
manufacturer’s instructions. Briefly, the reagent was mixed with
either the Cignal AP-1 reporter (SABiosciences, Germany) or the
control reporters, TATA (negative control) and CMV (positive
control), along with the Renilla luciferase reporter
(Promega, Fitchburg, WI, USA) and the mixture was incubated at room
temperature for 20 min. Following incubation of the cells with the
transfection mixture for 16 h, selected cell groups were further
treated with phorbol ester
12-O-tetradecanoylphorbol-13-acetate (TPA, 100 ng/ml;
Sigma-Aldrich) for 6 h. All cells were then treated with 5, 10 or
500 μM ATRA or ATPR for 24 h. After the cells were washed twice
with PBS, they were incubated in passive lysis buffer for 15 min
with continuous shaking, and the luciferase assay reagent II was
added. Firefly luciferase activity was determined with a SpectraMax
190 microplate luminometer (Molecular Devices). The Stop & Glo
reagent (Promega) was then added to measure the Renilla
luciferase activity, as an internal control for the transfection
activity assay. The reporter activity was determined by normalizing
the values obtained for the firefly luciferase activity to those
obtained for the Renilla luciferase reporter activity. The
experiment was performed 3 times.
Statistical analyses
Normally-distributed continuous variables were
compared by a one-way analysis of variance (ANOVA). When a
significant difference between groups was observed, comparisons of
means were performed using the Bonferroni type I error adjustment.
Data are presented as the means ± SD. All statistical assessments
are derived from two-sided tests, with a P-value of 0.05 considered
to indicate a statistically significant difference. Statistical
analyses were performed using SPSS 15.0 software (SPSS Inc.,
Chicago, IL, USA).
Results
Determination of the binding affinity of
ATPR to RARs
As the receptor mediating the effects of ATPR
remains unknown, ligand binding assays were undertaken to examine
the binding affinity of ATPR to 2 RARs, RAR-α and RXR-α. The
binding affinities of 2 well-known RA analogs, 9-cis-RA and
ATRA, were used as the positive controls. 9-cis-RA bound to
both receptors with high affinity (0.34 nM to RAR-α and 0.37 nM to
RXR-α) (Fig. 1A and B). ATRA
preferentially bound to RAR-α (0.23 nM) (Fig. 1C) as compared to RXR-α (19 nM)
(Fig. 1D). Furthermore, ATPR
preferentially bound to RXR-α (0.04 nM) (Fig. 1F) as compared to RAR-α (20.96 nM)
(Fig. 1E).
Suppression of gastric cancer cell growth
by ATPR
To determine whether ATPR suppresses cancer cell
proliferation, 3 gastric cancer cell lines, AGS, MKN-74 and SC-M1,
were cultured in the presence of 5, 10 or 500 μM ATPR, ATRA or DMSO
(vehicle control) for 3 or 6 days. As illustrated in Fig. 2, ATRA and ATPR inhibited growth of
AGS, MKN-74 and SC-M1 cells in a dose-dependent manner. At 3 days,
ATPR induced a significantly greater inhibitory effect on cell
growth compared to ATRA, with the exception of the 10 μM-treated
AGS cells, in which both ATPR and ATRA exerted comparable
inhibitory effects (all P<0.05) (Fig. 2A). At 6 days, a greater cell
growth inhibitory effect was only observed in the MKN-74 cells
treated with 5 and 10 μM ATPR and in the SC-M1 cells treated with
500 μM ATRA (P<0.05) (Fig. 2B and
C).
Alteration of cell cycle progression by
ATPR
To determine whether ATRA or ATPR alters the cell
cycel progression, the AGS and MKN-74 cells were treated with 10 or
50 μM ATRA or ATPR for 24 h prior to PI staining and flow
cytometric analysis. Whereas ATRA did not significantly alter the
cell cycle distribution of the AGS and MKN-74 cells (Fig. 3A and B, respectively), 50 μM ATPR
significantly increased the population of AGS and MKN-74 cells at
the subG1 phase, suggesting the induction of apoptosis in this
treatment group (both P<0.05). Furthermore, 50 μM ATPR
significantly reduced the percentage of cells at the G0/G1 and G2/M
phases in both cell lines (both P<0.05).
