Introduction
Hyperuricemia is a consistent and independent risk
factor for hypertension, metabolic syndrome, fatty liver, diabetes
and kidney diseases (1–3). Tubulointerstitial inflammation and
injury are commonly detected in hyperuricemia-induced chronic renal
injury, with increased macrophage and T cell infiltration observed
in the tubulointerstitium of the kidneys (4,5).
As some patients with hyperuricemia present with urate crystal
deposition in the tubules and interstitium, it was previously
assumed that hyperuricemia causes tubular injury through the
precipitation of urate in the form of crystals in the kidneys, in a
manner similar to which it causes gout. In gout, urate crystals
cause inflammation by stimulating leukocytes to produce the
pro-inflammatory cytokine, interleukin-1β (IL-1β). IL-1 production
occurs when the urate particles are ingested by phagocytes through
the Toll like receptor 4 (TLR4) activation pathway, which
facilitates the formation of the NACHT, LRR and PYD
domains-containing protein 3 [NALP3; also known as NOD-like
receptor family, pyrin domain containing 3 (NALP3) and cryopyrin]
inflammasome, thus promoting pro-caspase-1 activation, further
activating caspase-1; activated caspase-1 then cleaves pro-IL-1β
into its active form IL-1β and, thus, initiates downstream
inflammatory processes (6,7).
Accumulating evidence has demonstrated that urate crystal-induced
inflammation is a paradigm of innate immunity. Innate
immunity-related components, including TLR4, NALP3, ASC, caspase-1
and IL-1β are essential in the development of gouty inflammation
(8). However, recent findings
suggest that the presence of elevated soluble serum uric acid (UA)
levels represent the presence of low-grade systemic inflammation
even in the absence of gout (9).
A recent ultrasound study also suggested that ultrasonographic
changes suggestive of gouty arthritis in joints and tendons may
occur in patients with asymptomatic hyperuricaemia who have never
had an episode of gout (10).
Moreover, as urate crystal is as less likely to be deposited in the
kidneys as in the joints, these recent findings suggest that, not
only urate crystals, but also soluble UA may account for the injury
in patients with hyperuricemia. However, whether soluble UA can
also lead to the TLR4-mediated activation of innate immunity
remains unknown.
Soluble UA may have direct pro-inflammatory effects
as shown by previous studies, including stimulating the production
of C-reactive protein (CRP) and monocyte chemotactic protein-1
(MCP-1) in vascular cells through the activation of nuclear
factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK)
(11,12), inducing endothelial dysfunction
with mitochondrial alterations and decreased intracellular
adenosine triphosphate (ATP) concentrations (13), exerting pro-oxidative effects
mediated in part by the activation of the lactaldehyde reductase
(NADPH) oxidase system and stimulating mitochondrial oxidative
stress in vascular cells or adipocytes (14). In the kidneys, observational
studies have also observed renal injuries induced by soluble UA
without urate crystal formation induced by inflammation (15–18). In clinical observations, not only
hyperuricemia, but also a higher normal level of UA has been shown
to increase the risk of the early progression of renal function
loss in patients with type 1 diabetes (19). In hyperuricemic rats, renal
disease has been shown to progress rapidly without the presence of
urate crystals in the kidneys (18). In vitro findings have also
suggested that soluble UA actively participates in pro-inflammatory
processes in vascular smooth muscle cells and mesangial cells
(11,15,16). However, these studies failed to
illustrate the mechanisms of soluble UA-induced renal injury and
did not investigate the upstream events of the pro-inflammatory
effects of soluble UA.
As kidney diseases, including some metabolic renal
injuries, often manifest with renal immune dysregulation (20), it is possible that soluble UA may
directly induce renal tubular injury without urate crystal
deposition. More importantly, the TLR4-mediated activation of
innate immunity may be involved as an upstream event for the
pro-inflammatory effects of soluble UA. Therefore, we hypothesized
that soluble UA may cause innate immune injury through the
TLR4-dependent pathway, and may thus promote the formation of the
NALP3 inflammasome, the activation of caspase-1, the expression of
IL-1β and the overproduction of downstream inflammatory cytokines.
In order to verify this hypothesis, in the present study, we
incubated human primary renal proximal tubule epithelial cells
(PTECs) with soluble UA and then measured the expression levels of
TLR4, the NALP3 inflammasome, caspase-1 and IL-1β, as well as those
of the inflammatory marker, intercellular adhesion molecule-1
(ICAM-1). We also used the TLR4-specific inhibitor, TAK242, to
block the effects of TLR4 so as to determine whether the effects of
soluble UA on PTECs are TLR4-dependent.
Materials and methods
Reagents
Human primary renal PTECs and their culture medium
were purchased from ScienCell (San Diego, CA, USA). UA was
purchased from Sigma (St. Louis, MO, USA). The TLR4 inhibitor,
TAK242, was purchased from Chembest Research Laboratories Ltd.
(Shanghai, China). The MTT assay kit was from Amresco (Solon, OH,
USA). Reagents for real-time PCR were purchased from Takara (Kyoto,
Japan). Enzyme immunoassay kits for the detection of IL-1β were
purchased from eBioscience (San Diego, CA, USA). Anti-TLR4 (Cat.
no. ab22048) and anti-CIAS1/NALP3 (Cat. no. ab16097) antibodies
were from Abcam (Cambridge, UK). Anti-caspase-1 (Cat. no. sc-515)
and anti-ICAM-1 (Cat. no. sc-8439) antibodies were from Santa Cruz
Biotechnology (Dallas, TX, USA). Anti-mouse (Cat. no. A0216) and
anti-rabbit (Cat. no. A0208) secondary antibodies were from
SinoBios (Shanghai, China).
Cell culture
Human primary renal PTECs were cultured in
epithelial cell medium, which contains 500 ml of basal medium, 50
ml of fetal bovine serum, 5 ml of epithelial cell growth supplement
and 5 ml of penicillin/streptomycin solution. The cells were
incubated at 37°C in 5% CO2 and 95% air. In all the
experiments, there was a ‘growth arrest’ period of 24 h in
serum-free medium prior to stimulation.
Preparation of soluble UA
UA was dissolved in 1 M NaOH at a concentration of
50 mg/ml as previously described (21). The solution was examined free of
mycoplasma and filtered (22 μm pore size) before use.
Crystals were not detectable (polarizing microscopy), nor did they
develop during cell incubation.
Viability of PTECs under increasing
concentrations of soluble UA
To examine cell viability following treatment with
various concentrations of soluble UA, the growth-arrested human
primary renal PTECs were seeded in 96-well plates
(0.25×105 cells/well) and exposed to soluble UA (0–800
μg/ml) for 24, 48 and 72 h, respectively. The effects of
treatment with TAK242 on cell viability (0.25–2 μM) were
also examined. Subsequently, the cytotoxic effects of these
stimulations on the human primary renal PTECs were examined by MTT
assay. Briefly, 20 μl MTT solution were added to each well,
and the cells were incubated at 37°C in 5% CO2 and 95%
air for 4 h. The reaction was terminated by the addition of 150
μl DMSO, and the absorbance was measured at 570 nm by an
ELISA reader. All results were expressed as percentage changes in
absorbance compared with those of the medium control (defined as
human primary renal PTECs incubated with plain culture medium).
