Introduction
Systemic lupus erythematosus (SLE) is a typical
systemic autoimmune disease, involving diffuse connective tissues
(1) and is characterized by
immune inflammation. SLE has a complex pathogenesis (2), involving genetic, immunologic and
environmental factors. Thus, it may result in damage to multiple
tissues and organs, especially the kidneys (3). SLE arises from a combination of
heritable and environmental influences.
Epigenetics, the study of changes in gene expression
that occur without changes in the DNA sequence, have been suggested
to underlie age-related dysfunction and associated disorders
(5). The major epigenetic
mechanisms include DNA methylation, histone modifications and
microRNAs. Recent findings (4)
have shown that epigenetic abnormalities are closely correlated
with the pathogenesis of SLE. Epigenetic studies may provide clues
to elucidate the pathogenesis of SLE and develop new strategies to
treat this disease.
DNA hydroxymethylation (5-hydroxymethylcytosine,
5-hmC) (6,7) is a newly described epigenetic
modification. It is an oxidative product of the well-known DNA
methylation (5-methylcytosine, 5-mC) and catalyzed by the ten
eleven translocation (TET) family of enzymes (8), a family of enzymes dependent on
2-oxoglutarate and Fe(II) in vitro and in vivo.
The methylation of cytosine-guanine dinucleotides
(CpG) with C (9) is a common
epigenetic modification in mammals and is also widespread in
animals and plants. As an important epigenetic modification, 5-mC
regulates genomic functions, such as gene transcription,
X-chromosome inactivation, imprinting, genetic mutation and
chromosome stability (10–12).
5-mC is only one component of a dynamic epigenetic regulatory
network of DNA modifications that also includes 5-hmC,
5-formylcytosine and 5-carboxylcytosine. The reversible methylation
of N6-methyladenosine in RNA has also been demonstrated (13).
5-hmC was first found in bacteriophage DNA in 1952.
It was utilized several decades ago, only after its recent
identification in DNA from murine brain and stem cells rendered
5-hmC a major focus of epigenomic investigations (14). The lower affinity of
methyl-binding proteins to 5-hmC compared with 5-mC suggests that
this modification may have a distinct role in gene expression
regulation. However, 5-hmC is also involved in the DNA
demethylation process (15,16).
To obtain a deeper understanding of the role of
5-hmC with regard to the onset of SLE, we generated genome-wide
maps of 5-hmC in patients with SLE and healthy controls by
performing hydroxymethyl-DNA immunoprecipitation followed by
massively parallel sequencing with an Illumina Genome Analyzer
(hMeDIP-chip).
Materials and methods
Patients and controls
Whole blood samples from 15 SLE patients and 15
normal controls were obtained from the 181st Hospital of Guilin
(China), between January and September, 2011. The SLE diagnoses
were confirmed based on pathology and clinical evidence following
the American Rheumatism Association classification criteria
(1987).
Written informed consent was obtained from all the
subjects or their guardians. The use of biopsy material for studies
beyond routine diagnosis was approved by the local ethics
committee. This study abides by the Helsinki Declaration on ethical
principles for medical research involving human subjects.
Genomic DNA extraction and
fragmentation
Blood samples were obtained from SLE patients (n=15,
5 μl per subject pooled into one blood sample) and normal
controls (n=15, 5 μl per subject pooled into one blood
sample). Genomic DNA (gDNA) was extracted from the SLE patients and
normal control blood samples using a DNeasy Blood & Tissue kit
(Qiagen, Fremont, CA, USA). The purified gDNA was then quantified
and its quality assessed using a Nanodrop ND-1000 (Table I). The genomic DNA from each
sample pool was sonicated to ~200–1000 bp using a Bioruptor
sonicator (Diagenode, Denville, NJ, USA) on the ‘Low’ setting for
10 cycles of 30 sec ‘ON’ and 30 sec ‘OFF’. The gDNA and each
sheared DNA sample were analyzed on an agarose gel.
 | Table IDNA quantification and quality
assurance by NanoDrop spectrophotometer. |
Table I
DNA quantification and quality
assurance by NanoDrop spectrophotometer.
| Sample ID | OD260/2 80
ratio | OD260/2 30
ratio | Conc.
(ng/μl) | Volume
(μl) | Total amount
(ng) |
|---|
| Control | 1.81 | 2.05 | 41.60 | 100 | 4160.00 |
| SLE | 1.63 | 1.89 | 32.38 | 100 | 3238.00 |
GO analysis of differentially expressed
5-hmC
To investigate the specific functions of the
differentially expressed 5-hmC in the developmental process of SLE,
the 5-hmC targets of each differentially expressed 5-hmC were
identified by GO categories. The GO categories are derived from
gene ontology, which comprise three structured networks of defined
terms that describe gene product attributes.
Pathway analysis of differentially
expressed 5-hmC
Pathway analysis is a functional analysis mapping
genes to KEGG pathways. To evaluate the effect of SNP-to-gene
mapping strategy on pathway analysis, we also mapped SNPs to genes
within differentially expressed 5-hmC.
Immunoprecipitation
One microgram of the sonicated genomic DNA was used
for immunoprecipitation using a mouse monoclonal
anti-5-hydroxymethylcytosine antibody (Diagenode). Prior to
immunoprecipitation, the spike-in control sequences were mixed with
the genomic DNA fragments. The DNA was then heat-denatured at 94°C
for 10 min, rapidly cooled on ice, and immunoprecipitated with 1
μl of primary antibody overnight at 4°C with rocking
agitation in 400 μl of immunoprecipitation buffer (0.5% BSA
in PBS). To recover the immunoprecipitated DNA fragments, 200
μl of anti-mouse IgG magnetic beads were added and incubated
for an additional 2 h at 4°C with agitation. After
immunoprecipitation, a total of five immunoprecipitation washes
were performed with ice-cold immunoprecipitation buffer. The washed
beads were resuspended in TE buffer with 0.25% SDS and 0.25 mg/ml
proteinase K for 2 h at 65°C and then allowed to cool to room
temperature. The hMeDIP DNA fragments were purified using Qiagen
MinElute columns (Qiagen).
