Introduction
Microglia are resident macrophage-like cells in the
brain, and have been suggested to play a major role in host defense
and tissue repair in the central nervous system (CNS) (1,2).
Under pathological conditions, activated microglia release
pro-inflammatory mediators, including nitric oxide (NO),
prostaglandin E2 (PGE2), reactive oxygen
species (ROS) and pro-inflammatory cytokines (2,3).
The overproduction of these inflammatory mediators and cytokines
causes severe forms of various neurodegenerative diseases, such as
Alzheimer’s disease (AD), cerebral ischemia, multiple sclerosis and
trauma. Not surprisingly, activated microglia have been shown to be
a major cellular source of pro-inflammatory and/or cytotoxic
factors that cause neuronal damage in the CNS (4–6).
Thus, a decrease in the number of pro-inflammatory mediators and
cytokines in microglial cells may attenuate the severity of these
disorders (7–9).
Flavonoids are polyphenolic compounds that are
present in high concentrations in fruits and vegetables (10–12). They have multiple pharmacological
activities, such as antioxidant, anti-inflammatory,
immunomodulatory and antitumor effects (13–15). A number of studies have
demonstrated that flavonoids protect against neuronal cell death,
enhance existing neuronal function and stimulate neuronal
regeneration (16–18). Among these flavonoids,
7,8-dihydroxyflavone (7,8-DHF) is a selective tyrosine kinase
receptor B (TrkB) agonist that can cross the blood-brain barrier
(19–21). In a recent study, this flavonoid
was demonstrated to have neurotrophic activities in various
neurological diseases, such as stroke and Parkinson’s disease
(19). Although several studies
have indicated that 7,8-DHF has antioxidant and anti-inflammatory
activities (21–24), to our knowledge, no studies to
date have investigated the molecular mechanisms underlying its
anti-inflammatory effects in microglial cells.
In the present study, we investigated the inhibitory
effects of 7,8-DHF on the production of inflammatory mediators and
the mechanisms through which it induces these anti-inflammatory
effects on lipopolysaccharide (LPS)-stimulated murine BV2
microglial cells.
Materials and methods
Materials
7,8-DHF was obtained from Professor Jin Won Hyun of
Jeju National University (Jeju, Korea). LPS, Tween-20, bovine serum
albumin (BSA) and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO,
USA). Rabbit anti-inducible nitric oxide synthase (iNOS),
cyclooxygenase (COX)-2, nuclear factor-κB (NF-κB) p65 and IκB-α
polyclonal antibodies were purchased from Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA). Antibodies against lamin B,
extracellular signal-regulated kinase (ERK), phosphorylated
(p)-ERK, p38 mitogen-activated protein kinase (MAPK), p-p38 MAPK,
c-Jun N-terminal kinase (JNK), and p-JNK were purchased from Cell
Signaling Technology (Danvers, MA, USA). The peroxidase-labeled
donkey anti-rabbit immunoglobulin and peroxidase-labeled sheep
anti-mouse immunoglobulin were purchased from Amersham Corp.
(Arlington Heights, IL, USA). Dulbecco’s modified Eagle’s medium
(DMEM) containing l-glutamine (200 mg/l), fetal bovine serum (FBS),
penicillin and streptomycin, Triton X-100, and all other chemicals
were purchased from Gibco (Grand Island, NY, USA).
Cell culture
BV2 microglial cells were maintained in DMEM
supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml
streptomycin at 37°C in a humidified incubator with 5%
CO2. Confluent cultures were passaged by trypsinization.
Cells used in the experiments were washed twice with warm DMEM
(without phenol red) and treated in serum-free medium for at least
4 h prior to the treatments. In all experiments, cells were treated
with various concentrations of 7,8-DHF for the indicated times
prior to the addition of LPS (0.5 μg/ml).
Cell viability assay
Cell viability was measured based on the formation
of blue formazan that was metabolized from colorless MTT by
mitochondrial dehydrogenases, which are active only in live cells.
In brief, BV2 cells (3×105 cells/well) were seeded and
treated with various reagents for the indicated periods of time.
After the various treatments, the medium was removed and the cells
were incubated with 0.5 mg/ml of MTT solution. Following incubation
for 2 h at 37°C and 5% CO2, the supernatant was removed
and the formation of formazan was measured at 540 nm using a
microplate reader (Dynatech MR-7000; Dynatech Laboratories,
Chantilly, VA, USA).
NO production
Concentrations of NO in the culture supernatants
were determined by measuring nitrite, which is a major stable
product of NO, using Griess reagent (Sigma-Aldrich Chemical Co.).