Total RNA extraction and real-time PCR
for the quantification of TLR4 and ICAM-1 gene expression
The growth-arrested human primary renal PTECs were
incubated 100 μg/ml of soluble UA for 4 h. Total cellular
RNA was extracted using the NucleoSpin RNA II total RNA extraction
kit. The quality of the extracted RNA was monitored by formaldehyde
agarose gel electrophoresis. Aliquots of each RNA extraction were
reverse transcribed simultaneously into cDNA using the OneStep
RT-PCR kit (Takara, Tokyo, Japan) according to the manufacturer’s
instructions. Each real-time PCR reaction was performed in a total
volume of 25 μl in duplicate using the SYBR®
Premix Ex Taq™ kit (Takara, Kyoto, Japan) and the Fast Real-Time
PCR system 7500 (Applied Biosystems Inc., Foster City, CA, USA).
Primers and probe sets for human TLR4 and ICAM-1 were designed from
known sequences in GenBank and the primer sequences are listed in
Table I. GAPDH was used as an
endogenous control to normalize the amount of cDNA added to each
reaction (ΔCT), and the mean ΔCT value of the
control samples was used as the calibrator to calculate the
ΔΔCT value. The quantification of each transcript was
calculated using the comparative CT method. In this
method, the relative quantity of the target mRNA, normalized to the
endogenous control and relative to the calibrator, is equal to
2−ΔΔCT.
 | Table IPrimer sequences and size of PCR
products. |
Table I
Primer sequences and size of PCR
products.
| Genes | 5′→3′ sequences of
PCR primers | Size of products
(bp) | Accession
number |
|---|
| TLR4-f | ACA AGT GAT GTT TGA
TGG ACC TCT | 283 | U88880 |
| TLR4-r | TCT TGA ATG TTC TGT
TTC TGA GGA G | | |
|
ICAM-1-f | TTG AAC CCC ACA GTC
ACC TAT | 189 | NM_000201 |
|
ICAM-1-r | CCT CTG GCT TCG TCA
GAA TCA | | |
| GAPDH-f | ATG GGG AAG GTG AAG
GTC G | 118 | X01677 |
| GAPDH-r | GGG GTC ATT GAT GGC
AAC AAT A | | |
ELISA of IL-1β protein synthesis in cell
culture supernatants
The growth-arrested human primary renal PTECs were
incubated with 100 μg/ml soluble UA for 48 h. Cell culture
supernatants were collected and stored at −70°C until use in the
protein assay. The protein levels of IL-1β in the culture
supernatants were determined by commercial assay kits. The
procedures were carried out according to the manufacturer’s
instructions.
Western blot analysis for the protein
expression of TLR4, the NALP3 inflammasome and caspase-1
After harvesting the cell culture supernatant as
described above, the remaining cells were lysed with lysis buffer
containing protease inhibitor cocktails (Sigma). Ten micrograms of
total protein extracted from 106 cells were
electrophoresed through a 12% SDS-PAGE gel before being transferred
onto polyvinylidene difluoride (PVDF) membranes. After blocking for
1 h at room temperature in blocking buffer [5% bovine serum albumin
in Tris-buffered saline (TBS) with 0.05% Tween-20 (TBST)], the
membranes were incubated overnight with mouse anti-TLR4 (3:500),
mouse anti-CIAS1/NALP3 (1:1,000), rabbit anti-caspase-1 p10 (1:500)
and rabbit anti-GAPDH (1:10,000) in TBST. The membranes were washed
and incubated for 1 h at room temperature with a peroxidase-labeled
goat anti-rabbit (Cat. no. A0208) or goat anti-mouse (Cat. no.
A0216) immunoglobulin (SinoBios). After further washing, the
membranes were detected by ECL chemiluminescence (Amersham
Pharmacia Biotech, Arlington Heights, IL, USA).
ICAM-1 detection by
fluorescence-activated cell sorting (FACS) analysis
After an incubation of 48 h with 100 μg/ml of
soluble UA, the human primary renal PETCs were detached with 0.25%
trypsin in 1 mM ethylenediamine tetraacetic acid-4Na, washed twice
with PBS buffer, pH 7.2, and then incubated with fluorescein
isothiocyanate-conjugated anti-human ICAM-1 (1:50; Santa Cruz
Biotechnology) for 1 h in an ice-bath. The expression of ICAM-1 was
detected using a FACScan (BD FACS Caljbur; BD Bioscience, San Jose,
CA, USA). Data were compared as the geometric mean (GMean) values
of the fluorescence intensity of the ICAM-1-positive cells. The
same experiment was repeated 3 times.
Statistical analysis
All data are expressed as the means ± SD unless
otherwise specified. Statistical analysis was performed using SPSS
v.19.0 for Windows (SPSS, Inc., Chicago, IL, USA). Intergroup
differences for continuous variables were assessed by multivariate
ANOVA. A value of P<0.05 was considered to indicate a
statistically significant difference.
Results
Viability of human primary renal PTECs
and pH value of the medium following incubation with soluble UA and
TAK242
The viability of the human primary renal PTECs
cultured with serial dilutions of soluble UA and TAK242 (TLR4
inhibitor) for 24, 48 and 72 h was examined by MTT assay. The
results revealed that soluble UA (Fig. 1A) from 0 to 800 μg/ml and
TAK242 (Fig. 1B) from 0.25 to 2
μM did not affect the cell viability at all the time points
examined (P>0.05). The pH values of the human primary renal
PTECs in the culture medium following the addition of soluble UA
were examined using a pH meter (Merck, Darmstadt, Germany). The pH
values were all within the range of 7.1 to 7.4.
