Introduction
Hereditary retinal degeneration is the most common
form of genetic eye diseases causing irreversible blindness.
Mutations in almost 200 genes are associated with hereditary
retinal degeneration (RetNet: http://www.sph.uth.tmc.edu/Retnet/sum-dis.htm). Of
these diseases, retinitis pigmentosa (RP) is the most common type
and refers to various forms of progressive retinal degeneration
with predominantly impaired rod photoreceptors. RP is clinically
and genetically highly heterogeneous. Currently, mutations in at
least 62 genes were associated with autosomal dominant, autosomal
recessive, or X-linked RP. Mutations in all these genes may be
responsible for approximately half of the cases of RP (1–3).
The causes for the remaining half of RP cases await identification.
The development of high-throughput techniques for gene analysis has
revealed an increasing number of new genes that are associated with
RP (4,5). Additionally, a number of genes known
to cause other retinopathy or extraocular diseases have been
recently reported to also be responsible for RP, such as mutation
in ADAMTS18, CYP4V2, or OFD1 (6–8).
In the present study, exome sequencing on 157
families with RP detected mutations in the RAB geranylgeranyl
transferase holoenzyme component A gene (the CHM gene) in
six families. These mutations are known to cause choroideremia
(9–11). No other causative mutations in the
61 of the 62 RP-associated genes were detected in the six families
by exome sequencing. Sanger sequencing confirmed four novel and two
known mutations in the CHM gene in the six families.
Clinical data of the six probands showed certain characteristics of
RP and some atypical indications of choroideremia. The result
emphasizes the importance of a comprehensive analysis of clinical
data and potential genes for hereditary diseases with overlapping
phenotypes (12) or among the
list requiring differential diagnosis.
Materials and methods
Patients
Probands with initial diagnosis of RP from 157
families were recruited at the Eye Hospital of the Zhongshan
Ophthalmic Center (Guangzhou, China). The diagnosis of RP was based
on night blindness beginning in early childhood, decreasing visual
acuity with age, and fundus changes as previously described
(13). Written informed consent
that followed the tenets of the Declaration of Helsinki was
obtained from each participating individual or their guardians
prior to the study. This study was approved by the Institutional
Review Board of the Zhongshan Ophthalmic. Genomic DNA was prepared
from venous leukocytes.
Exome sequencing
Exome sequencing was performed by BGI Shenzhen, as
previously described (14). The
exome sequencing, genotype calling, and SNP calling were conducted
as previously described (15).
Exome capture was carried out using a NimbleGen SeqCap EZ Exome
(44M) array (Roche, Basil, Switzerland). Exon-enriched DNA
fragments were sequenced by the Illumina Genome Analyzer II
(Illumina, Inc., San Diego, CA, USA). The average sequencing depth
was set at 60-fold. The SOAP aligner was used to align the
sequencing reads to UCSC hg19 (16,17). The likelihood of possible
genotypes in target regions was calculated using SOAPsnp (18). Data were reviewed for all the
genes known to be associated with hereditary retinal disease.
Probands with a hemizygous mutation in the CHM gene but
without other causative mutations in the 61 of the 62 RP-associated
genes were selected in this study.
Sanger sequencing
Sanger sequencing was used to verify the variants in
CHM detected by exome sequencing. The fragments with
variants found in patients were amplified by the polymerase chain
reaction with the primers listed in Table I. The sequences of the amplicons
were determined with an ABI Big Dye Terminator cycle sequencing kit
v3.1 on an ABI3130 Genetic Analyzer (both from Applied Biosystems,
Foster City, CA, USA). Sequencing results from patients and
controls were aligned using the SeqManII program of the Lasergene
package (DNAStar, Inc., Madison, WI, USA). Variants in available
family members were also analyzed. Novel variants were then
evaluated in 96 control individuals. The mutations were described
in accordance with the nomenclature for the description of sequence
variants (HGVS: http://www.hgvs.org/mutnomen/).
 | Table IPrimers used for mutation confirmation
by Sanger sequencing for the CHM gene. |
Table I
Primers used for mutation confirmation
by Sanger sequencing for the CHM gene.