Cells (5×105 cells/ml) were stimulated in 24-well plates
for 24 h, and then 100 μl of each culture medium was mixed with an
equal volume of Griess reagent (1% sulfanilamide/0.1%
N-(1-naphthyl)-ethylenediamine dihydrochloride/2.5%
H3PO4). Nitrite levels were determined using
an ELISA plate reader at 540 nm, and nitrite concentrations were
calculated by reference to a standard curve generated by known
concentrations of sodium nitrite (25).
RNA isolation and RT-PCR
Total RNA was isolated using TRIzol reagent
(Invitrogen, Carlsbad, CA, USA). Total RNA (1.0 μg) obtained from
the cells was reverse-transcribed using M-MLV reverse transcriptase
(Promega, Madison, WI, USA) to produce cDNAs. The iNOS,
COX-1, COX-2, IL-1β and TNF-α genes
were amplified from the cDNA by PCR. The PCR primers were as
follows: mouse iNOS (5′-ATG TCC GAA GCA AAC ATC AC-3′ and 5′-TAA
TGT CCA GGA AGT AGG TG-3′); COX-2 (5′-CAG CAA ATC CTT GCT GTT CC-3′
and 5′-TGG GCA AAG AAT GCA AAC ATC-3′); IL-1β (5′-ATG GCA ACT GTT
CCT GAA CTC AAC T-3′ and 5′-TTT CCT TTC TTA GAT ATG GAC AGG AC-3′);
and TNF-α (5′-ATG AGC ACA GAA AGC ATG ATC-3′ and 5′-TAC AGG CTT GTC
ACT CGA ATT-3′). Following amplification, the PCR reactions were
electrophoresed on 1% agarose gels.
Protein extraction and western blot
analysis
The cells were washed 3 times with
phosphate-buffered saline (PBS) and lysed in lysis buffer (1%
Triton X-100, 1% deoxycholate and 0.1% NaN3) containing
protease inhibitor cocktail tablets (Roche Diagnostics GmbH,
Mannheim, Germany). In a parallel experiment, nuclear proteins were
prepared using nuclear extraction reagents (Pierce, Rockford, IL,
USA) according to the manufacturer’s instructions. Equal amounts of
protein were separated on SDS-polyacrylamide gels, transferred onto
nitrocellulose membranes (Schleicher and Schuell, Keene, NH, USA)
by electroblotting, and subsequently blocked in 5% bovine serum
albumin (BSA)-Tris-buffered saline Tween-20 (TBST, 100 mM Tris, pH
8.0, 150 mM NaCl and 0.1% Tween-20) for 1 h at room temperature.
Following incubation with the appropriate primary antibodies for 1
h, the membranes were incubated for 1 h at room temperature with
secondary antibodies conjugated to horseradish peroxidase.
Following 3 washes in TBST, immunoreactive bands were visualized
using an ECL detection system (Pierce).
Cytokine assays
The levels of IL-1β and TNF-α were measured by ELISA
kits (R&D Systems, Minneapolis, MN, USA) according to the
manufacturer’s instructions. Briefly, the BV2 microglial cells
(5×105 cells/ml) were plated in 24-well plates and
pre-treated with the indicated concentrations of 7,8-DHF for 1 h
prior to treatment with 0.5 μg/ml LPS for 24 h. One hundred
microliters of culture-medium supernatants were collected for
determination of the IL-1β and TNF-α concentrations by ELISA, as
previously described (26).
NF-κB luciferase assay
A total of 1×106 BV2 cells were
transfected with 2 μg NF-κB-luciferase reporter plasmids (BD
Biosciences, San Jose, CA, USA) using Lipofectamine according to
the manufacturer’s instructions (Gibco). Following incubation with
DNA-Lipofectamine mixtures, the cells were pre-incubated in the
presence or absence of 7,8-DHF for 1 h prior to being stimulated
with LPS for 0.5 or 1 h. The cells were then washed twice with PBS
and lysed with reporter lysis buffer (Promega). After vortexing and
centrifugation at 12,000 × g for 1 min at 4°C, the supernatant was
stored at −70°C for use in the luciferase assay. After 20 μl of the
cell extract was mixed with 100 μl of the luciferase assay reagent
at room temperature, the mixture was measured on a microplate
luminometer LB96V (Perkin-Elmer, Foster City, CA, USA).
Statistical analyses
Data values represent the means ± standard deviation
(SD). Statistical significance was determined using an analysis of
variance that was followed by a Student’s t-test. A value of
P<0.05 was considered to indicate a statistically significant
difference.
Results
7,8-DHF attenuates NO and PGE2
production in LPS- stimulated BV2 microglial cells
To evaluate the inhibitory effects of 7,8-DHF on
LPS-stimulated NO production in BV2 microglial cells, NO levels in
the cell culture medium were measured by Griess assay. For this
experiment the, BV2 microglial cells were pre-treated with various
concentrations of 7,8-DHF (10–70 μM) for 1 h prior to being
stimulated with LPS (0.5 μg/ml). According to the NO detection
assay, LPS alone was able to markedly induce NO production by the
cells. Pre-treatment with 7,8-DHF significantly repressed the
levels of NO production in LPS-stimulated BV2 microglial cells in a
concentration-dependent manner (Fig.