DNA labeling and array hybridization
For DNA labeling, the NimbleGen Dual-Color DNA
Labeling kit was used according to the manufacturer’s instructions
as detailed in the NimbleGen hMeDIP-chip protocol (NimbleGen
Systems, Inc., Madison, WI, USA). DNA (1 μg) from each
sample was incubated for 10 min at 98°C with 1 OD of Cy5-9mer
primer (IP sample) or Cy3-9mer primer (Input sample). Then, 100
pmol of deoxynucleoside triphosphates and 100 units of the Klenow
fragment (New England Biolabs, Beverly, MA, USA) were added, and
the mixture was incubated at 37°C for 2 h. The reaction was stopped
by adding 0.1X volume of 0.5 MEDTA, and the labeled DNA was
purified by isopropanol/ethanol precipitation. The microarrays were
hybridized at 42°C for 16–20 h with Cy3/5-labeled DNA in Nimblegen
hybridization buffer/hybridization component A in a hybridization
chamber (Hybridization System - Nimblegen Systems, Inc.). Following
hybridization, washing was performed using the Nimblegen Wash
Buffer kit (Nimblegen Systems, Inc.). For array hybridization,
Roche NimbleGen’s Promoter plus CpG Island Array was used, which is
a 385K array containing 28,226 CpG islands and well-characterized
promoter regions (approximately −800 to +200 bp relative to the
TSSs) that were completely covered by ~385,000 probes.
Quantitative RT-PCR verification of
5-hmC
The DNA was reverse transcribed to cDNA using
gene-specific primers (Table
II). The cycle parameters for the PCR reactions were 95°C for
10 min followed by 40 cycles of a denaturing step at 95°C for 10
sec and an annealing/extension step at 60°C for 60 sec. The
relative amount of each gene was described using the equation
2-ΔCt, where ΔCt = (CtmRNA-CtU6). The genes analyzed included
TREX1, CDKN1A and CDKN1B.
| Table IIReverse-transcription and RT-qPCR
primers. |
Table II
Reverse-transcription and RT-qPCR
primers.
| Gene name | RT-qPCR
primers | Annealing
temperature (°C) | Product length
(bp) |
|---|
| TREX1 | F:
5′-GTGTTCCAAGTGCTGCCAAA-3′ | | |
| R:
5′-CATAAAGAGCGTGGGCTACATAC-3′ | 60 | 245 |
| CDKN1A | F:
5′-AGCCTTCCTCACATCCTCCTT-3′ | | |
| R:
5′-GACGGCCAGAAAGCCAATC-3′ | 60 | 207 |
| CDKN1B | F:
5′-GCCAGCCAGAGCAGGTTT-3′ | | |
| R:
5′-GATTGACACGGCGAGTCTATTT-3′ | 60 | 224 |
Results
hMeDIP-chip
Using specific antibodies, we performed hMeDIP-chip
(17) on two samples: SLE
patients and normal controls. To determine the 5-hmC status of a
comprehensive set of human promoters, we enriched the DNA from
whole blood samples for hydroxymethylated DNA using hMeDIP-chip
methodology combined with microarray detection. The selected
platform was a single array design that included 28,226 CpG islands
and all the Ref gene promoter regions (approximately −800 to +200
bp relative to the TSSs) that were completely covered by ~385,000
probes. The median probe spacing was 101 bp.
DMR analysis using the MEDME method
To accurately quantify the CpG 5-hmC levels, we used
a new analytical methodology, MEDME (modeling experimental data
with hMeDIP enrichment), to improve the evaluation and
interpretation of the hMeDIP-derived 5-hmC estimates. MEDME
utilizes the absolute 5-hmC score (AHS) as the value for DNA
hydroxymethylation, which is calculated based on the weighted count
of the hydroxymethylated CpG dinucleotides in a 1 kb window
centered at each probe. The AHS has been verified to be a more
accurate and sensitive measurement of 5-hmC levels than the
log-ratio. The MEDME method also provides a relative 5-hmC score
(RHS) that normalizes the AHS to the total number of CpGs
represented by CpGw. This method allows investigators to obtain a
relative measurement of the 5-hmC that is independent of the CpG
density of the corresponding region. The RMS is especially useful
when comparing regions with different CpG densities.
Promoter classes in relation to CpG
frequency
Approximately 70% of human genes are associated with
promoter CpG islands, whereas the remaining promoters tend to be
depleted in CpGs. The presence of 5-hmC in promoter regions is
associated with high levels of transcription, which is consistent
with a role for 5-hmC in the maintenance and promotion of gene
expression. This effect is also partially dependent on the CpG
density of the promoter. Based on the CpG density, the CpG ratio
and length of the CpG-rich region, the promoters are subdivided
into three classes: high (HCP), low (LCP), and intermediate (ICP)
CpG density.
These classes are defined as follows: i)
High-CpG-density promoters (HCP) are promoters containing a 500-bp
interval within the region from 0.7 kb upstream to 0.2 kb
downstream of the TSS with a GC percentage ≥55% and a CpG
observed-to-expected ratio (O/E) ≥0.6. ii) Low-CpG-density
promoters (LCP) are promoters containing no 500 bp interval with a
CpG O/E ≥0.4. iii) Intermediate-CpG-density promoters (ICP) include
the remaining promoters that were not classified as HCP or LCP.
CpG island 5-hmC
Mammalian genomes are punctuated by DNA sequences
that contain an atypically high frequency of CpG sites termed CpG
islands (CGIs). These sequences are characterized as ≥200 bp in
length with a GC content of 50% and a CpG O/E of 0.6.