Effects of ATPR on apoptosis-related
protein expression in MKN-74 cells
To evaluate the hypothesis that ATPR induces
apoptosis in MKN-74 cells, the expression of the apoptosis-related
proteins, cyclin E, Bcl-2 and Bax, in response to 10 and 50 μM ATPR
treatment was assessed by western blot analysis. The treatment of
MKN-74 cells with 50 μM ATPR reduced the cyclin E level and
upregulated Bax expression compared with the DMSO-treated group
(Fig. 4A). The Bcl-2/Bax ratio
was significantly reduced following treatment with 50 μM ATPR as
compared with the DMSO-treated group (P<0.05) (Fig. 4B); however, this effect was not
observed with 10 μM ATPR.
Suppressive effects of ATPR on AP-1
reporter activity
As a number of RA analogs suppress tumor growth
through the inhibition of AP-1 activity (4,5),
the effects of ATPR on AP-1 activity in MNK-74 and SC-M1 cells were
determined using a reporter gene construct with tandem repeats of
the AP-1 binding site upstream of the firefly luciferase gene.
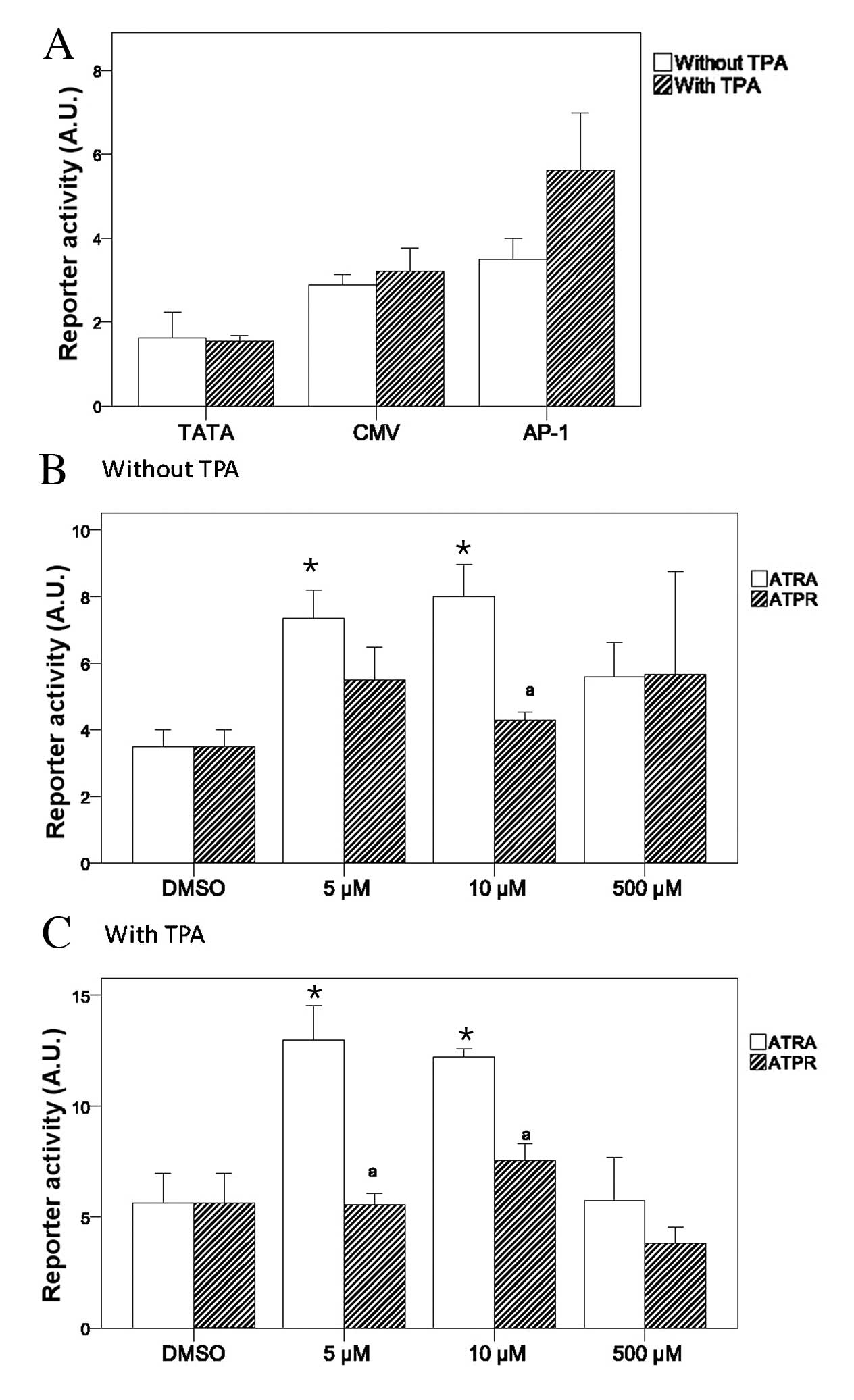
Compared with the reporter activities of the TATA
and CMV groups, the basal and TPA-induced AP-1 reporter activities
were relatively higher in the MNK-74 cells (Fig. 5A). Furthermore, the treatment of
MKN74 cells with TPA for 24 h significantly increased the AP-1
reporter activity as compared with the corresponding
non-TPA-treated group (P<0.05) (Fig. 5A). Comparable effects on the AP-1
reporter activity were observed upon treatment with various
concentrations of ATPR and ATRA in the absence of TPA (Fig. 5B). However, in the presence of
TPA, both 5 and 10 μM ATPR significantly suppressed AP-1 reporter
activity as compared with ATRA (Fig.