| Proband | Primers and sequences
(5′→3′) | Size of amplicons
(bp) | Annealing temp
(°C) |
|---|
| RP31 | F1:
atggatcaggttttgctgct
R1: aagctgatgcccagttacaa | 397 | 58–65 |
| RP229 | F2:
ctgcctacggaggatgagtc
R2: gggcccagatactgttttca | 337 | 58–65 |
| RP263 | F3:
aattaacccccaacctccaa
R3: aagctcaaaaagaggccaca | 383 | 58–65 |
| RP285, RP304 | F4:
ccacctatgtcctttgtgagg
R4: aatggagtgttgccataccg | 291 | 58–65 |
| RP359 | F5:
caccatgacttgctcagctc
R5: cccacatgtttaggcagaca | 376 | 58–65 |
Results
Whole exome sequencing detected six hemizygous
mutations in CHM in six of the 157 families with RP. Sanger
sequencing confirmed the six mutations in CHM (Fig. 1). Of the six, four were novel and
two were known mutations (Table
II). Four of the six resulted in truncation of the encoded
proteins and the remaining two were predicted to eliminate the
splicing sites (Table II).
Potential pathogenic mutations in the 62 RP-associated genes were
not detected in the six families.
| Table IIHemizygous mutations in the
CHM gene and their associated clinical data. |
Table II
Hemizygous mutations in the
CHM gene and their associated clinical data.
| Proband ID | CHM variation | Gender | Age (year) at | First symptom | Visual acuity
(right; left) | Fundus changes | ERG responses |
|---|
| |
|
|
|---|
| Nucleotide
change | Effect | Database | | exam | onset | rod | cone |
|---|
| RP31 |
c.[703-1G>A];[0] | SSA | Known [34] | M | 23 | 5 | NB | 0.3;0.3 | AV, CS, BSP | NA | NA |
| RP229 |
c.[558_559delTT];[0] | T186TfsX12 | Novel | M | 34 | 7 | NB | 0.1;0.6 | AV, CS, BSP | ND | ND |
| RP263 |
c.[1166+2T>G];[0] | SSA | Novel | M | 22 | EC | NB | 0.7;0.7 | AV, WSP | ND | ND |
| RP285 |
c.[966delA];[0] | E322DfsX3 | Novel | M | 34 | EC | NB | 0.2;0.3 | SPP | NA | NA |
| RP304 |
c.[964G>T];[0] |
E322* | Novel | M | 24 | EC | NB | 0.5;0.5 | BSP | NA | NA |
| RP359 |
c.[1584_1587delTGTT];[0] | F528FfsX8 | Known [34] | M | 21 | EC | NB | 0.4;0.03 | AV, CS, BSP | ND | ND |
For the patients with the CHM mutations,
three were singleton cases without a family history of retinal