1A).
As PGE2 represents another important
inflammatory mediator that is produced from the conversion of
arachidonic acid by COXs, we then evaluated the inhibitory effects
of 7,8-DHF on PGE2 levels present in the supernatant by
ELISA under the same conditions. The amount of PGE2
present in the culture medium increased from the initial levels
after 24 h of exposure to LPS alone. A marked repression was
observed following the administration of 7,8-DHF (Fig. 1B).
In order to exclude the cytotoxic effects of 7,8-DHF
on the growth of BV2 microglial cells, the cells were exposed to
various concentrations of 7,8-DHF for 24 h in the presence or
absence of LPS, and cell viability was then measured by MTT assay.
The concentrations of 7,8-DHF used to inhibit NO and
PGE2 production did not affect cell viability (Fig. 2). These results clearly indicated
that the inhibition of NO and PGE2 production in
LPS-stimulated BV2 cells was not due to a cytotoxic action of
7,8-DHF.
7,8-DHF inhibits LPS-stimulated iNOS and
COX-2 expression in LPS-stimulated BV2 microglial cells
We performed RT-PCR and western blot analyses to
determine whether the inhibition of NO and PGE2
production by 7,8-DHF in LPS-stimulated BV2 cells was associated
with the decreased levels of iNOS and COX-2 expression. The levels
of iNOS and COX-2 proteins were markedly induced after 24 h of
exposure to LPS, and 7,8-DHF significantly inhibited iNOS and COX-2
protein expression in the LPS-stimulated BV2 microglial cells in a
concentration-dependent manner (Fig.
3A). Next, to determine whether or not 7,8-DHF suppresses the
LPS-mediated induction of iNOS and COX-2 at the transcriptional
level, the effects of 7,8-DHF on iNOS and COX-2 mRNA expression
were evaluated. The RT-PCR data showed that the reduction in
iNOS and COX-2 mRNAs correlated with the reduction in
the corresponding protein levels (Fig. 3B). These results suggested that
the 7,8-DHF-induced reductions in the expression of iNOS and
COX-2 were the cause of the inhibition of NO and
PGE2 production.
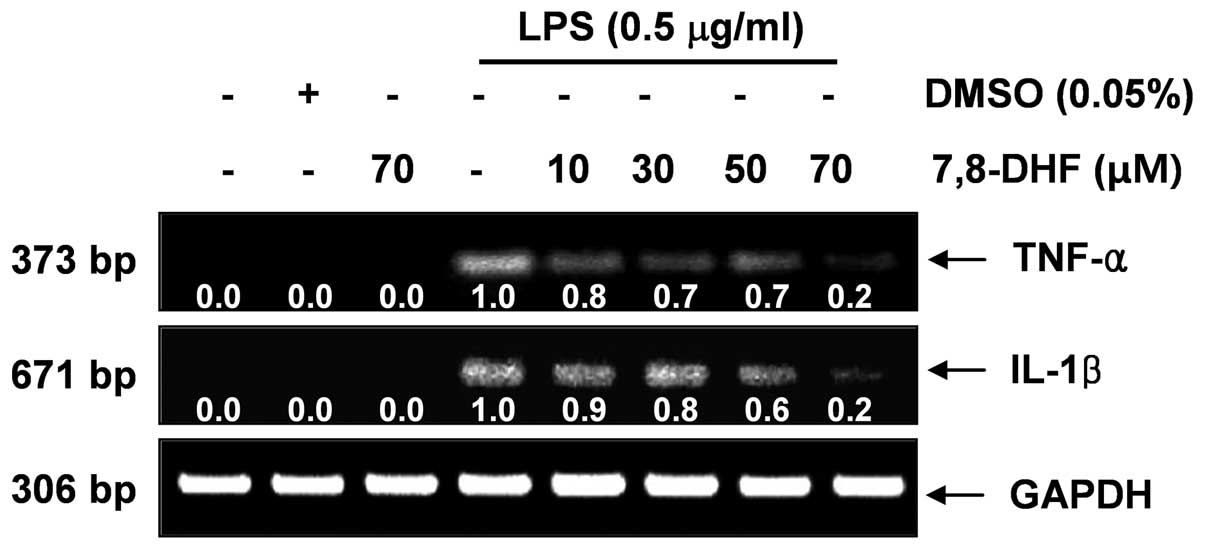
7,8-DHF suppresses LPS-induced IL-1β and
TNF-α production in LPS-stimulated BV2 microglial cells
Next, we analyzed the effects of 7,8-DHF on the
production of pro-inflammatory cytokines, such as IL-1β and TNF-α.