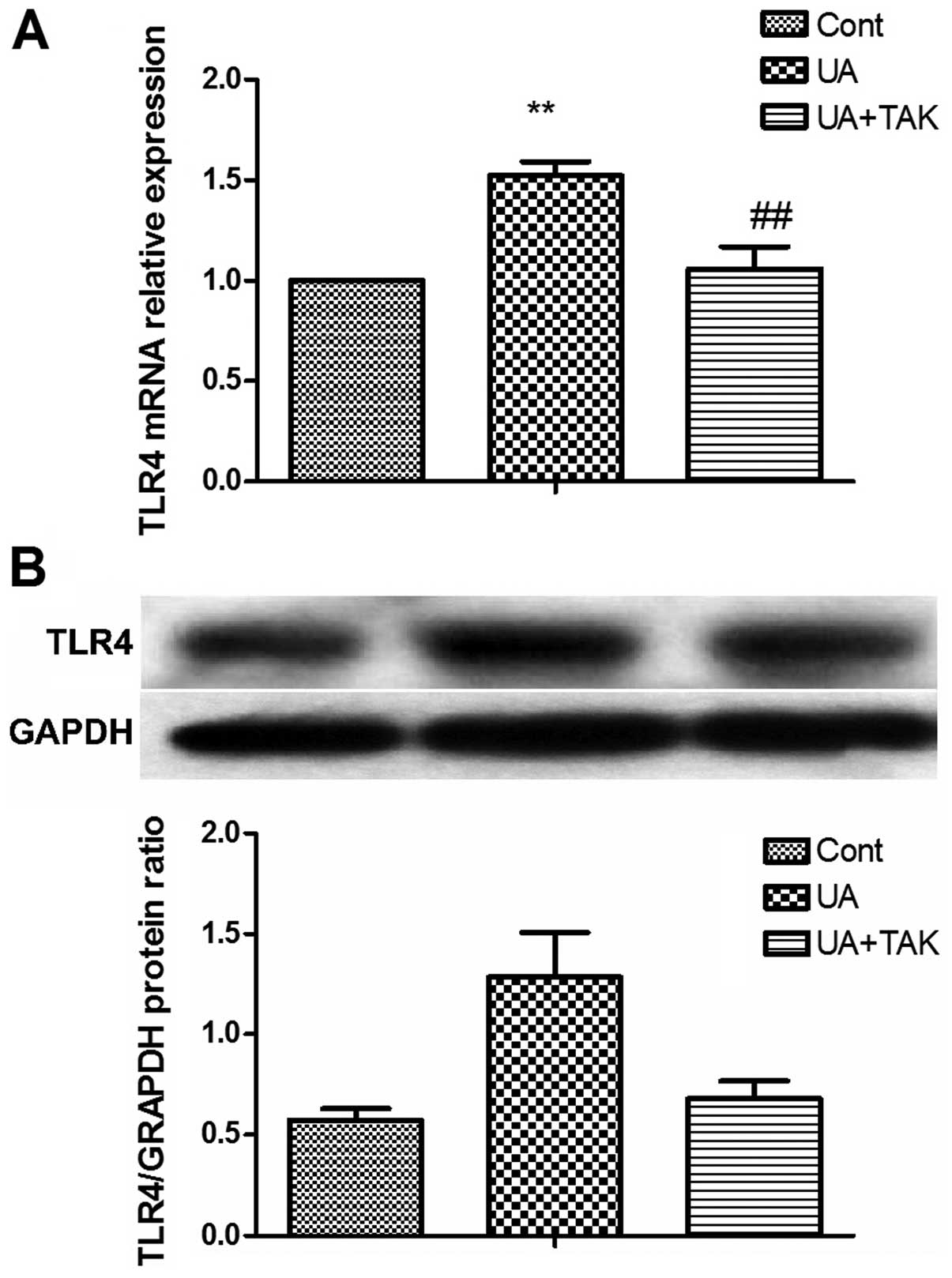
Soluble UA increases TLR4 expression in
human primary renal PTECs
To determine whether the innate immunity of the
human primary renal PTECs by soluble UA was activated, the
expression of TLR4, a typical membrane receptor for innate immunity
(22), was examined following
treatment with 100 μg/ml soluble UA for 4 h (gene
expression) or 48 h (protein expression) with or without 1 h
pre-incubation with the TLR4 inhibitor, TAK242 (1 μM). The
results from real-time PCR and western blot analysis revealed that
treatment with soluble UA significantly upregulated the TLR4 gene
(Fig. 2A; P<0.01) and protein
(Fig. 2B; P<0.01) expression
in the human primary renal PTECs. TAK242 reversed this upregulation
of soluble UA-induced TLR4 gene (Fig.
2A; P<0.01) and protein expression (Fig. 2B; P<0.01) in the human primary
renal PTECs.
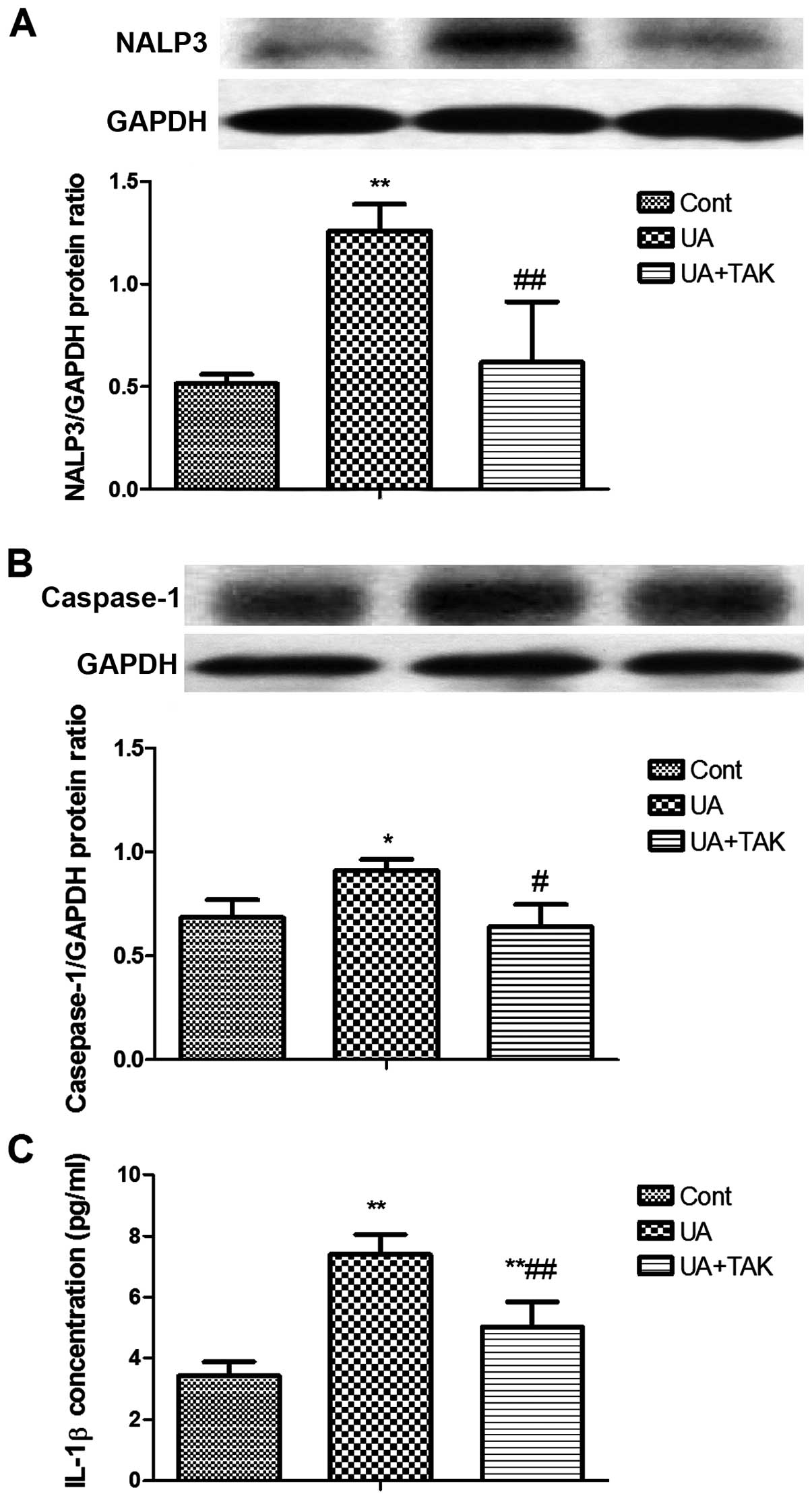
Induction of TLR4-dependent NALP3
expression by soluble UA in human primary renal PTECs
The formation of the NALP3 inflammasome is initiated
upon TLR4 activation (22). Thus,
in order to examine the soluble UA-induced activation of innate
immunity following tubular damage, NALP3 expression was measured by
western blot analysis after the human primary renal PTECs were
incubated with 100 μg/ml soluble UA for 48 h. TAK242 (1
μM) was added to the human primary renal PTECs 1 h prior to
stimulation with soluble UA. Soluble UA significantly enhanced
NALP3 protein expression in the human primary renal PTECs (Fig. 3A; P<0.01). TAK242 reversed this
soluble UA-induced increase in NALP3 protein expression in the
human primary renal PTECs (Fig.