degeneration while the remaining three had a family history
consistent with an X-linked trait. All the probands had experienced
night blindness since early childhood, with gradually reduced
visual acuity later in life. Fundus observation revealed retinal
degeneration with pigmentary disturbance. A review of the fundus
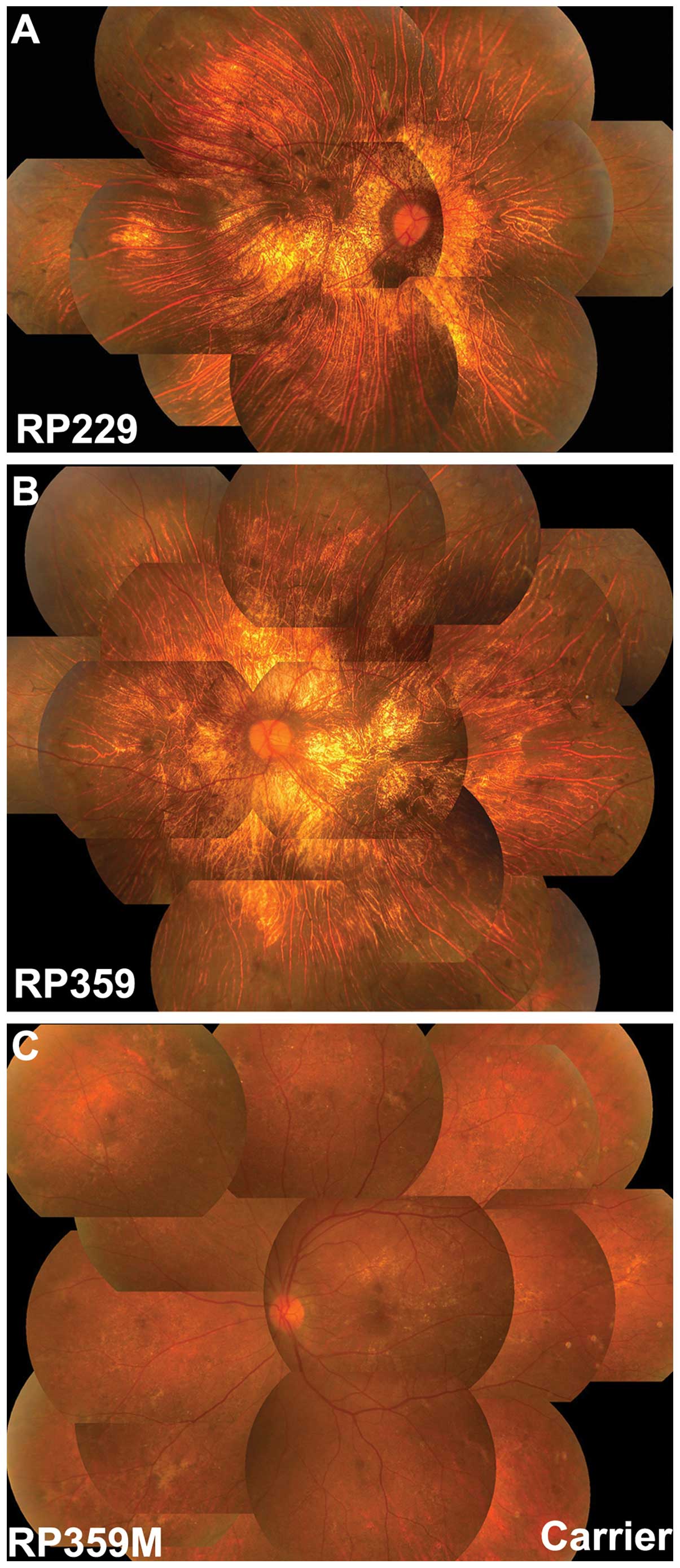
images obtained showed fundus changes consistent with choroideremia
(Fig. 2) although typical
indications of choroideremia, such as chorioretinal scalloped
atrophy in the mid-peripheral fundus with preserved macula, was not
recorded in these patients (Table
II). Electroretinography recordings on three probands showed no
appreciable responses of cones and rods in both eyes.
Available family members were analyzed in family
RP359, where the mother (RP359M) of the proband had a heterozygous
c.1584_1587delTGTT mutation. The mother had normal visual acuity
without night blindness, but had a number of crystalline-like spots
in the macular area and irregular retinal dystrophy in the
mid-peripheral retina (Fig. 2).
Electroretinography showed normal rod responses and mildly reduced
cone responses.
Discussion
The present study identified six hemizygous
CHM mutations in six of the 15 unrelated families with
initial diagnosis of RP, initially analyzed by exome sequencing and
confirmed by Sanger sequencing. All six mutations resulted in
truncation (or loss of function) that is generally observed in
CHM mutations. Initial clinical diagnosis of the six
probands was RP while subsequent re-evaluation of the clinical data
suggested atypical form of choroideremia.
Mutations in CHM are known to cause choroideremia
alone (9,19–37). At least 147 mutations in CHM have
been previously reported in patients with choroideremia, including
2 missense mutations, 39 nonsense mutations, 25 splicing mutations,
32 small deletions, 9 small insertions, 5 small indels, 31 gross
deletions, 1 gross insertions/duplications, and 3 complex
rearrangements, based on the HGMD® Professional 2012.4
(https://portal.biobase-international.com/hgmd/pro/gene.php?gene=CHM).