5C).
In the SC-M1 cells, the basal AP-1 reporter activity
appeared similar to that of the CMV group, and TPA failed to
significantly increase it (Fig.
6A). In the absence of TPA, 5 or 10 μM ATRA significantly
enhanced the AP-1 reporter activity compared with the corresponding
DMSO-treated group (Fig. 6B).
Although 10 μM ATPR significantly reduced the AP-1 reporter
activity as compared with the same concentration of ATRA, the
reduced AP-1 activity was comparable to that observed in the
corresponding DMSO-treated group (Fig. 6B). In the presence of TPA, 5 or 10
μM ATRA significantly enhanced the AP-1 reporter activity (Fig. 6C). Although 5 and 10 μM ATPR
reduced AP-1 reporter activity as compared with the corresponding
ATRA-treated groups, the reduction was again comparable to that
observed in the corresponding DMSO-treated group (Fig. 6C).
Discussion
The effect of a novel RA analog, ATPR, on gastric
cancer cell growth was evaluated in the present study. At 3 days,
ATPR inhibited the growth of MKN-74 and SC-M1 cells to a greater
extent compared with ATRA. ATPR further significantly increased the
population of cells at the subG1 phase and reduced the percentage
of cells at the G0/G1 and G2/M phases. The MKN-74 cells treated
with 50 μM ATPR showed a reduction in cyclin E levels and an
increased Bax protein expression. A reduction in TPA-induced AP-1
activity was also observed in the cells treated with ATPR.
As opposed to 9-cis-RA that bound to both
RAR-α and RXR-α with high affinity, and ATRA that preferentially
bound to RAR-α, binding assays revealed that ATPR preferentially
binds RXR-α. As previously described, the effects of retinoids can
be mediated by either RAR-RXR heterodimers or RXR-RXR homodimers
(3,22). The effects of a set of novel
selective RA analogs were examined by Kizaki et al (22); ligands that specifically activated
RAR/RXR heterodimers not only inhibited growth, but also induced
the differentiation of human leukemic cells, as opposed to analogs
that activated RXR homodimers alone (22). In human glioblastoma cell lines,
the inhibition of cell proliferation was observed upon ATRA
treatment, highlighting the importance of RAR activation in these
cells (23). Further studies are
required to determine whether ATPR-bound RXR-α reduces gastric
cancer cell growth and inhibits AP-1 activity through the induction
of RXR homodimerization and subsequent transrepression or through
heterodimerization with RAR, followed by RARE transactivation.
In the present study, ATPR inhibited gastric cancer
cell growth to a greater extent compared with ATRA. The
anti-proliferative effects of ATPR are consistent with those
reported in previous in vivo and in vitro studies
(24). ATPR reduced the cyclin E
level in MKN-74 cells, and altered cell cycle progression in the
AGS and MKN-74 cells; however, no such effects were observed
following treatment with ATRA. Wu et al (25) observed differences in cell cycle
progression upon treatment with ATRA in BGC-823 gastric cancer
cells. We speculate that this difference in results between the
studies is due to differences in the examined cell types
(differential RAR expression), as well as in the concentration of
ATRA used. In vivo analyses revealed that ATRA inhibited the
growth and metastasis of gastric cancer cell xenografts, as well as
the expression of proteins associated with metastasis, including
nm23, mts1/p16 and ICAM-1 (25).
Furthermore, reduced microvessel formation was observed with ATRA
treatment (25), which may be
related to inhibition, by ATRA, of the mRNA and protein expression
of the vascular endothelial growth factor, observed in gastric
cancer cells (26).