The levels of IL-1β and TNF-α production were markedly increased in
the culture medium of LPS-stimulated BV2 microglial cells (Fig. 4). Pre-treatment with 7,8-DHF
resulted in a significant decrease in the release of these
pro-inflammatory cytokines in a concentration-dependent manner. In
a parallel experiment, using RT-PCR, we investigated the effects of
7,8-DHF on LPS-induced IL-1β and TNF-α mRNA
expression. IL-1β and TNF-α mRNA transcription also
decreased following treatment with 7,8-DHF (Fig. 5). These results suggest that
7,8-DHF is effective in the suppression of pro-inflammatory
cytokine production through the alteration of the transcription
levels of IL-1β and TNF-α in activated microglial cells.
7,8-DHF blocks LPS-induced NF-κB activity
in LPS-stimulated BV2 microglial cells
NF-κB is one of the important transcription factors
that regulate the gene expression of pro-inflammatory mediators;
therefore, we wished to determine whether 7,8-DH affects NF-κB
activity. The results from immunoblot analysis shown in Fig. 6A revealed that the amount of NF-κB
p65 in the nucleus was markedly increased following exposure to LPS
alone. LPS-induced p65 levels in the nuclear fractions were reduced
by 7,8-DHF pre-treatment (Fig.
7A). In addition, IκB-α was markedly degraded at 15 min
following exposure to LPS; however this LPS-induced IκB-α
degradation was significantly reversed by 7,8-DHF. These results
suggest that 7,8-DHF inhibits NF-κB activation in BV2 microglial
cells through the suppression of IκB degradation and the nuclear
translocation of NF-κB.
We then tried to confirm the inhibition of
LPS-induced NF-κB activation by 7,8-DHF using a luciferase assay.
For this experiment, the BV2 cells transfected with
NF-κB-luciferase reporter plasmids were pre-treated with 7,8-DHF
for 1 h and stimulated with LPS for 0.5 or 1 h, and then luciferase
activity was measured. LPS markedly enhanced NF-κB activity up to
approximately 8-fold over the basal level, while 7,8-DHF
significantly inhibited the LPS-induced increase in NF-κB activity
(Fig. 6B). Taken together, the
above findings show that the anti-inflammatory effects of 7,8-DHF
in LPS-stimulated BV2 cells involve the NF-κB pathway.
7,8-DHF reduces the LPS-induced
phosphorylation of MAPKs in LPS-stimulated BV2 microglial
cells
MAPK pathways are known to be important for the
expression of pro-inflammatory mediators and cytokines. Therefore,
MAPKs act as specific targets for inflammatory responses. To
examine whether the inhibition of inflammation by 7,8-DHF is
mediated through MAPK pathways, we examined the effects of 7,8-DHF
on the LPS-induced phosphorylation of p38 MAPK, ERK and JNK in BV2
microglial cells by western blot analysis. 7,8-DHF attenuated the
LPS-induced phosphorylation of these kinases (Fig. 7). By contrast, the levels of total
MAPK proteins were unaffected by either LPS or 7,8-DHF treatment.
These results suggest that the activation of MAPKs may be involved
in the inhibitory effects of 7,8-DHF on LPS-induced
pro-inflammatory mediators in BV2 microglial cells.
Discussion
The present results reveal that 7,8-DHF inhibits the
levels of the pro-inflammatory mediators, iNOS and COX-2, induced
by LPS, as well as the production of cytokines, such as IL-1β and
TNF-α in activated murine BV2 microglial cells through the blockade
of NF-κB and of MAPK signaling pathways.
Inflammation in the brain caused by activated
microglia plays an important role in the pathology of
neurodegenerative disorders, such as AD, cerebral ischemia,
multiple sclerosis and amyotrophic lateral sclerosis (7,27).
Neuroinflammation with prolonged microglial activation leads to an
increase in pro-inflammatory mediators and neurotoxic compounds,
including NO, PGE2, ROS, complement factors and
pro-inflammatory cytokines such as IL-1β, IL-6 and TNF-α, and
subsequently contributes to neuronal dysfunction and neuronal loss
(28,29). Thus, the suppression of
neuroinflammation and microglial activation process would
theoretically attenuate the progression of neurodegenerative
diseases. Accordingly, the inhibition of pro-inflammatory mediators
and cytokines by 7,8-DHF shown in the present study may play a
beneficial role in the treatment of neurodegenerative diseases.