CpG islands can be grouped into three classes based
on their distance to RefSeq annotated genes: i) Promoter islands
occur from approximately −10 to +0.5 kb around the transcription
start site. ii) Intragenic islands occur from 0.5 kb downstream of
the transcription start site to the site of transcription
termination. iii) Intergenic islands include all other CpG islands
that were not classified as being in the promoter or intragenic
category (Fig. 1).
Genome-wide profiling of promoter DNA
5-hmC
Based on the data obtained, we examined the CpG
content in the pool of hydroxymethylated promoters compared to
non-hydroxymethylated promoters that exhibited significant
differences in 5-hmC levels between the SLE patients and normal
controls. We found that 65.95% of hydroxymethylated genes belonged
to the HCP cluster, which is similar to the average occurrence of
HCP genes genome-wide (67.82%) (Fig.
2A). Similarly, 69.85% of the non-hydroxymethylated genes were
associated with HCPs (Fig. 2A). A
detailed analysis of the distribution of the hydroxymethylated
probes over these promoters, which contained at least one CpG
island by definition, indicated that 75.21% of the HCP genes had a
hydroxymethylated probe that overlapped with the CpG island itself
(Figs. 1 and 2C). By contrast, ~45% of the ICP and LCP
genes were characterized as hydroxymethylated genes (Fig. 2B). We conclude that DNA
hydroxymethylation in the blood of SLE patients primarily occurs at
HCP promoters or at nonpromoter-CpG islands within HCP genes.
GO analysis of differentially expressed
5-hmC
To investigate the specific functions of the
differentially expressed 5-hmC in the developmental process of SLE,
the 5-hmC targets of each differentially expressed 5-hmC were
identified by GO categories. The GO categories were derived from
gene ontology, comprising three structured networks of defined
terms that describe gene product attributes. The P-value denotes
the significance of GO term enrichment in the differentially
expressed 5-hmC list. Thus, the lower the P-value, the more
significant the GO term, with P≤0.05 being recommended.
In terms of the GO database, the differentially
expressed proteins encoded by these genes were divided into three
categories: biological process, cell component and molecular
function (Fig. 3). Through GO
analysis for differentially expressed 5-hmC genes, we found
that 71 differentially expressed 5-hmC genes with annotation
terms being linked to the GO biological process categories, 30
being linked to the cell component and 20 being linked to the
molecular function, with P<0.01. Details of the cell component
categories, molecular function ontology, biological process
ontology are presented in Table
III.
| Table IIIFunctional analysis of genetic
differences of 5-hmC (P<0.01). |
Table III
Functional analysis of genetic
differences of 5-hmC (P<0.01).
| GO ID | Term | Gene (n) | P-value | Enrichment
score |
|---|
| Molecular
function |
| GO:0005488 | Binding | 975 | 8.47676E-06 | 5.071770331 |
| GO:0005515 | Protein
binding | 574 | 0.0006979 | 3.156206958 |
| GO:0008601 | Protein phosphatase
type 2A regulator activity | 6 | 0.002390223 | 2.621561587 |
| GO:0005275 | Amine transmembrane
transporter activity | 14 | 0.002741565 | 2.562001469 |
| GO:0046943 | Carboxylic acid
transmembrane transporter activity | 16 | 0.003032561 | 2.518190484 |
| GO:0008198 | Ferrous iron
binding | 5 | 0.003048552 | 2.515906422 |
| GO:0031267 | Small GTPase
binding | 19 | 0.003112164 | 2.506937561 |
| GO:0050662 | Coenzyme
binding | 26 | 0.003164007 | 2.499762629 |
| GO:0005342 | Organic acid
transmembrane transporter activity | 16 | 0.003738627 | 2.427287911 |
| GO:0019903 | Protein phosphatase
binding | 12 | 0.003795492 | 2.420731959 |
| GO:0004683 |
Calmodulin-dependent protein kinase
activity | 6 | 0.004182328 | 2.37858187 |
| GO:0015248 | Sterol transporter
activity | 5 | 0.004284399 | 2.368110104 |
| GO:0015370 | Solute:sodium
symporter activity | 10 | 0.005096459 | 2.292731449 |
| GO:0015293 | Symporter
activity | 19 | 0.005283676 | 2.277063859 |
| GO:0003677 | DNA binding | 210 | 0.00545348 | 2.263326261 |
| GO:0017016 | Ras GTPase
binding | 17 | 0.005511619 | 2.258720783 |
| GO:0003676 | Nucleic acid
binding | 290 | 0.005672365 | 2.246235825 |
| GO:0036094 | Small molecule
binding | 229 | 0.00603527 | 2.219303294 |
| GO:0000166 | Nucleotide