AP-1 transactivation is associated with cell growth,
proliferation and survival, and the transrepression of its activity
in the absence of RARE transactivation is a well-characterized
anticancer retinoid effect (4,27,28). In the present study, both ATPR and
ATRA inhibited basal and TPA-induced AP-1 activity, in agreement
with previous reports on BGC-823 gastric cancer cells (29). However, this effect was not
observed in the MKN-45 cells, which are negative for RAR-β
expression, suggesting that RAR-β may act as the mediator of
retinoid-induced AP-1 transrepression (29). Alternatively, in the SC-M1 cells,
lower concentrations of ATRA actually increased AP-1 activity.
In the present study, ATPR altered cell cycle
progression by inducing apoptosis (i.e., increased the population
of cells at the subG1 phase). Quantification of the levels of the
apoptosis-related proteins, Bax and Bcl-2, indicated that ATPR
reduced the Bcl-2/Bax ratio. However, no such effects were observed
following treatment with ATRA (data not shown). Bcl-2 promotes cell
survival and is overexpressed in many tumor types (30), and its inhibition by small
molecules may be beneficial for cancer treatment (30,31). The reduced Bcl-2/Bax ratio is
associated with the activation of caspase-3 and subsequent
apoptosis (32,33). In our study, in addition to the
reduced Bcl-2/Bax ratio, a lower cyclin E level was observed upon
treatment with 50 μM ATPR. The cyclin E level not only reflects the
proliferative state of cells, but may also determine the extent of
apoptosis (34). Specifically,
p18-cyclin E, which results from the N-terminal truncation of
cyclin E in tumor cells, can induce apoptosis independently of Cdk2
(34). Further studies are
required to assess whether the reduced cyclin E level following
ATPR treatment results in a concomitant increase in the p18-cyclin
E level. Although the present study did not directly assess the
effects of ATPR on apoptosis (which will be the subject of a future
study e.g., by TUNEL assay), its effects on cell cycle progression
and on the Bcl-2/Bax ratio suggest that it induces the apoptosis of
gastric cancer cells.
In addition to influencing cell growth and
apoptosis, RA analogs, including ATPR, can induce cell
differentiation in embryonic and adult tissues (1,2,24).
Similarly, RA and a number of its analogs inhibit tumor progression
by inducing cell differentiation (16), possibly through
phosphatidylinositol 3-kinase (PI3K) signaling (35). Although the present study did not
analyze the effects of ATPR on gastric cancer cell differentiation,
this will be analyzed in future studies using an in vivo
model of gastric cancer. The effects of ATPR on tumor formation and
morphology will also be assessed using in vivo tumor
xenografts.
In conclusion, we demonstrate that ATPR inhibits
cancer cell proliferation, induces cell cycle arrest and inhibits
the activity of AP-1 to a greater extent compared with ATRA. These
data form the basis of future analyses aiming to determine its
efficacy in vivo.
Acknowledgements
The present study was supported by the following
grants: National ‘Major Drug Discovery’ Major Science and
Technology Funding of China (no. 2011ZX09401-021); Natural Science
Foundation of the Department of Education, Anhui Province (no.
KJ2010A171); Natural Science Foundation of the Bureau of Health,
Anhui Province (no. 09C157).
Abbreviations:
|
ATPR
|
4-amino-2-trifluoromethyl-phenyl
retinate
|
|
ATRA
|
all-trans retinoic acid
|
|
RA
|
retinoic acid
|
|
RAR-α
|
retinoic acid receptor-α
|
|
RXR-α
|
retinoid X receptor-α
|
|
RARE
|
RA response elements
|
|
AP-1
|
activator protein-1
|
|
TPA
|
12-O-tetradecanoylphorbol-
13-acetate
|
References
|
1
|
Campo-Paysaa F, Marlétaz F, Laudet V and
Schubert M: Retinoic acid signaling in development: tissue-specific
functions and evolutionary origins. Genesis. 46:640–656. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mark M, Ghyselinck NB and Chambon P:
Function of retinoic acid receptors during embryonic development.
Nucl Recept Signal. 7:e0022009.PubMed/NCBI
|
|
3
|
McKenna NJ and O’Malley BW: Combinatorial
control of gene expression by nuclear receptors and coregulators.
Cell. 108:465–474. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li JJ, Dong Z, Dawson MI and Colburn NH:
Inhibition of tumor promoter-induced transformation by retinoids
that transrepress AP-1 without transactivating retinoic acid
response element. Cancer Res. 56:483–489. 1996.PubMed/NCBI
|
|
5
|
Liu S, Wu Q, Chen ZM and Su WJ: The effect
pathway of retinoic acid through regulation of retinoic acid
receptor alpha in gastric cancer cells. World J Gastroenterol.