Among pro-inflammatory mediators released by
microglia, NO and PGE2 are the main cytotoxic mediators
participating in the innate immune response in mammals. NO is
synthesized from three different isoforms of NOS: endothelial NOS
(eNOS), neuronal NOS (nNOS) and iNOS. Among these, iNOS is not
usually expressed in the brain. However, activated microglial cells
are a major cellular source of iNOS in the brain. The excessive
release of NO by activated microglial cells correlates with the
progression of neurodegenerative disorders (30,31). COXs are the enzymes that catalyze
the conversion of arachidonic acid to prostaglandin H2
(PGH2), which is the precursor of a variety of
biologically active mediators, such as PGE2,
prostacyclin and thromboxane A2 (32). COXs exist as two major isozymes:
COX-1, a constitutive COX, and COX-2, an isoform that is induced
during the response to many stimulants and is activated at the site
of the inflammation (33).
Several studies have reported that COX-2 is associated with
cytotoxicity in brain diseases as the inhibition of COX-2 induction
and/or activity reduces brain injury following ischemia and
attenuates the progression of neurodegenerative disorders (34–36). Thus, agents that inhibit the
production of these inflammatory mediators have been previously
considered as potential candidates for anti-inflammatory agents.
The results of this study demonstrated that 7,8-DHF inhibited NO
and PGE2 production through the suppression of iNOS and
COX-2 expression, respectively, which appears to be due to the
suppression of these genes at the transcriptional level (Figs. 1 and 3). The inhibitory effects of 7,8-DHF on
the LPS-induced production of NO and PGE2 were not due
to the cytotoxicity of 7,8-DHF, as assessed by MTT assays (Fig. 2).
Our results also revealed that 7,8-DHF significantly
attenuated the production of pro-inflammatory cytokines, including
TNF-α and IL-1β (Fig. 4). Since
the neuroinflammatory response in activated microglia produces
elevated levels of these cytokines, and they have been shown to
induce neuronal cell damage, suppressing their production is
important for the prevention of neurodegenerative diseases
(37–39). TNF-α is primarily produced by
activating monocytes, macrophages and T cells. The major producers
of TNF-α in the brain are microglial cells, and they may play a
role in many pathological conditions in the brain (40,41). Moreover, TNF-α overexpression has
been implicated in the pathogenesis of several human CNS disorders
(3,42,43). IL-1β is also a potent
pro-inflammatory cytokine that acts through the IL-1 receptors
found on numerous cell types, including neurons and microglia. This
cytokine is an important mediator of neuroimmune interactions that
participate directly in neurodegeneration (44,45). IL-1β may interact with other
molecules that are either released or induced in response to
damage, or it may affect only compromised neurons. Thus, the
inhibition of cytokine production or function serves as a key
mechanism in the control of neurodegeneration. Taken together, our
results indicate that 7,8-DHF could be a promising therapeutic
candidate for neurodegenerative diseases caused by microglial
activation in the brain.
Various intracellular signaling pathways are
involved in the modulation of inflammatory responses. NF-κB and
MAPK pathways are amongst the most important signaling molecules
involved in the production of pro-inflammatory mediators and
cytokines (46,47). NF-κB, as a result of its key role
in several pathologic conditions, is a major drug target in a
variety of diseases. NF-κB is also a primary regulator of genes
that are involved in the production of pro-inflammatory cytokines
and enzymes involved in the inflammatory process. It is well known
that the blockade of NF-κB transcriptional activity in the
microglial nucleus can also suppress the expression of iNOS, COX-2
and pro-inflammatory cytokines (48,49). In unstimulated cells, NF-κB is
retained in the cytoplasm by binding to IκB inhibitors. Upon
activation by stimuli, IκB is sequentially phosphorylated by IκB
kinases and then degraded by the proteasome (50,51). This process provides for the
availability of free NF-κB in the cytoplasm and allows for the
translocation of NF-κB proteins from the cytoplasm to the nuclei.
In this study, 7,8-DHF significantly attenuated LPS-induced IκB-α
degradation, and inhibited the nuclear translocation of the p65
subunit of NF-κB and transcriptional activity of NF-κB in BV2
microglia (Fig. 6). Therefore,
the inhibition of NF-κB signaling pathways in microglial cells by
7,8-DHF may result in the downregulation of pro-inflammatory
mediators, resulting in an anti-inflammatory effect.
As major signaling pathways, MAPKs are known to be
involved in the LPS-induced production of COX-2 and iNOS through
the control of NF-κB activation in microglial cells (51,52). LPS has been demonstrated to
activate three types of MAPKs, including p38 MAPK, ERK and JNK in
microglia. According to accumulating data, MAPKs signaling pathways
are involved in LPS-induced IL-1β transcription and production in
microglia and other cells (53,54). Furthermore, COX-2 was upregulated
by IL-1β via MAPKs signaling pathways in various cell types
(7), and IL-1β induced iNOS and
NO production in C6 astrocytoma cells (54). Hence, in this study, we further
evaluated the effectd of 7,8-DHF on the activation of three MAPKs
induced by LPS in microglial cells. LPS increased the activation of
MAPKs, including p38 MAPK, ERK and JNK, whereas 7,8-DHF decreased
the LPS-induced activation of these kinases (Fig. 7). These results suggest that the
7,8-DHF-mediated attenuation of pro-inflammatory mediators and
cytokines is associated with the inactivation of MAPK signaling
pathways, suggesting that the inhibition of MAPKs by 7,8-DHF may
partially explain the anti-inflammatory mechanisms of 7,8-DHF.