binding | 215 | 0.006277361 | 2.202222878 |
| GO:0019902 | Phosphatase
binding | 15 | 0.008158707 | 2.088378687 |
| Cellular
component |
| GO:0044424 | Intracellular
component | 991 | 1.25802E-06 | 5.900311838 |
| GO:0005622 | Intracellular | 1011 | 1.96706E-06 | 5.706182597 |
| GO:0005737 | Cytoplasm | 751 | 1.1809E-05 | 4.927788392 |
| GO:0043226 | Organelle | 863 | 1.23569E-05 | 4.908089407 |
| GO:0043229 | Intracellular
organelle | 860 | 2.02157E-05 | 4.694310928 |
| GO:0044444 | Cytoplasmic
component | 556 | 0.000162517 | 3.789099975 |
| GO:0043227 | Membrane-bound
organelle | 773 | 0.000201161 | 3.696456379 |
| GO:0043231 | Intracellular
membrane-bound organelle | 771 | 0.000263573 | 3.579099179 |
| GO:0005634 | Nucleus | 503 | 0.000511564 | 3.291100377 |
| GO:0044422 | Organelle
component | 518 | 0.000698363 | 3.155918681 |
| GO:0044446 | Intracellular
organelle component | 511 | 0.000826581 | 3.082714383 |
| GO:0005815 | Microtubule
organizing center | 54 | 0.001500698 | 2.823706722 |
| GO:0044464 | Cell component | 1146 | 0.001599278 | 2.79607593 |
| GO:0005623 | Cell | 1146 | 0.00163091 | 2.78757007 |
| GO:0015630 | Microtubule
cytoskeleton | 86 | 0.001672538 | 2.776623893 |
| GO:0031988 | Membrane-bound
vesicle | 85 | 0.002151442 | 2.667270359 |
| GO:0000159 | Protein phosphatase
type 2A complex | 6 | 0.002383019 | 2.622872453 |
| GO:0005856 | Cytoskeleton | 163 | 0.002653136 | 2.576240518 |
| GO:0030312 | External
encapsulating structure | 5 | 0.003040318 | 2.517080953 |
| GO:0016023 | Cytoplasmic
membrane-bound vesicle | 82 | 0.003540727 | 2.450907507 |
| GO:0031410 | Cytoplasmic
vesicle | 85 | 0.005938359 | 2.226333534 |
| GO:0044428 | Nuclear
component | 215 | 0.006110243 | 2.213941539 |
| GO:0043228 | Non-membrane-bound
organelle | 265 | 0.006188271 | 2.208430644 |
| GO:0043232 | Intracellular
non-membrane-bound organelle | 265 | 0.006188271 | 2.208430644 |
| GO:0031982 | Vesicle | 88 | 0.006466493 | 2.189331194 |
| GO:0005874 | Microtubule | 37 | 0.007122082 | 2.147393031 |
| GO:0043240 | Fanconi anaemia
nuclear complex | 4 | 0.007554527 | 2.12179274 |
| GO:0005829 | Cytosol | 201 | 0.007559368 | 2.121514538 |
| GO:0008328 | Ionotropic
glutamate receptor complex | 6 | 0.008472321 | 2.071997622 |
| GO:0031974 | Membrane-enclosed
lumen | 228 | 0.009528722 | 2.020965349 |
| Biological
process |
| GO:0009987 | Cell process | 1046 | 4.20173E-07 | 6.376571542 |
| GO:0006259 | DNA metabolic
process | 93 | 0.000371782 | 3.429711695 |
| GO:0042136 | Neurotransmitter
biosynthetic process | 6 | 0.000405199 | 3.392331799 |
| GO:0051179 | Localization | 374 | 0.000503121 | 3.2983276 |
| GO:0033554 | Cell response to
stress | 114 | 0.000534186 | 3.272307489 |
| GO:0016043 | Cell component
organization | 344 | 0.000617972 | 3.209030892 |
| GO:0008219 | Cell death | 166 | 0.000636118 | 3.19646263 |
| GO:0016265 | Death | 166 | 0.000672212 | 3.172493732 |
| GO:0071840 | Cell component
organization or biogenesis | 352 | 0.000808943 | 3.092082017 |
| GO:0071705 | Nitrogen compound
transport | 29 | 0.000836139 | 3.077721402 |
| GO:0010950 | Positive regulation
of endopeptidase activity | 18 | 0.000934594 | 3.029376965 |
| GO:0012501 | Programmed cell
death | 152 | 0.000944228 | 3.024923241 |
| GO:0071702 | Organic substance
transport | 59 | 0.001039557 | 2.983151486 |
| GO:0021987 | Ccerebral cortex
development | 12 | 0.001156639 | 2.9368022 |
| GO:0006915 | Apoptotic
process | 150 | 0.001275381 | 2.894360003 |
| GO:0015697 | Quaternary ammonium
group transport | 6 | 0.001359721 | 2.866550106 |
| GO:0006308 | DNA catabolic
process | 13 | 0.001439981 | 2.841643257 |
| GO:0010952 | Positive regulation
of peptidase activity | 18 | 0.001648322 | 2.782957883 |
| GO:0030301 | Cholesterol
transport | 12 | 0.001863628 | 2.729640865 |
| GO:0015918 | Sterol
transport | 12 | 0.002166069 | 2.664327795 |
| GO:0021543 | Pallium
development | 15 | 0.002411739 | 2.617669728 |
| GO:0042632 | Cholesterol
homeostasis | 11 | 0.00280871 | 2.551493105 |
| GO:0055092 | Sterol
homeostasis | 11 | 0.00280871 | 2.551493105 |
| GO:0007169 | Transmembrane
receptor protein tyrosine kinase signaling pathway | 66 | 0.003092099 | 2.509746586 |
| GO:0006281 | DNA repair | 45 | 0.00309275 | 2.509655117 |
| GO:0006919 | Activation of
cysteine-type endopeptidase activity involved in apoptotic
process | 14 | 0.003187141 | 2.496598699 |