All but two of these (21,38)
are loss of function mutations. The six mutations identified in
this study include 1 nonsense, 3 small deletion, and 2 splicing
mutations, all of which are loss of function mutations.
Choroideremia and RP share several common features,
such as night blindness, constriction of the visual field,
gradually reduced visual acuity, and retinal degeneration, and may
be confused with each other (39). Previous studies have suggested
that ‘about 6% of individuals diagnosed with RP-related disorders
actually have choroideremia’ (39), while about one quarter of
clinically diagnosed choroideremia may actually be other diseases,
including RP (40).
Choroideremia, referring to the absence (-eremia) of choroid, is an
X-linked disease characterized by chorioretinal scalloped atrophy
initiated from the mid-peripheral fundus, with preservation of the
macula (20,40–42). However, these types of typical
fundus changes for choroideremia may not yet have developed at the
first visit to the ophthalmologists. Considering the great
variability in the appearance of the fundus in RP, choroideremia
without a typical fundus appearance may easily be diagnosed as
RP.
The typical manifestation for choroideremia [i.e.,
chorioretinal scalloped atrophy with preservation of the macula
(42,43)], was not found in the six probands
with CHM mutations in the present study. However, the fundus
changes of the six probands with CHM mutations were also
atypical compared to those seen in classic RP. Retinal pigmentary
degeneration with choroidal sclerosis has been visualized, not only
in choroideremia, but also in severe retinitis pigmentosa with the
PROM1 mutation (44) or in
Bietti crystalline corneoretinal dystrophy with the CYP4V2
mutation (45). The six patients
in the present study with atypical fundus changes may be
misdiagnosed as RP if phenotypic variation of choroideremia is not
obvious and systemic fundus examination is not performed.
In summary, six mutations leading to the truncation
of CHM were identified in six of 157 (4%) unrelated patients with
initial diagnosis of RP. The results of this study suggest that CHM
should be included as a candidate gene for atypical RP.
Additionally, choroideremia may be misdiagnosed as RP. These
findings emphasize that genes known to cause one form of retinal
degeneration may also be ideal candidates for other forms of
retinal degeneration, particularly for those with overlapping
phenotypes.
Acknowledgements
The authors would like to thank the patients for
their participation. This study was supported by the National
Natural Science Foundation of China (U1201221 to Q.Z.), ‘985
project’ of Sun Yat-sen University, and the Fundamental Research
Funds of State Key Laboratory of Ophthalmology.
References
|
1
|
Hartong DT, Berson EL and Dryja TP:
Retinitis pigmentosa. Lancet. 368:1795–1809. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
den Hollander AI, Black A, Bennett J and
Cremers FP: Lighting a candle in the dark: advances in genetics and
gene therapy of recessive retinal dystrophies. J Clin Invest.
120:3042–3053. 2010.PubMed/NCBI
|
|
3
|
Xu Y, Guan L, Shen T, Zhang J, Xiao X,
Jiang H, Li S, Yang J, Jia X, Yin Y, Guo X, Wang J and Zhang Q:
Mutations of 60 known causative genes in 157 families with
retinitis pigmentosa based on exome sequencing. Hum Genet. (In
press).
|
|
4
|
Züchner S, Dallman J, Wen R, et al:
Whole-exome sequencing links a variant in DHDDS to retinitis
pigmentosa. Am J Hum Genet. 88:201–206. 2011.PubMed/NCBI
|
|
5
|
Tucker BA, Scheetz TE, Mullins RF, et al:
Exome sequencing and analysis of induced pluripotent stem cells
identify the cilia-related gene male germ cell-associated kinase
(MAK) as a cause of retinitis pigmentosa. Proc Natl Acad Sci USA.