In conclusion, we found that 7,8-DHF significantly
attenuated the levels of neurotoxic pro-inflammatory mediators and
cytokines, including NO, PGE2, TNF-α and IL-1β in
LPS-stimulated microglial cells. The inhibitory action of 7,8-DHF
was accompanied by the attenuation of NF-κB activity through the
prevention of NF-κB translocation from the cytoplasm to the nucleus
and by the inhibition of IκB-degradation. In addition, levels of
phosphorylated MAPKs were significantly decreased by pre-treatment
with 7,8-DHF in LPS-stimulated microglial cells. These results
indicate that 7,8-DHF exerts its anti-inflammatory effects
inhibiting the activation of the NF-κB signaling pathway and the
phosphorylation of MAPKs. As a result of the findings presented in
this report, 7,8-DHF may ultimately prove useful in the treatment
of inflammatory diseases and may be an effective form of therapy
for the treatment of several neurodegenerative diseases which
accompany microglial activation.
Acknowledgements
This study was supported by the R&D program of
MKE/KEIT (10040391, Development of Functional Food Materials and
Device for Prevention of Aging-associated Muscle Function Decrease)
and Blue-Bio Industry Regional Innovation Center (RIC08-06-07) at
Dongeui University as a RIC program under MKE and Busan city,
Republic of Korea.
References
|
1
|
Napoli I and Neumann H: Microglial
clearance function in health and disease. Neuroscience.
158:1030–1038. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Loane DJ and Byrnes KR: Role of microglia
in neurotrauma. Neurotherapeutics. 7:366–377. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Meda L, Cassatella MA, Szendrei GI, Otvos
L Jr, Baron P, Villalba M, Ferrari D and Rossi F: Activation of
microglial cells by β-amyloid protein and interferon-γ. Nature.
373:647–650. 1995.
|
|
4
|
Eikelenboom P and van Gool WA:
Neuroinflammatory perspectives on the two faces of Alzheimer’s
disease. J Neural Transm. 111:281–294. 2004.PubMed/NCBI
|
|
5
|
Gao HM, Liu B, Zhang W and Hong JS: Novel
anti-inflammatory therapy for Parkinson’s disease. Trends Pharmacol
Sci. 24:395–401. 2003.
|
|
6
|
Liu B and Hong JS: Role of microglia in
inflammation-mediated neurodegenerative diseases: mechanisms and
strategies for therapeutic intervention. J Pharmacol Exp Ther.
304:1–7. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rock RB and Peterson PK: Microglia as a
pharmacological target in infectious and inflammatory diseases of
the brain. J Neuroimmune Pharmacol. 1:117–126. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kaur C and Ling EA: Antioxidants and
neuroprotection in the adult and developing central nervous system.
Curr Med Chem. 15:3068–3080. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Deng YY, Lu J, Ling EA and Kaur C: Role of
microglia in the process of inflammation in the hypoxic developing
brain. Front Biosci (Schol Ed). 3:884–900. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ramos S: Cancer chemoprevention and
chemotherapy: dietary polyphenols and signalling pathways. Mol Nutr
Food Res. 52:507–526. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guo W, Kong E and Meydani M: Dietary
polyphenols, inflammation, and cancer. Nut Cancer. 61:807–810.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen S: Natural products triggering
biological targets-a review of the anti-inflammatory phytochemicals
targeting the arachidonic acid pathway in allergy asthma and
rheumatoid arthritis. Curr Drug Targets. 12:288–301. 2011.