3A; P<0.01).
TLR4-dependent caspase-1 activation by
soluble UA in human primary renal PTECs
Caspase-1 is the activated form processed from
pro-caspase-1 by the downstream process of the NALP3 inflammasome
(23). In order to determine the
function of the NALP3 inflammasome, caspase-1 activation in the
human primary renal PTECs was examined by western blot analysis
following stimulation with 100 μg/ml soluble UA for 4 h. To
determine whether the activation of TLR4 is specific for soluble
UA-induced caspase-1 activation, the TLR4-specific inhibitor,
TAK242 (1 μM), was added to the human primary renal PTECs 1
h prior to stimulation with soluble UA. Soluble UA significantly
promoted caspase-1 activation in the human primary renal PTECs
(Fig. 3B; P<0.05), whereas
TAK242 reversed this soluble UA-induced activation of caspase-1 in
the human primary renal PTECs (Fig.
3B; P<0.05).
Induction of TLR4-dependent IL-1β protein
synthesis by soluble UA in human primary renal PTECs
IL-1β can be cleaved from pro-IL-1β by caspase-1
(24). In order to determine the
effects of activated caspase-1, IL-1β protein expression in the
human primary renal PTECs was examined by ELISA following
stimulation with 100 μg/ml UA for 48 h. To determine whether
the activation of TLR4 is specific for soluble UA-induced IL-1β
protein expression, the TLR4-specific inhibitor, TAK242 (1
μM), was added to the human primary renal PTECs 1 h prior to
stimulation with soluble UA. Soluble UA significantly upregulated
IL-1β protein expression in the human primary renal PTECs (Fig. 3C; P<0.01). TAK242 reversed this
soluble UA-induced increase in IL-1β protein expression (Fig. 3C; P<0.01).
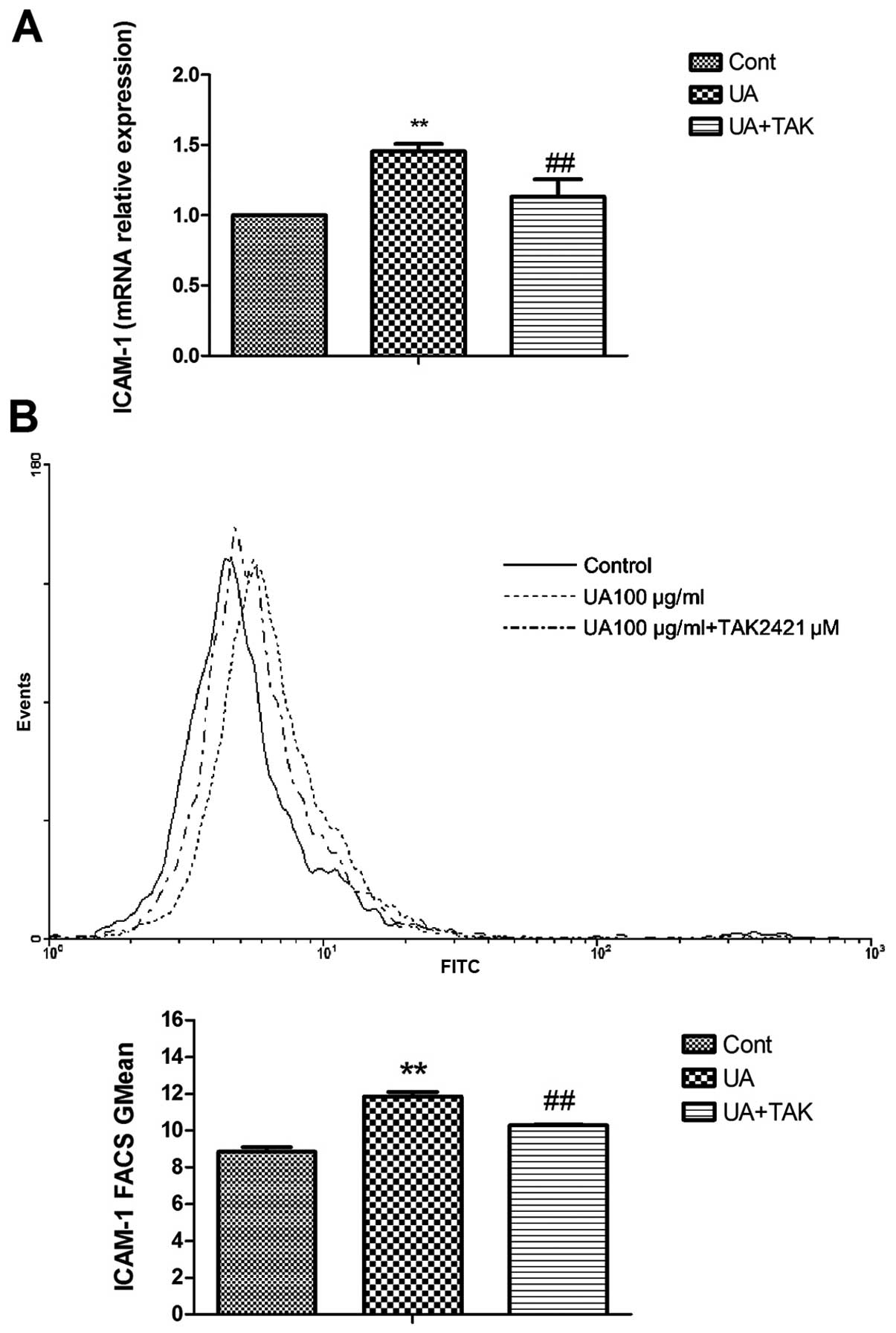
Induction of TLR4-dependent ICAM-1
expression by soluble UA in human primary renal PTECs
ICAM-1 is the downstream cytokine of IL-1β
production which facilitates the adhesion of monocytes/macrophages
to injured tubulointerstitial tissue (25). In order to determine the
downstream effect of IL-1β, the gene and protein expression levels
of ICAM-1 in the human primary renal PTECs were examined by
real-time PCR and FACS analysis following stimulation with 100
μg/ml soluble UA for 4 h (gene expression) or 48 h (protein
expression), respectively. To determine whether the activation of
TLR4 is specific for soluble UA-induced ICAM-1 expression, the
TLR4-specific inhibitor, TAK242 (1 μM), was added to the
human primary renal PTECs 1 h prior to stimulation with soluble UA.