7:662–666. 2001.PubMed/NCBI
|
|
6
|
Sundqvist A, Zieba A, Vasilaki E, Herrera
Hidalgo C, Söderberg O, Koinuma D, Miyazono K, Heldin CH, Landegren
U, Ten Dijke P and van Dam H: Specific interactions between Smad
proteins and AP-1 components determine TGFβ-induced breast cancer
cell invasion. Oncogene. 32:3606–3615. 2013.PubMed/NCBI
|
|
7
|
McKenna NJ: EMBO Retinoids 2011:
Mechanisms, biology and pathology of signaling by retinoic acid and
retinoic acid receptors. Nucl Recept Signal. 10:e0032012.PubMed/NCBI
|
|
8
|
Kilewer SA, Umesono K, Noonan DJ, Heyman
RA and Evans RM: Covergence of 9-cis retinoic acid and peroxisome
proliferator signaling pathways through heterodimer formation of
their receptors. Nature. 358:771–774. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dierov J, Sawaya BE, Prosniak M and
Gartenhaus RB: Retinoic acid modulates a bimodal effect on cell
cycle progression in human adult T-cell leukemia cells. Clin Cancer
Res. 5:2540–2547. 1999.PubMed/NCBI
|
|
10
|
Tillmanns TD, Kamelle SA, Guruswamy S,
Gould NS, Rutledge TL and Benbrook DM: Sensitization of cervical
cancer cell lines to low-dose radiation by retinoic acid does not
require functional p53. Gynecol Oncol. 97:142–150. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lehmann S, Ravn A, Carlsson L, Antunovic
P, Deneberg S, Möllgård L, Derolf AR, Stockelberg D, Tidefelt U,
Wahlin A, Wennström L, Höglund M and Juliusson G: Continuing high
early death rate in acute promyelocytic leukemia: a
population-based report from the Swedish Adult Acute Leukemia
Registry. Leukemia. 25:1128–1134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pagoni M, Garofalaki M, Panitsas F, Manola
K, Psarra K, Economopoulos P, Vourtsi A, Antoniades M, Gkirkas K,
Tzouvara E, Katis F, Prokopiou C, Tziotziou I, Balta A, Lemissiou
E, Tsirigotis P, Repoussis P and Harhalakis N: Acute promyelocytic
leukemia: an experience on 95 greek patients treated in the
all-trans-retinoic acid era. Mediterr J Hematol Infect Dis.
3:e20110532011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Park JH, Qiao B, Panageas KS, Schymura MJ,
Jurcic JG, Rosenblat TL, Altman JK, Douer D, Rowe JM and Tallman
MS: Early death rate in acute promyelocytic leukemia remains high
despite all-trans retinoic acid. Blood. 118:1248–1254. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Latagliata R, Petti MC, Fenu S, Mancini M,
Spiriti MA, Breccia M, Brunetti GA, Avvisati G, Lo Coco F and
Mandelli F: Therapy-related myelodysplastic syndrome-acute
myelogenous leukaemia in patients treated for acute promyelocytic
leukaemia: an emerging problem. Blood. 99:822–824. 2002. View Article : Google Scholar
|
|
15
|
Zompi S and Viguie F: Therapy-related
acute myeloid leukaemia and myelodysplasia after successful
treatment of acute promyelocytic leukaemia. Leuk Lymphoma.
43:275–280. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Simoni D, Rondanin R, Baruchello R,
Roberti M, Rossi M, Grimaudo S, D’Alessandro N, Invidiata FP and
Tolomeo M: Retinoic acid and analogs as potent inducers of
differentiation and apoptosis. New promising chemopreventive and
chemotherapeutic agents in oncology. Pure Appl Chem. 73:1437–1444.
2001. View Article : Google Scholar
|
|
17
|
Adamson PC: All-trans-retinoic acid
pharmacology and its impact on the treatment of acute promyelocytic
leukemia. Oncologist. 1:305–314. 1996.PubMed/NCBI
|
|
18
|
Allenby G, Bocquel MT, Saunders M, Kazmer
S, Speck J, Rosenberger M, Lovey A, Kastner P, Grippo JF, Chambon P
and Levin AA: Retinoic acid receptors and retinoid X receptors
interactions with endogenous retinoic acids. Proc Natl Acad Sci
USA. 90:30–34. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tang J, Yao J, Shi J, Xiao Q, Zhou J and
Chen F: Synthesis, characterization, drug-loading capacity and
safety of novel pH-independent amphiphilic amino acid copolymer
micelles. Pharmazie. 67:756–764. 2012.PubMed/NCBI
|
|
20
|
Ahn HK, Jang J, Lee J, Se Hoon P, Park JO,
Park YS, Lim HY, Kim KM and Kang WK: P21-activated kinase 4
overexpression in metastatic gastric cancer patients. Transl Oncol.