View Article : Google Scholar
|
|
13
|
García-Lafuente A, Guillamón E, Villares
A, Rostagno MA and Martínez JA: Flavonoids as anti-inflammatory
agents: implications in cancer and cardiovascular disease. Inflamm
Res. 58:537–552. 2009.PubMed/NCBI
|
|
14
|
González-Gallego J, García-Mediavilla MV,
Sánchez-Campos S and Tuñón MJ: Fruit polyphenols, immunity and
inflammation. Br J Nut. 104(Suppl 3): S15–S27. 2010.PubMed/NCBI
|
|
15
|
Magrone T and Jirillo E: Potential
application of dietary polyphenols from red wine to attaining
healthy ageing. Curr Top Med Chem. 11:1780–1796. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Juurlink BH and Paterson PG: Review of
oxidative stress in brain and spinal cord injury: suggestions for
pharmacological and nutritional management strategies. J Spinal
Cord Med. 21:309–334. 1998.PubMed/NCBI
|
|
17
|
Ramassamy C: Emerging role of polyphenolic
compounds in the treatment of neurodegenerative diseases: a review
of their intracellular targets. Eur J Pharmacol. 545:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
del Campos-Esparza MR and Torres-Ramos MA:
Neuroprotection by natural polyphenols: molecular mechanisms. Cent
Nerv Syst Agents Med Chem. 10:269–277. 2010.PubMed/NCBI
|
|
19
|
Jang SW, Liu X, Yepes M, Shepherd KR,
Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE and Ye K: A
selective TrkB agonist with potent neurotrophic activities by
7,8-dihydroxyflavone. Proc Natl Acad Sci USA. 107:2687–2692. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Andero R, Heldt SA, Ye K, Liu X, Armario A
and Ressler KJ: Effect of 7,8-dihydroxyflavone, a small-molecule
TrkB agonist, on emotional learning. Am J Psychiatry. 168:163–172.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen J, Chua KW, Chua CC, Yu H, Pei A,
Chua BH, Hamdy RC, Xu X and Liu CF: Antioxidant activity of
7,8-dihydroxyflavone provides neuroprotection against
glutamate-induced toxicity. Neurosci Lett. 499:181–185. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang R, Kang KA, Piao MJ, Ko DO, Wang ZH,
Chang WY, You HJ, Lee IK, Kim BJ, Kang SS and Hyun JW: Preventive
effect of 7,8-dihydroxyflavone against oxidative stress induced
genotoxicity. Biol Pharm Bull. 32:166–171. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kozics K, Valovicova Z and Slamenova D:
Structure of flavonoids influences the degree inhibition of
Benzo(a)pyrene-induced DNA damage and micronuclei in HepG2 cells.
Neoplasma. 58:516–524. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Park HY, Kim GY, Hyun JW, Hwang HJ, Kim
ND, Kim BW and Choi YH: 7,8-Dihydroxyflavone exhibits
anti-inflammatory properties by downregulating the NF-κB and MAPK
signaling pathways in lipopolysaccharide-treated RAW264.7 cells.
Int J Mol Med. 29:1146–1152. 2012.PubMed/NCBI
|
|
25
|
Kyung J, Kim D, Park D, Yang YH, Choi EK,
Lee SP, Kim TS, Lee YB and Kim YB: Synergistic anti-inflammatory
effects of Laminaria japonica fucoidan and Cistanche
tubulosa extract. Lab Anim Res. 28:91–97. 2012. View Article : Google Scholar
|
|
26
|
Lee SH, Kim DW, Eom SA, Jun SY, Park M,
Kim DS, Kwon HJ, Kwon HY, Han KH, Park J, Hwang HS, Eum WS and Choi
SY: Suppression of 12-O-tetradecanoylphorbol-13-acetate
(TPA)-induced skin inflammation in mice by transduced Tat-Annexin
protein. BMB Rep. 45:354–359. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
McGeer PL, Yasojima K and McGeer EG:
Inflammation in Parkinson’s disease. Adv Neurol. 86:83–89.
2001.
|
|
28
|
Leone S, Ottani A and Bertolini A: Dual
acting anti-inflammatory drugs. Curr Topics Med Chem. 7:265–275.
2007. View Article : Google Scholar
|
|
29
|
Mariani MM and Kielian T: Microglia in
infectious diseases of the central nervous system. J Neuroimmune
Pharmacol. 4:448–461. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Saha RN and Pahan K: Regulation of
inducible nitric oxide synthase gene in glial cells. Antioxid Redox
Signal. 8:929–947. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brown GC and Neher JJ: Inflammatory
neurodegeneration and mechanisms of microglial killing of neurons.
Mol Neurobiol. 41:242–247. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hawkey CJ: COX-2 inhibitors. Lancet.
353:307–314. 1999. View Article : Google Scholar
|
|
33
|
Vane JR, Mitchell JA, Appleton I,
Tomlinson A, Bishop-Bailey D, Croxtall J and Willoughby DA:
Inducible isoforms of cyclooxygenase and nitric-oxide synthase in
inflammation. Proc Natl Acad Sci USA. 91:2046–2050. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Giovannini MG, Scali C, Prosperi C,
Bellucci A, Pepeu G and Casamenti G: Experimental brain
inflammation and neurodegeneration as model of Alzheimer’s disease:
protective effects of selective COX-2 inhibitors. Int J
Immunopathol Pharmacol. 16(Suppl 2): S31–S40. 2003.
|
|
35
|
Minghetti L and Pocchiari M:
Cyclooxygenase-2, prostaglandin E2, and microglial
activation in prion diseases. Int Rev Neurobiol. 82:265–275. 2007.