As indicated in Fig. 4, soluble
UA significantly upregulated ICAM-1 gene (Fig. 4A; P<0.05) and protein
expression on the cell surface (Fig.
4B; P<0.01). TAK242 reversed this soluble UA-induced
increase in ICAM-1 gene and protein expression on the cell surface
(Fig. 4; P<0.05).
Discussion
Serum UA levels are elevated in chronic kidney
disease and closely correlate with the progression of renal disease
(26). However, whether soluble
UA actively participates in renal injury is incompletely
understood, which substantially limits our clinical understanding
of this recently revisited disease at the entity. In the present
study, we found that soluble UA significantly induced the
upregulation of pro-inflammatory cytokine ICAM-1 expression with
TLR4-NALP3-caspase-1-IL-1β signaling pathway activation in cultured
human primary renal PTECs, indicating that soluble UA may
participate in the development and progression of renal disease
through the TLR4-mediated activation of innate immunity.
Our results are indicative as we selected human
primary renal PTECs, which share most of the properties of the
human situation compared with other immortalized human renal
tubular cells or primary cells from other species. Studies on the
effects of UA on tubular cells have mainly used rat (4,27–29) and rabbit (30) tubular cells or the human proximal
tubule epithelial cell line, HK-2 (29,31). In the studies on rat renal
proximal tubular cells, UA was shown to possess pro-inflammatory
and pro-fibrotic properties (4,27,28). One study in 2007 found that UA
inhibited rat renal proximal tubule cell proliferation through at
least two signaling pathways involving protein kinase C, MAPK,
cytosolic phospholipase A2 and NF-κB (30). Later, UA was shown to induce cell
apoptosis by regulating apoptotic proteins (29) in the human renal tubular cell
line, HK-2, to induce epithelial-to-mesenchymal transition
(27) and to increase fibronectin
synthesis (28) in rat renal
epithelial cells. These studies have elegantly demonstrated the
downstream effects of UA; however, they failed to provide the
upstream information for the mechanisms through which UA triggers
these pro-inflammatory cascades. In the present study, we observed
the upregulation of ICAM-1, an important pro-inflammatory cytokine
mediating the interaction of resident renal cells to immune cells
(32). The soluble UA-induced
overepxression of ICAM-1 was dependent on TLR4 activation, as the
TLR4 inhibitor reversed this soluble UA-induced upregulation of
ICAM-1, suggesting that TLR4-mediated immunity is UA-dependent and
may be capable of leading to the activatio of other
pro-inflammatory cascades.
Innate immunity has recently been found to be great
interest even in several metabolic diseases (20). It is closely associated with
disease initiation and progression; the participation of the innate
and the adaptive immune response in mechanisms that contribute to
inflammation in cardiovascular disease has been reported in
atherosclerosis and hypertension (33). The involvement of immune
dysregulation in cardiac, vascular and renal changes in
hypertension has been demonstrated in experimental models (34). Renal epithelial cells are also
immune privileged and are surrounded by a dense network of immune
cells, which provides an environment for the communication of
tubular cells with immune cells. In fact, renal epithelial cells
share many phenotypic and functional characteristics with
mononuclear phagocytes, such as the secretion of chemokines in
response to direct stimulation with TLR ligands (35) and the expression of major
histocompatibility complex (MHC) I and II, as well as
co-stimulatory molecules. There are even data suggesting that
proximal tubules can present antigen to T cells (36).
The urate crystal form of UA, monosodium urate (MSU)
crystals, has been suggested to be a so-called ‘danger signal’ that
alerts the immune system to cell injury and helps to trigger both
innate and adaptive immune responses (7). The triggered immune responses
initiate the production of pro-inflammatory cytokines and then
facilitate the communication between immune cells and local tissue
cells. It is only found in immune cells and soluble UA directly
activates T cells without antigen presentation (21). In the present study, we provide
further evidence in that soluble UA modulates the innate immune
injury with the formation of the TLR4-dependent NALP3 inflammasome,
resulting in caspase-1 activation, IL-1β and ICAM-1 production in
human primary renal PTECs. Innate immunity has been shown to
trigger inflammation in a number of diseases (37) and, thus, our findings suggest that
the many pro-inflammatory effects of UA may be initiated by the
activation of innate immunity through TLR4. Targeting the innate
immunity pathway may provide an alternative for the treatment of
diseases with inflammation.
TLRs are typical membrane receptors for innate
immunity and are the first line of defense during the innate immune
response (38). Studies have
documented the TLR4-mediated pro-inflammatory effects in podocytes
(39), oxidative stress in
mesangial cells (40) and renal
tubular damage (41,42) in proximal tubular cells under the
stimulation of high glucose, lipopolysaccharide and lipids. TLR4
has also been recognized as a receptor for UA and plays a
significant role in amplifying inflammatory effects (6). In peripheral blood from patients
with acute gout arthritis, the NF-κB level and IL-1β production
were markedly reduced after TLR4 blockade with anti-TLR4 antibody
(43), suggesting the key role of
TLR4 in gout. TLR4 signaling, including the adaptor molecule
myeloid differentiation factor 88 (MyD88) and the inflammasome
complex, actively participates in mediating experimental
tubule-interstitial nephritis (44). In experimental renal
transplantation, local TLR4 activation by endogenous ligands may be
a pathological link from unspecific primary damage to subsequent
chemokine release, and the infiltration and activation of immune
cells, leading to the deterioration of renal function and the
induction of renal fibrosis, in which UA may play an important role
(45). However, to the best of
our knowledge, whether TLR4 is an alternative pathway for UA
activation in renal tubular cells and whether it is a link between
metabolic injury to subsequent inflammatory cytokine release has
not been previously examined. In the present study, TLR4 expression
was elevated by soluble UA and the TLR4 inhibitor, TAK242,
significantly blocked the downstream NALP3 inflammasome, caspase-1
activation, IL-1β and ICAM-1 synthesis induced by soluble UA,
indicating the TLR4-dependent effects of soluble UA. The
inflammatory cytokine, ICAM-1, was also blocked by the TLR4
inhibitor, suggesting that TLR4 may be such a bridge to link
metabolic damage to inflammatory injury.