| GO:0097202 | Activation of
cysteine-type endopeptidase activity | 14 | 0.003187141 | 2.496598699 |
| GO:0007049 | Cell cycle | 131 | 0.003246836 | 2.488539594 |
| GO:0006950 | Response to
stress | 254 | 0.003252788 | 2.487744307 |
| GO:0060317 | Cardiac epithelial
to mesenchymal transition | 5 | 0.003278753 | 2.484291311 |
| GO:0015837 | Amine
transport | 24 | 0.003396431 | 2.468977217 |
| GO:0010033 | Response to organic
substance | 156 | 0.003703461 | 2.431392194 |
| GO:0043280 | Positive regulation
of cysteine-type endopeptidase activity involved in apoptotic
process | 16 | 0.003962 | 2.402085511 |
| GO:2001056 | Positive regulation
of cysteine-type endopeptidase activity | 16 | 0.003962 | 2.402085511 |
| GO:0010941 | Regulation of cell
death | 119 | 0.004400697 | 2.35647858 |
| GO:0034641 | Cellular nitrogen
compound metabolic process | 465 | 0.004692655 | 2.328581332 |
| GO:0008629 | Induction of
apoptosis by intracellular signals | 15 | 0.004728501 | 2.32527652 |
| GO:0071842 | Cellular component
organization at the cellular level | 268 | 0.0048397 | 2.315181593 |
| GO:0032677 | Regulation of
interleukin-8 production | 8 | 0.004962569 | 2.304293443 |
| GO:0006807 | Nitrogen compound
metabolic process | 473 | 0.005208958 | 2.283249159 |
| GO:0042981 | Regulation of
apoptotic process | 115 | 0.005273137 | 2.277930908 |
| GO:0071294 | Cell response to
zinc ion | 4 | 0.005446325 | 2.26389645 |
| GO:0006139 |
Nucleobase-containing compound metabolic
process | 431 | 0.005604946 | 2.251428588 |
| GO:0006810 | Transport | 302 | 0.005935568 | 2.22653772 |
| GO:0071841 | Cell component
organization or biogenesis at the cellular level | 275 | 0.00611302 | 2.213744154 |
| GO:0051716 | Cell response to
stimulus | 413 | 0.006239288 | 2.204864975 |
| GO:0032365 | Intracellular lipid
transport | 5 | 0.006266841 | 2.20295135 |
| GO:0040007 | Growth | 76 | 0.006279845 | 2.20205109 |
| GO:0044272 | Sulfur compound
biosynthetic process | 11 | 0.006528232 | 2.185204402 |
| GO:0043067 | Regulation of
programmed cell death | 115 | 0.006686996 | 2.174768905 |
| GO:0007167 | Enzyme-linked
receptor protein signaling pathway | 83 | 0.006693713 | 2.174332938 |
| GO:0030168 | Platelet
activation | 28 | 0.007338694 | 2.134381237 |
| GO:0015695 | Organic cation
transport | 7 | 0.007484604 | 2.125831149 |
| GO:0014066 | Regulation of
phosphatidylinositol 3-kinase cascade | 9 | 0.007501308 | 2.124863025 |
| GO:0050794 | Regulation of the
cell process | 631 | 0.007609603 | 2.118638012 |
| GO:0044260 | Cellular
macromolecule metabolic process | 530 | 0.007722583 | 2.11223743 |
| GO:0042157 | Lipoprotein
metabolic process | 15 | 0.00781696 | 2.10696213 |
| GO:0051234 | Establishment of
localization | 305 | 0.007999858 | 2.096917735 |
| GO:0003203 | Endocardial cushion
morphogenesis | 4 | 0.008030603 | 2.095251834 |
| GO:0042149 | Cell response to
glucose starvation | 4 | 0.008030603 | 2.095251834 |
| GO:0045540 | Regulation of
cholesterol biosynthetic process | 4 | 0.008030603 | 2.095251834 |
| GO:0043065 | Positive regulation
of apoptotic process | 61 | 0.008093257 | 2.091876684 |
| GO:0035556 | Intracellular
signal transduction | 173 | 0.008130715 | 2.089871248 |
| GO:0032637 | Interleukin-8
production | 8 | 0.008361973 | 2.077691232 |
| GO:0090304 | Nucleic acid
metabolic process | 365 | 0.00864538 | 2.063215898 |
| GO:0046686 | Response to cadmium
ion | 7 | 0.009025274 | 2.044539606 |
| GO:0006586 | Indolalkylamine
metabolic process | 6 | 0.009194049 | 2.036493163 |
| GO:0009225 | Nucleotide-sugar
metabolic process | 6 | 0.009194049 | 2.036493163 |
| GO:0042430 | Indole-containing
compound metabolic process | 6 | 0.009194049 | 2.036493163 |
| GO:0006974 | Response to DNA
damage stimulus | 61 | 0.009681709 | 2.014047956 |
| GO:0043068 | Positive regulation
of programmed cell death | 61 | 0.009681709 | 2.014047956 |
Pathway analysis of differentially
expressed 5-hmC
Pathway analysis is a functional analysis mapping
genes to KEGG pathways. The P-value (EASE-score, Fisher P-value or
Hypergeometric P-value) denotes the significance of the pathway
correlated with the following conditions: the lower the P-value,
the more significant the pathway, with P=0.05 as the cut-off value.
In order to evaluate the influence of SNP-to-gene mapping strategy
on the pathway analysis, we mapped SNPs to genes within
differentially expressed 5-hmC.