4:345–349. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li CJ, Chu CY, Huang LH, Wang MH, Sheu LF,
Yeh JI and Hsu HY: Synergistic anticancer activity of triptolide
combined with cisplatin enhances apoptosis in gastric cancer in
vitro and in vivo. Cancer Lett. 319:203–213. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kizaki M, Dawson MI, Heyman R, Elster E,
Morosetti R, Pakkala S, Chen DL, Ueno H, Chao W, Morikawa M, Ikeda
Y, Heber D, Pfahl M and Koeffler HP: Effects of novel retinoid X
receptor-selective ligands on myeloid leukemia differentiation and
proliferation in vitro. Blood. 87:1977–1984. 1996.PubMed/NCBI
|
|
23
|
Zang C, Wächter M, Liu H, Posch MG, Fenner
MH, Stadelmann C, von Deimling A, Possinger K, Black KL, Koeffler
HP and Elstner E: Ligands for PPARgamma and RAR cause induction of
growth inhibition and apoptosis in human glioblastomas. J
Neurooncol. 65:107–118. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang N, Ge JF, Pan CX, Peng XQ, Chen HH,
Wang XQ, Tang J, Hu W and Chen FH: Anti-tumor effect of
4-Amino-2-Trifluoromethyl-Phenyl Retinate on human breast cancer
MCF-7 cells via up-regulation of retinoid receptor-induced gene-1.
Biomed Pharmacother. 67:687–692. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu Q, Chen YQ, Chen ZM, Chen F and Su WJ:
Effects of retinoic acid on metastasis and its related proteins in
gastric cancer cells in vivo and in vitro. Acta Pharmacol Sin.
23:835–841. 2002.PubMed/NCBI
|
|
26
|
Zhang JP, Chen XY and Li JS: Effects of
all-trans-retinoic on human gastric cancer cells BGC-823. J Dig
Dis. 8:29–34. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Agadir A, Shealy YF, Hill DL and Zhang X:
Retinyl methyl ether down-regulates activator protein 1
transcriptional activation in breast cancer cells. Cancer Res.
57:3444–3450. 1997.PubMed/NCBI
|
|
28
|
Wan H, Dawson MI, Hong WK and Lotan R:
Enhancement of Calu-1 human lung carcinoma cell growth in
serum-free medium by retinoids: dependence on AP-1 activation, but
not on retinoid response element activation. Oncogene.
15:2109–2118. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu Q, Chen ZM and Su WJ: Anticancer effect
of retinoic acid via AP-1 activity repression is mediated by
retinoic acid receptor alpha and beta in gastric cancer cells. Int
J Biochem Cell Biol. 34:1102–1114. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cory S and Adams JM: Killing cancer cells
by flipping the Bcl-2/Bax switch. Cancer Cell. 8:5–6. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Reed JC: Proapoptotic multidomain
Bcl-2/Bax-family proteins: mechanisms, physiological roles, and
therapeutic opportunities. Cell Death Differen. 13:1378–1386. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tacar O, Sriamornsak P and Dass CR:
Doxorubicin: an update on anticancer molecular action, toxicity and
novel drug delivery systems. J Pharm Pharmacol. 65:157–170. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Albano F, Arcucci A, Granato G, Romano S,
Montagnani S, De Vendittis E and Ruocco MR: Markers of
mitochondrial dysfunction during the diclofenac-induced apoptosis
in melanoma cell lines. Biochimie. 95:934–945. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mazumder S, Plesca D and Almasan A: A
jekyll and hyde role of cyclin E in the genotoxic stress response:
switching from cell cycle control to apoptosis regulation. Cell
Cycle. 6:1437–1442. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
López-Carballo G, Moreno L, Masiá S, Pérez
P and Barettino D: Activation of the phosphatidylinositol
3-kinase/Akt signaling pathway by retinoic acid is required for
neural differentiation of SH-SY5Y human neuroblastoma cells. J Biol
Chem. 277:25297–25304. 2002.
|