Extensive examination of the entering of UA into
tubular cells has revealed the presence of urate transporters on
both sides of tubular cells which mediate the absorption and
excretion of UA (46), as well as
many of the detrimental effects of UA (4,27).
However, in a previous study, the inhibitor of urate transporter
could not completely block the pro-inflammatory effects of UA
(28), suggesting the activation
of other systems which may also account for the injury. The innate
immune response has also been demonstrated to participate in
recognition, uptake and the responses of cells to MSU crystals in
immune cells. Recognition of the naked MSU crystal by TLR2 and TLR4
has been shown to promote the ingestion of naked MSU crystal by
phagocytes (6,47). As the uptake of urate by urate
transporters has not been examined for its association with innate
immunity, it is possible that TLR4 may also participate in soluble
UA uptake and play a role in soluble UA-induced renal injury.
Therefore, in the present study, we used 1 μM of the TLR4
inhibitor, TAK242, which has been shown to fully block TLR4
signaling, in order to examine the specific effects of TLR4. TAK242
reversed the NALP3 activation and caspase-1 overexpression induced
by soluble UA, suggesting the full dependence of NALP3 and
caspase-1 activation on TLR4 following stimulation with soluble UA.
These findings support our hypothesis that TLR4 mediates soluble UA
uptake and initiates the downstream injury induced by soluble
UA.
Nevertheless, unlike the complete blockade of the
NALP3 inflammasome and caspase-1 by TAK242, TAK242 only partially
inhibited the upregulation of IL-1β following incubation with
soluble UA. It has been reported that apart from the classical
cleavage pattern for IL-1β by caspase-1, IL-1β can also be cleaved
from pro-IL-1β by protease (48)
which has been detected in both the proximal and distal renal
tubules in normal renal tissue, as well as in patients with tubular
disorders (49). Alternatively,
cathepsin D has also been suggested as another possible means of
IL-1β processing, as shown in the acidosis driven damage-associated
molecular patterns (DAMPs) in cultured primary mouse glial cells
(50). It is possible that there
are other pathways available for IL-1β processing during soluble
UA-induced tubular injury and the urate transporter may play a
role; however, this requires further investigation.
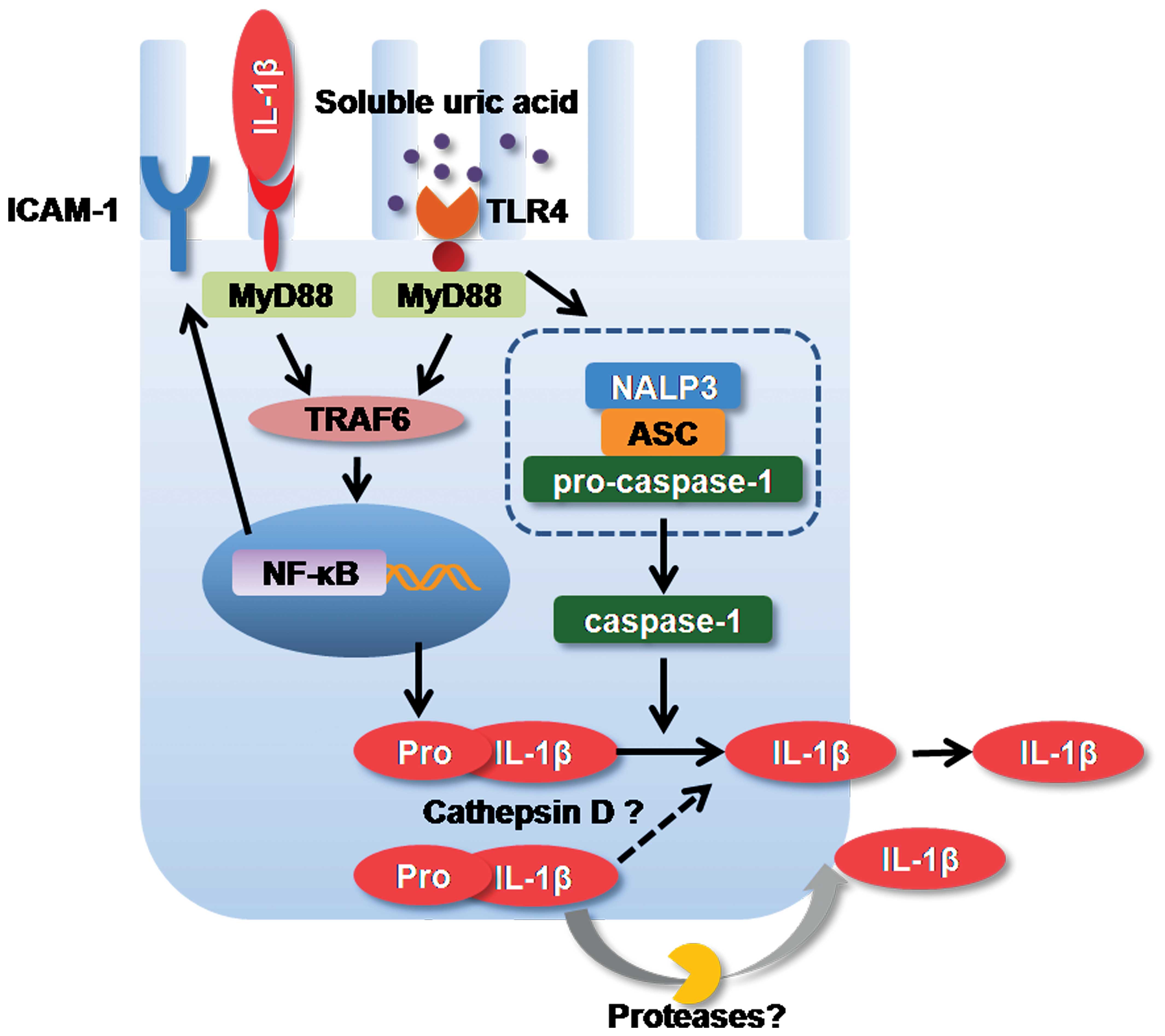
In conclusion, our present findings suggest that
soluble UA induces the formation of the NALP3 inflammasome,
caspase-1 activation, IL-1β expression and ICAM-1 synthesis in
human primary renal PTECs through a TLR4-dependent pathway, leading
to the activation of innate immunity and the induction of
pro-inflammatory cytokine production in human primary renal PTECs,
as illustrated in Fig. 5.