108:E569–E576. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Peluso I, Conte I, Testa F, et al: The
ADAMTS18 gene is responsible for autosomal recessive early onset
severe retinal dystrophy. Orphanet J Rare Dis. 8:162013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Webb TR, Parfitt DA, Gardner JC, et al:
Deep intronic mutation in OFD1, identified by targeted genomic
next-generation sequencing, causes a severe form of X-linked
retinitis pigmentosa (RP23). Hum Mol Genet. 21:3647–3654. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang Y, Guo L, Cai SP, et al: Exome
sequencing identifies compound heterozygous mutations in CYP4V2 in
a pedigree with retinitis pigmentosa. PLoS One. 7:e336732012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cremers FP, van de Pol DJ, van Kerkhoff
LP, Wieringa B and Ropers HH: Cloning of a gene that is rearranged
in patients with choroideraemia. Nature. 347:674–677. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rak A, Pylypenko O, Niculae A, Pyatkov K,
Goody RS and Alexandrov K: Structure of the Rab7:REP-1 complex:
insights into the mechanism of Rab prenylation and choroideremia
disease. Cell. 117:749–760. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Seabra MC, Brown MS and Goldstein JL:
Retinal degeneration in choroideremia: deficiency of rab
geranylgeranyl transferase. Science. 259:377–381. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Berger W, Kloeckener-Gruissem B and
Neidhardt J: The molecular basis of human retinal and vitreoretinal
diseases. Prog Retin Eye Res. 29:335–375. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang Q, Zulfiqar F, Xiao X, et al: Severe
autosomal recessive retinitis pigmentosa maps to chromosome
1p13.3-p21.2 between D1S2896 and D1S457 but outside ABCA4. Hum
Genet. 118:356–365. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiao X, Li S, Guo X and Zhang Q: A novel
locus for autosomal dominant congenital motor nystagmus mapped to
1q31–q32.2 between D1S2816 and D1S2692. Hum Genet. 131:697–702.
2012.PubMed/NCBI
|
|
15
|
Li Y, Vinckenbosch N, Tian G, et al:
Resequencing of 200 human exomes identifies an excess of
low-frequency non-synonymous coding variants. Nat Genet.
42:969–972. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li R, Li Y, Kristiansen K and Wang J:
SOAP: short oligonucleotide alignment program. Bioinformatics.
24:713–714. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li R, Yu C, Li Y, et al: SOAP2: an
improved ultrafast tool for short read alignment. Bioinformatics.
25:1966–1967. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li R, Li Y, Fang X, et al: SNP detection
for massively parallel whole-genome resequencing. Genome Res.
19:1124–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chi JY, MacDonald IM and Hume S: Copy
number variant analysis in CHM to detect duplications underlying
choroideremia. Ophthalmic Genet. 34:229–233. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Coussa RG and Traboulsi EI: Choroideremia:
a review of general findings and pathogenesis. Ophthalmic Genet.
33:57–65. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Esposito G, De Falco F, Tinto N, et al:
Comprehensive mutation analysis (20 families) of the choroideremia
gene reveals a missense variant that prevents the binding of REP1
with Rab geranylgeranyl transferase. Hum Mutat. 32:1460–1469. 2011.
View Article : Google Scholar
|
|
22
|
Forsythe P, Maguire A, Fujita R, Moen C,
Swaroop A and Bennett J: A carboxy-terminal truncation of 99 amino
acids resulting from a novel mutation (Arg555→stop) in the CHM gene
leads to choroideremia. Exp Eye Res. 64:487–490. 1997.PubMed/NCBI
|
|
23
|
Fujiki K, Hotta Y, Hayakawa M, et al:
REP-1 gene mutations in Japanese patients with choroideremia.
Graefes Arch Clin Exp Ophthalmol. 237:735–740. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Garcia-Hoyos M, Lorda-Sanchez I,
Gómez-Garre P, et al: New type of mutations in three Spanish
families with choroideremia. Invest Ophthalmol Vis Sci.
49:1315–1321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hotta Y, Fujiki K, Hayakawa M, et al: A
hemizygous A to CC base change of the CHM gene causing
choroideremia associated with pinealoma. Graefes Arch Clin Exp
Ophthalmol. 235:653–655. 1997. View Article : Google Scholar
|
|
26
|
Iino Y, Fujimaki T, Fujiki K and Murakami
A: A novel mutation (967–970+2)delAAAGGT in the choroideremia gene
found in a Japanese family and related clinical findings. Jpn J
Ophthalmol. 52:289–297. 2008.
|
|
27
|
Itabashi T, Wada Y, Kawamura M, Sato H and
Tamai M: Clinical features of Japanese families with a 402delT or a
555–556delAG mutation in choroideremia gene. Retina. 24:940–945.