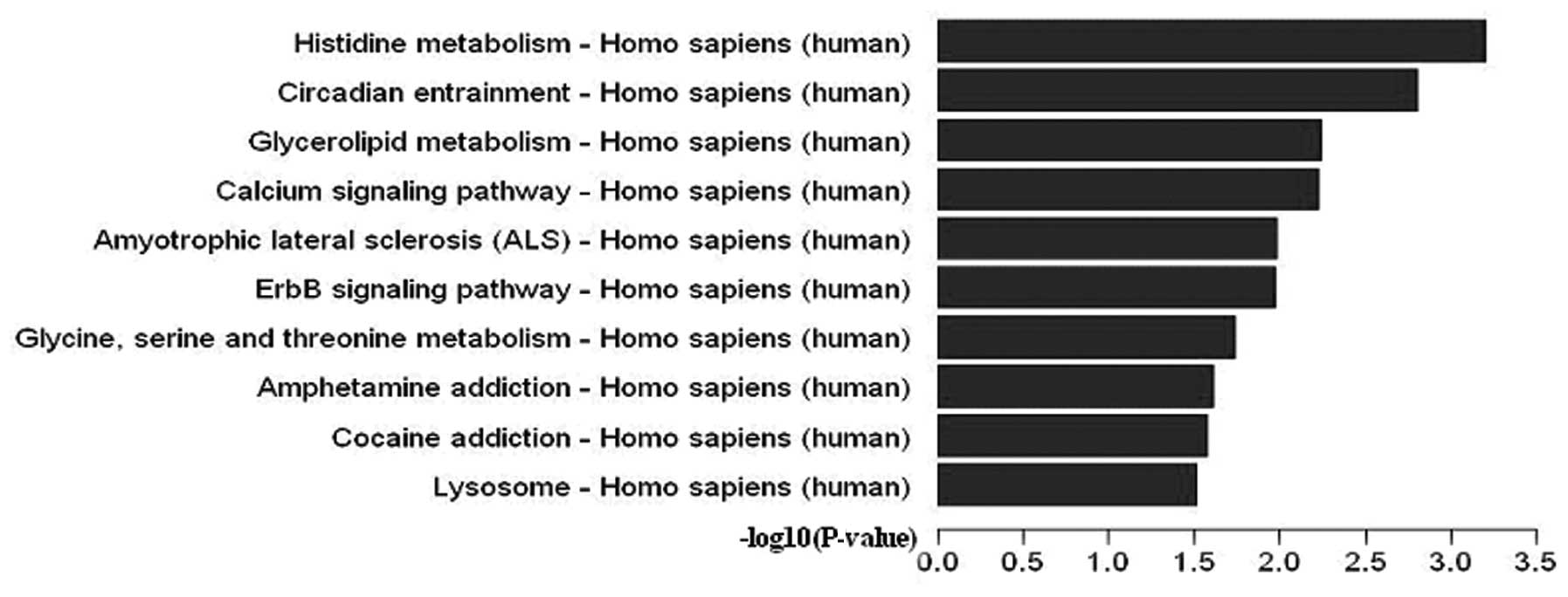
In terms of the Pathway database, 17 pathways were
significant (P<0.05). Differentially expressed 5-hmC is shown in
Fig. 4, while details of the
pathways are present in Table
IV. Furthermore, the CDKN1A and CDKN1B genes
contributed to the ErbB (P=0.01073062), P13-Akt (P=0.04341327), and
HIF-1 (P=0.04345306) signaling pathways.
| Table IVTransduction pathway analysis in
5-hmC differences genes. |
Table IV
Transduction pathway analysis in
5-hmC differences genes.
KEGG
Pathways | Fisher (n) | Gene
P-value | Enrichment
score | Genes |
|---|
| Histidine
metabolism - Homo sapiens | 0.000632576 | 8 | 3.198888 | ACY3,
ALDH1B1, ALDH3A2, ALDH3B1, HAL,
HEMK1, MAOA, WBSCR22 |
| Circadian
entrainment - Homo sapiens | 0.001569437 | 16 | 2.804256 | ADCY4,
ADCYAP1R1, CACNA1G, CAMK2D, GNAS,
GNG13, GRIA1, GRIN2A, GRIN2C,
GRIN2D, KCNJ5, MAPK1, NOS1,
PER1, PRKG1, RYR2 |
| Glycerolipid
metabolism - Homo sapiens | 0.005711304 | 10 | 2.243265 | AGPAT3,
ALDH1B1, ALDH3A2, CEL, DGKH,
DGKQ, DGKZ, GPAM, LIPG,
PPAP2A |
| Calcium signaling
pathway - Homo sapiens | 0.005990646 | 23 | 2.222526 | ADCY4,
ADRA1D, ADRB2, ATP2B3, CACNA1G,
CAMK2D, CAMK4, CD38, CYSL-TR1,
EGFR, GNAS, GRIN2A, GRIN2C, GRIN2D,
HTR7, MYLK2, MYLK3, NOS1, P2RX1,
PDE1B, PHKG2, PLCD4, RYR2 |
| Amyotrophic lateral
sclerosis (ALS) - Homo sapiens | 0.0104193 | 9 | 1.982162 | APAF1,
CYCS, DERL1, GRIA1, GRIN2A,
GRIN2C, GRIN2D, NEFH, NOS1 |
| ErbB signaling
pathway - Homo sapiens | 0.01073062 | 13 | 1.969375 | CAMK2D,
CDKN1A, CDKN1B, CRK, CRKL, EGFR,
ELK1, MAPK1, NCK1, NRG2, NRG3,
PIK3R3, PTK2 |
| Glycine, serine and
threonine metabolism - Homo sapiens | 0.01823201 | 7 | 1.739166 | BHMT,
CTH, DAO, GNMT, MAOA, PSPH,
SDSL |
| Amphetamine
addiction - Homo sapiens | 0.02439724 | 10 | 1.612659 | CAMK2D,
CAMK4, CREB3L2, GNAS, GRIA1,
GRIN2A, GRIN2C, GRIN2D, MAOA,
TH |
| Cocaine addiction -
Homo sapiens | 0.02669436 | 8 | 1.57358 | BDNF,
CREB3L2, GNAS, GRIN2A, GRIN2C,
GRIN2D, MAOA, TH |
| Lysosome - Homo
sapiens | 0.03098124 | 15 | 1.508901 | ABCA2,
AP3M1, AP3S2, AP4B1, ARSA,
ATP6V0B, ATP6V1H, CLTA, CTNS,
CTSL2, HEXB, IGF2R, NPC1, NPC2,
SLC17-A5 |
| Glycerophospholipid
metabolism - Homo sapiens | 0.03160531 | 12 | 1.50024 | AGPAT3,
CHAT, DGKH, DGKQ, DGKZ, ETNK1,
GNPAT, GPAM, LYPLA1, PEMT,
PPAP2A, TAZ |
| Fanconi anemia
pathway - Homo sapiens | 0.03646938 | 8 | 1.438072 | BLM,