View Article : Google Scholar
|
|
36
|
Choi SH, Aid S and Bosetti F: The distinct
roles of cyclooxygenase-1 and −2 in neuroinflammation: implications
for translational research. Trends Pharmacol Sci. 30:174–181.
2009.
|
|
37
|
Boka G, Anglade P, Wallach D, Javoy-Agid
F, Agid Y and Hirsch EC: Immunocytochemical analysis of tumor
necrosis factor and its receptors in Parkinson’s disease. Neurosc
Lett. 172:151–154. 1994.PubMed/NCBI
|
|
38
|
Hunot S, Dugas N, Faucheux B, Hartmann A,
Tardieu M, Debre P, Agid Y, Dugas B and Hirsch EC:
FcepsilonRII/CD23 is expressed in Parkinson’s disease and induces,
in vitro, production of nitric oxide and tumor necrosis
factor-alpha in glial cells. J Neurosci. 19:3440–3447.
1999.PubMed/NCBI
|
|
39
|
De Nardin E: The role of inflammatory and
immunological mediators in periodontitis and cardiovascular
disease. Ann Periodontol. 6:30–40. 2001.PubMed/NCBI
|
|
40
|
Sawada M, Kondo N, Suzumura A and
Marunouchi T: Production of tumor necrosis factor-alpha by
microglia and astrocytes in culture. Brain Res. 491:394–397. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Botchkina GI, Meistrell ME III, Botchkina
IL and Tracey KJ: Expression of TNF and TNF receptors (p55 and p75)
in the rat brain after focal cerebral ischemia. Mol Med. 3:765–781.
1997.PubMed/NCBI
|
|
42
|
Akassoglou K, Bauer J, Kassiotis G,
Pasparakis M, Lassmann H, Kollias G and Probert L: Oligodendrocyte
apoptosis and primary demyelination induced by local TNF/p55TNF
receptor signaling in the central nervous system of transgenic
mice: models for multiple sclerosis with primary
oligodendrogliopathy. Am J Pathol. 153:801–813. 1998. View Article : Google Scholar
|
|
43
|
Sriram K, Matheson JM, Benkovic SA, Miller
DB, Luster MI and O’Callaghan JP: Mice deficient in TNF receptors
are protected against dopaminergic neurotoxicity: implications for
Parkinson’s disease. FASEB J. 16:1474–1476. 2002.PubMed/NCBI
|
|
44
|
Gougeon PY, Lourenssen S, Han TY, Nair DG,
Ropeleski MJ and Blennerhassett MG: The pro-inflammatory cytokines
IL-1β and TNFα are neurotrophic for enteric neurons. J Neurosci.
33:3339–3351. 2013.
|
|
45
|
Rothwell N, Allan S and Toulmond S: The
role of interleukin 1 in acute neurodegeneration and stroke:
pathophysiological and therapeutic implications. J Clin Invest.
100:2648–2652. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Haddad JJ: The role of inflammatory
cytokines and NF-kappaB/MAPK signaling pathways in the evolution of
familial Mediterranean fever: current clinical perspectives and
potential therapeutic approaches. Cell Immunol. 260:6–13. 2009.
View Article : Google Scholar
|
|
47
|
Hamilton T, Novotny M, Pavicic PJ Jr,
Herjan T, Hartupee J, Sun D, Zhao C and Datta S: Diversity in
post-transcriptional control of neutrophil chemoattractant cytokine
gene expression. Cytokine. 52:116–122. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Baldwin AS Jr: The NF-kappa B and I kappa
B proteins: new discoveries and insights. Ann Rev Immunol.
14:649–683. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Moon DO, Choi YH, Kim ND, Park YM and Kim
GY: Anti-inflammatory effects of beta-lapachone in
lipopolysaccharide-stimulated BV2 microglia. Int Immunopharmacol.
7:506–514. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sarkar FH and Li Y: NF-kappaB: a potential
target for cancer chemoprevention and therapy. Front Biosci.
13:2950–2959. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wertz IE and Dixit VM: Signaling to
NF-kappaB: regulation by ubiquitination. Cold Spring Harb Perspect
Biol. 2:a0033502010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mattson MP and Camandola S: NF-kappaB in
neuronal plasticity and neurodegenerative disorders. J Clin Invest.
107:247–254. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Nakajima K and Kohsaka S: Characterization
of brain microglia and the biological significance in the central
nervous system. Adv Neurol. 60:734–743. 1993.PubMed/NCBI
|
|
54
|
Kim YJ, Hwang SY, Oh ES, Oh S and Han IO:
IL-1beta, an immediate early protein secreted by activated
microglia, induces iNOS/NO in C6 astrocytoma cells through p38 MAPK
and NF-kappaB pathways. J Neurosci Res. 84:1037–1046. 2006.
View Article : Google Scholar : PubMed/NCBI
|