2004.
|
|
28
|
McTaggart KE, Tran M, Mah DY, Lai SW,
Nesslinger NJ and MacDonald IM: Mutational analysis of patients
with the diagnosis of choroideremia. Hum Mutat. 20:189–196. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nesslinger N, Mitchell G, Strasberg P and
MacDonald IM: Mutation analysis in Canadian families with
choroideremia. Ophthalmic Genet. 17:47–52. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Perez-Cano HJ, Garnica-Hayashi RE and
Zenteno JC: CHM gene molecular analysis and X-chromosome
inactivation pattern determination in two families with
choroideremia. Am J Med Genet A. 149A:2134–2140. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ponjavic V, Abrahamson M, Andreasson S, et
al: Phenotype variations within a choroideremia family lacking the
entire CHM gene. Ophthalmic Genet. 16:143–150. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sankila EM, Tolvanen R, van den Hurk JA,
Cremers FP and de la Chapelle A: Aberrant splicing of the CHM gene
is a significant cause of choroideremia. Nat Genet. 1:109–113.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schwartz M, Rosenberg T, van den Hurk JA,
van de Pol DJ and Cremers FP: Identification of mutations in Danish
choroideremia families. Hum Mutat. 2:43–47. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
van Bokhoven H, Schwartz M, Andréasson S,
et al: Mutation spectrum in the CHM gene of Danish and Swedish
choroideremia patients. Hum Mol Genet. 3:1047–1051. 1994.
|
|
35
|
Yip SP, Cheung TS, Chu MY, et al: Novel
truncating mutations of the CHM gene in Chinese patients with
choroideremia. Mol Vis. 13:2183–2193. 2007.PubMed/NCBI
|
|
36
|
Zhou Q, Liu L, Xu F, et al: Genetic and
phenotypic characteristics of three Mainland Chinese families with
choroideremia. Mol Vis. 18:309–316. 2012.PubMed/NCBI
|
|
37
|
Lin Y, Liu X, Luo L, et al: Molecular
analysis of the choroideremia gene related clinical findings in two
families with choroideremia. Mol Vis. 17:2564–2569. 2011.PubMed/NCBI
|
|
38
|
Sergeev YV, Smaoui N, Sui R, et al: The
functional effect of pathogenic mutations in Rab escort protein 1.
Mutat Res. 665:44–50. 2009. View Article : Google Scholar
|
|
39
|
MacDonald IM, Smaoui N and Seabra MC:
Choroideremia. GeneReviews® [Internet]. Pagon RA, Adam
MP, Bird TD, Dolan CR, Fong CT, Smith RJH and Stephens K:
University of Washington; Seattle: pp. 1993–2014. 2003
|
|
40
|
Lee TK, McTaggart KE, Sieving PA, et al:
Clinical diagnoses that overlap with choroideremia. Can J
Ophthalmol. 38:364–372. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Roberts MF, Fishman GA, Roberts DK, et al:
Retrospective, longitudinal, and cross sectional study of visual
acuity impairment in choroideraemia. Br J Ophthalmol. 86:658–662.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mura M, Sereda C, Jablonski MM, MacDonald
IM and Iannaccone A: Clinical and functional findings in
choroideremia due to complete deletion of the CHM gene. Arch
Ophthalmol. 125:1107–1113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
MacDonald IM, Russell L and Chan CC:
Choroideremia: new findings from ocular pathology and review of
recent literature. Surv Ophthalmol. 54:401–407. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang Q, Zulfiqar F, Xiao X, et al: Severe
retinitis pigmentosa mapped to 4p15 and associated with a novel
mutation in the PROM1 gene. Hum Genet. 122:293–299. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xiao X, Mai G, Li S, Guo X and Zhang Q:
Identification of CYP4V2 mutation in 21 families and overview of
mutation spectrum in Bietti crystalline corneoretinal dystrophy.
Biochem Biophys Res Commun. 409:181–186. 2011. View Article : Google Scholar
|