FANCB, FANCD2, FANCE, FANCF,
RPA2, STRA13, WDR48 |
| Alcoholism -
Homo sapiens | 0.03755879 | 20 | 1.425288 | BDNF,
CAMK4, CREB3L2, GNAS, GNG13,
GRIN2A, GRIN2C, GRIN2D, H2AFV,
H2BFM, HIST1H3F, HIST1H4D, HIST2H2AB,
HIST2H2BF, HIST2H3D, HIST3H3, MAOA,
MAPK1, NTRK2, TH |
| PI3K-Akt signaling
pathway - Homo sapiens | 0.04341327 | 34 | 1.362377 | BCL2L11,
CDKN1A, CDKN1B, CHAD, COL6A1,
CREB3L2, EGFR, EPOR, FASLG,
FGF1, FLT1, GNG13, HSP90AA1,
HSP90B1, INSR, ITGAV, ITGB4,
JAK2, KITLG, LAMA5, LAMC3,
MAPK1, PIK3R3, PPP2R1A, PPP2R2B,
PPP2R3A, PPP2R5E, PRKCZ, PTEN,
PTK2, SGK1, THEM4, TLR2,
YWHAZ |
| HIF-1 signaling
pathway - Homo sapiens | 0.04345306 | 13 | 1.36198 | ALDOA,
CAMK2D, CDKN1A, CDKN1B, EGFR,
FLT1, IFNGR1, INSR, MAPK1, PGK1,
PIK3R3, SLC2A1, TF |
| DNA replication -
Homo sapiens | 0.04358268 | 6 | 1.360686 | MCM2,
PCNA, POLA2, RFC2, RNASEH2C,
RPA2 |
| Focal adhesion -
Homo sapiens | 0.04412979 | 22 | 1.355268 | BCAR1,
CHAD, COL6A1, CRK, CRKL, EGFR,
ELK1, FLNC, FLT1, ITGAV, ITGB4,
LAMA5, LAMC3, MAPK1, MYL12A,
MYLK2, MYLK3, PIK3R3, PPP1R12B,
PTEN, PTK2, VCL |
Comparison of 5-hmC status between SLE
patients and normal controls
By applying the analysis procedure described above
to the sequencing results, we found that 1,701 gene promoter
regions showed significantly different levels of 5-hmC in the SLE
patients compared with the normal controls. Of these genes, 884
exhibited increased 5-hmC and 817 exhibited decreased 5-hmC
(Fig. 5A). The CpG islands of
3,826 genes showed significant differences in 5-hmC levels in the
SLE patients compared with the normal controls. Of these genes,
2,034 exhibited increased 5-hmC and 1,792 exhibited decreased 5-hmC
(Fig. 5B).
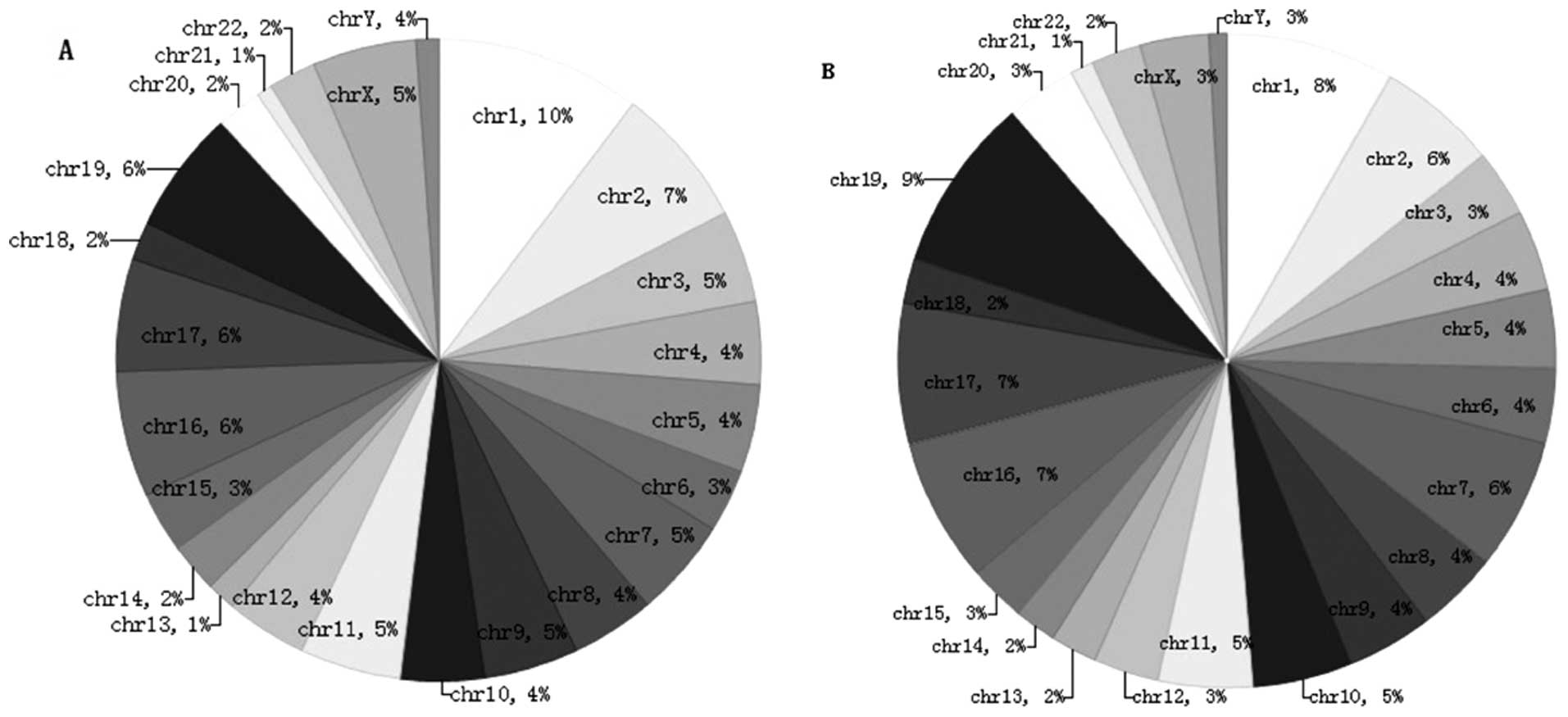
Pie chart A shows the chromosomal locations of the
884 genes that were hyper-hydroxymethylated within the promoter
region in the SLE patients compared with the normal controls
(clockwise from chromosome 1 to the X and Y sex chromosomes). The
percentage of genes hyper-hydroxymethylated on chromosome 1 was 10%
(Fig. 6A). Pie chart B shows the
chromosomal locations of the 2,034 genes that were
hyper-hydroxymethylated within the CpG islands in the SLE patients
compared with the normal controls (clockwise from chromosome 1 to
the X and Y sex chromosomes). The percentage of genes
hyper-hydroxymethylated on chromosome 29 was 9% (Fig. 6B).