However, further studies on soluble UA using animal models and
renal biopsy samples from patients with hyperuricemia are warranted
to verify these findings in vivo.
Acknowledgments
The results of the present study have been presented
in the form of a poster at the World Congress of Nephrology 2013
(May 31 to June 4, Hong Kong). The present study was financially
suported by the National Natural Science Foundation of China (grant
no. 30900684/C140405) and the Shanghai Medical Guide of Science and
Technology Projects (grant no. 114119a6200).
References
|
1
|
Macisaac RJ, Ekinci EI and Jerums G:
Markers of and risk factors for the development and progression of
diabetic kidney disease. Am J Kidney Dis. 63(Suppl 2): S39–S62.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rizzo M, Obradovic M, Labudovic-Borovic M,
et al: Uric acid metabolism in pre-hypertension and the metabolic
syndrome. Curr Vasc Pharmacol. 12:572–585. 2014. View Article : Google Scholar
|
|
3
|
Johnson RJ, Nakagawa T, Sanchez-Lozada LG,
Shafiu M, Sundaram S, Le M, Ishimoto T, Sautin YY and Lanaspa MA:
Sugar, uric acid, and the etiology of diabetes and obesity.
Diabetes. 62:3307–3315. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou Y, Fang L, Jiang L, Wen P, Cao H, He
W, Dai C and Yang J: Uric acid induces renal inflammation via
activating tubular NF-κB signaling pathway. PLoS One. 7:e397382012.
View Article : Google Scholar
|
|
5
|
Kang DH, Nakagawa T, Feng L, Watanabe S,
Han L, Mazzali M, Truong L, Harris R and Johnson RJ: A role for
uric acid in the progression of renal disease. J Am Soc Nephrol.
13:2888–2897. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu-Bryan R, Scott P, Sydlaske A, Rose DM
and Terkeltaub R: Innate immunity conferred by Toll-like receptors
2 and 4 and myeloid differentiation factor 88 expression is pivotal
to monosodium urate monohydrate crystal-induced inflammation.
Arthritis Rheum. 52:2936–2946. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rock KL, Kataoka H and Lai JJ: Uric acid
as a danger signal in gout and its comorbidities. Nat Rev
Rheumatol. 9:13–23. 2013. View Article : Google Scholar :
|
|
8
|
Ghaemi-Oskouie F and Shi Y: The role of
uric acid as an endogenous danger signal in immunity and
inflammation. Curr Rheumatol Rep. 13:160–166. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Grainger R, McLaughlin RJ, Harrison AA and
Harper JL: Hyperuricaemia elevates circulating CCL2 levels and
primes monocyte trafficking in subjects with inter-critical gout.
Rheumatology (Oxford). 52:1018–1021. 2013. View Article : Google Scholar
|
|
10
|
Pineda C, Amezcua-Guerra LM, Solano C,
Rodriguez-Henríquez P, Hernández-Díaz C, Vargas A, Hofmann F and
Gutiérrez M: Joint and tendon subclinical involvement suggestive of
gouty arthritis in asymptomatic hyperuricemia: an ultrasound
controlled study. Arthritis Res Ther. 13:R42011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kanellis J, Watanabe S, Li JH, et al: Uric
acid stimulates monocyte chemoattractant protein-1 production in
vascular smooth muscle cells via mitogen-activated protein kinase
and cyclooxygenase-2. Hypertension. 41:1287–1293. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kang DH, Park SK, Lee IK and Johnson RJ:
Uric acid-induced C-reactive protein expression: implication on
cell proliferation and nitric oxide production of human vascular
cells. J Am Soc Nephrol. 16:3553–3562. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sánchez-Lozada LG, Lanaspa MA,
Cristóbal-García M, García-Arroyo F, Soto V, Cruz-Robles D,
Nakagawa T, Yu MA, Kang DH and Johnson RJ: Uric acid-induced
endothelial dysfunction is associated with mitochondrial
alterations and decreased intracellular ATP concentrations. Nephron
Exp Nephrol. 121:e71–e78. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sautin YY, Nakagawa T, Zharikov S and
Johnson RJ: Adverse effects of the classic antioxidant uric acid in
adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am
J Physiol Cell Physiol. 293:C584–C596. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Albertoni G, Maquigussa E, Pessoa E,
Barreto JA, Borges F and Schor N: Soluble uric acid increases
intracellular calcium through an angiotensin II-dependent mechanism
in immortalized human mesangial cells. Exp Biol Med (Maywood).
235:825–832. 2010. View Article : Google Scholar
|
|
16
|
Convento MS, Pessoa E, Dalboni MA, Borges
FT and Schor N: Pro-inflammatory and oxidative effects of
noncrystalline uric acid in human mesangial cells: contribution to
hyperuricemic glomerular damage. Urol Res. 39:21–27. 2011.
View Article : Google Scholar
|
|
17
|
Borges FT, Dalboni MA, Michelacci YM and
Schor N: Noncrystalline uric acid inhibits proteoglycan and
glycosaminoglycan synthesis in distal tubular epithelial cells
(MDCK). Braz J Med Biol Res. 43:957–963. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mazzali M, Hughes J, Kim YG, Jefferson JA,
Kang DH, Gordon KL, Lan HY, Kivlighn S and Johnson RJ: Elevated
uric acid increases blood pressure in the rat by a novel
crystal-independent mechanism. Hypertension. 38:1101–1106. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ficociello LH, Rosolowsky ET, Niewczas MA,
et al: High-normal serum uric acid increases risk of early
progressive renal function loss in type 1 diabetes: results of a
6-year follow-up. Diabetes Care. 33:1337–1343. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Imig JD and Ryan MJ: Immune and
inflammatory role in renal disease. Compr Physiol. 3:957–976.
2013.PubMed/NCBI
|
|
21
|
Webb R, Jeffries M and Sawalha AH: Uric
acid directly promotes human T-cell activation. Am J Med Sci.
337:23–27. 2009. View Article : Google Scholar
|
|
22
|
Shi Y, Mucsi AD and Ng G: Monosodium urate
crystals in inflammation and immunity. Immunol Rev. 233:203–217.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kanneganti TD, Ozören N, Body-Malapel M,
et al: Bacterial RNA and small antiviral compounds activate
caspase-1 through cryopyrin/Nalp3. Nature. 440:233–236. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brough D and Rothwell NJ:
Caspase-1-dependent processing of pro-interleukin-1beta is
cytosolic and precedes cell death. J Cell Sci. 120:772–781. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Witkowska AM and Borawska MH: Soluble
intercellular adhesion molecule-1 (sICAM-1): an overview. Eur
Cytokine Netw. 15:91–98. 2004.PubMed/NCBI
|
|
26
|
Johnson RJ, Nakagawa T, Jalal D,
Sánchez-Lozada LG, Kang DH and Ritz E: Uric acid and chronic kidney
disease: which is chasing which. Nephrol Dial Transplant.
28:2221–2228. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ryu ES, Kim MJ, Shin HS, Jang YH, Choi HS,
Jo I, Johnson RJ and Kang DH: Uric acid-induced phenotypic
transition of renal tubular cells as a novel mechanism of chronic
kidney disease. Am J Physiol Renal Physiol. 304:F471–F480. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang Z, Xiaohua W, Lei J, Ruoyun T,
Mingxia X, Weichun H, Li F, Ping W and Junwei Y: Uric acid
increases fibronectin synthesis through upregulation of lysyl
oxidase expression in rat renal tubular epithelial cells. Am J
Physiol Renal Physiol. 299:F336–F346. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Quan H, Peng X, Liu S, Bo F, Yang L, Huang
Z, Li H, Chen X and Di W: Differentially expressed protein profile
of renal tubule cell stimulated by elevated uric acid using SILAC
coupled to LC-MS. Cell Physiol Biochem. 27:91–98. 2011.PubMed/NCBI
|
|
30
|
Han HJ, Lim MJ, Lee YJ, Lee JH, Yang IS
and Taub M: Uric acid inhibits renal proximal tubule cell
proliferation via at least two signaling pathways involving PKC,
MAPK, cPLA2, and NF-kappaB. Am J Physiol Renal Physiol.
292:F373–F381. 2007. View Article : Google Scholar
|
|
31
|
Chen W, Roncal-Jimenez C, Lanaspa M,
Gerard S, Chonchol M, Johnson RJ and Jalal D: Uric acid suppresses
1 alpha hydroxylase in vitro and in vivo. Metabolism. 63:150–160.
2014. View Article : Google Scholar
|
|
32
|
Hua S: Targeting sites of inflammation:
Intercellular adhesion molecule-1 as a target for novel
inflammatory therapies. Front Pharmacol. 4:1272013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schiffrin EL: The immune system: role in
hypertension. Can J Cardiol. 29:543–548. 2013. View Article : Google Scholar
|
|
34
|
Schiffrin EL: Immune mechanisms in
hypertension and vascular injury. Clin Sci (Lond). 126:267–274.
2014. View Article : Google Scholar
|
|
35
|
Tsuboi N, Yoshikai Y, Matsuo S, Kikuchi T,
Iwami K, Nagai Y, Takeuchi O, Akira S and Matsuguchi T: Roles of
toll-like receptors in C-C chemokine production by renal tubular
epithelial cells. J Immunol. 169:2026–2033. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hato T, El-Achkar TM and Dagher PC:
Sisters in arms: myeloid and tubular epithelial cells shape renal
innate immunity. Am J Physiol Renal Physiol. 304:F1243–F1251. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Newton K and Dixit VM: Signaling in innate
immunity and inflammation. Cold Spring Harb Perspect Biol. 4:42012.
View Article : Google Scholar
|
|
38
|
Sloane JA, Blitz D, Margolin Z and
Vartanian T: A clear and present danger: endogenous ligands of
Toll-like receptors. Neuromolecular Med. 12:149–163. 2010.
View Article : Google Scholar :
|
|
39
|
Cha JJ, Hyun YY, Lee MH, et al: Renal
protective effects of toll-like receptor 4 signaling blockade in
type 2 diabetic mice. Endocrinology. 154:2144–2155. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee IT, Shih RH, Lin CC, Chen JT and Yang
CM: Role of TLR4/NADPH oxidase/ROS-activated p38 MAPK in VCAM-1
expression induced by lipopolysaccharide in human renal mesangial
cells. Cell Commun Signal. 10:332012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lin M, Yiu WH, Wu HJ, Chan LY, Leung JC,
Au WS, Chan KW, Lai KN and Tang SC: Toll-like receptor 4 promotes
tubular inflammation in diabetic nephropathy. J Am Soc Nephrol.
23:86–102. 2012. View Article : Google Scholar :
|
|
42
|
Cheng A, Dong Y, Zhu F, Liu Y, Hou FF and
Nie J: AGE-LDL activates Toll like receptor 4 pathway and promotes
inflammatory cytokines production in renal tubular epithelial
cells. Int J Biol Sci. 9:94–107. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Qing YF, Zhang QB, Zhou JG and Jiang L:
Changes in toll-like receptor (TLR)4-NFκB-IL1β signaling in male
gout patients might be involved in the pathogenesis of primary
gouty arthritis. Rheumatol Int. 34:213–220. 2014. View Article : Google Scholar
|
|
44
|
Correa-Costa M, Braga TT, Semedo P, et al:
Pivotal role of Toll-like receptors 2 and 4, its adaptor molecule
MyD88, and inflammasome complex in experimental tubule-interstitial
nephritis. PLoS One. 6:e290042011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bergler T, Hoffmann U, Bergler E, Jung B,
Banas MC, Reinhold SW, Krämer BK and Banas B: Toll-like receptor 4
in experimental kidney transplantation: early mediator of
endogenous danger signals. Nephron Exp Nephrol. 121:e59–e70. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Anzai N and Endou H: Urate transporters:
an evolving field. Semin Nephrol. 31:400–409. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Cronstein BN and Terkeltaub R: The
inflammatory process of gout and its treatment. Arthritis Res Ther.
8(Suppl 1): S32006. View
Article : Google Scholar : PubMed/NCBI
|
|
48
|
van de Veerdonk FL and Netea MG: New
insights in the immunobiology of IL-1 family members. Front
Immunol. 4:1672013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Manea M, Tati R, Karlsson J, Békássy ZD
and Karpman D: Biologically active ADAMTS13 is expressed in renal
tubular epithelial cells. Pediatr Nephrol. 25:87–96. 2010.
View Article : Google Scholar
|
|
50
|
Edye ME, Lopez-Castejon G, Allan SM and
Brough D: Acidosis drives damage-associated molecular pattern
(DAMP)-induced interleukin-1 secretion via a caspase-1-independent
pathway. J Biol Chem. 288:30485–30494. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chen Z, Liu Y, Sun B, et al:
Polyhydroxylated metallofullerenols stimulate IL-1beta secretion of
macrophage through TLRs/MyD88/NF-κB pathway and NLRP3
inflammasome activation. Small. 10:2362–2372. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Palová-Jelínková L, Dáňová K, Drašarová H,
et al: Pepsin digest of wheat gliadin fraction increases production
of IL-1β via TLR4/MyD88/TRIF/MAPK/NF-κB signaling pathway and an
NLRP3 inflammasome activation. PloS One. 8:e624262013. View Article : Google Scholar
|