The 5-hmC modifications of 15 selected genes are
shown in Table V. The selected
genes showed the greatest differences. Of these genes, we selected
three prime repair exonuclease 1 (TREX1), cyclin-dependent kinase
inhibitor 1A (p21, Cip1; CDKN1A), and cyclin-dependent kinase
inhibitor 1B (p27, Kip1; CDKN1B) for verification. The microarray
data were consistent with the RT-qPCR results (Table VI) showing that TREX1,
CDKN1A, and CDKN1B exhibited significantly increased
levels of 5-hmC. The three genes showed the largest differences in
5-hmC levels and may therefore be associated with SLE.
| Table VThe 15 selected genes with
hydroxymethylation alterations between SLE and normal controls,
identified by hmeDIP-seq. |
Table V
The 15 selected genes with
hydroxymethylation alterations between SLE and normal controls,
identified by hmeDIP-seq.
| Peak ID | Gene name | Peak region | Peak score | Peak M-value |
|---|
| 4636 | RTN1 | Chr14:
59263472-59264121 | 4.71 |
1.26471321387763 |
| 3845 |
S100A7L2 | Chr1:
151678931-151679801 | 4.55 |
0.967562504015608 |
| 5554 | ZNRF4 | Chr19:
5406466-5407310 | 4.48 |
1.10626932760703 |
| 5087 | CPNE7 | Chr16:
88168557-88169204 | 4.37 |
1.37675784921923 |
| 7131 | FZD3 | Chr8:
28406910-28407744 | 4.35 |
1.00487584265558 |
| 4370 | LAYN | Chr11:
110916352-110917416 | 4.32 |
0.86128687613001 |
| 6211 | LZTR1 | Chr22:
19665877-19666606 | 4.32 |
1.21336961342519 |
| 3824 | PRMT6 | Chr1:
107400534-107401083 | 4.23 |
0.934008953032345 |
| 7529 | VMA21 | ChrX:
150315981-150316530 | 4.13 |
1.16048247400229 |
| 6560 | FREM3 | Chr4:
144840477-144841631 | 4.09 |
1.18891146363541 |
| 3911 | HIST3H3 | Chr1:
226679229-226679963 | 4.08 |
1.33700123117401 |
| 6204 | GNB1L | Chr22:
18221904-18222633 | 4.07 |
0.77963334561276 |
| 6355 | TREX1 | Chr3:
48482232-48484048 | 2.7 |
0.450419940777759 |
| 6777 | CDKN1A | Chr6:
36754464-36763087 | 2.66 |
1.58157470937086 |
| 679 | CDKN1B | Chr12:
12761568-12766572 | 2.03 |
0.6574848262603 |
| Table VIRT-qPCR verification results. |
Table VI
RT-qPCR verification results.
| Sample | Input-IP
| Input-neg
| IP/neg |
|---|
| Input (Ct) | IP(Ct) | % | Input (Ct) | Neg (Ct) | % |
|---|
| TREX1 |
| SLE | 26.628 | 29.827 | 2.178 | 26.628 | NA | NA | – |
| Normal
controls | 25.648 | 37.884 | 0.004 | 25.648 | NA | NA | – |
| CDKN1A |
| SLE | 23.375 | 28.664 | 0.512 | 23.375 | NA | NA | – |
| Normal
controls | 22.38 | NA | NA | 25.648 | 22.38 | NA | – |
| CDKN1B |
| SLE | 26.474 | 33.43 | 0.161 | 26.474 | NA | NA | – |
| Normal
controls | 27.284 | 33.512 | 0.267 | 27.284 | NA | NA | – |
Discussion
The 5-hmC modification has been identified in
mammalian DNA (6), but its
broader role in epigenetics remains to be resolved. Early evidence
suggests a few putative mechanisms that have potentially important
implications (18): i) Conversion
of methylcytosine (5-mC) to 5-hmC may displace methyl-binding
proteins (MBPs). MeCP2, for instance, does not bind to 5-hmC. ii)
5-hmC may induce demethylation by interfering with the methylation
maintenance function of DNMT1 during cell division. iii) 5-hmC may
have its own specific binding proteins that alter the chromatin
structure or DNA methylation patterns.
5-hmC was previously observed; however, little is
known regarding its subtle interrelationship with other epigenetic
modifications and potential functional significance in human
disease. In this study, we selected 5-hmC as the target, performed
an investigation using hMeDIP-chip, and investigated the hypothesis
that 5-hmC is associated with the pathogenesis of SLE. We mainly
analyzed the levels of 5-hmC in SLE patients and normal controls.
The identified candidate genes with significant differences in
5-hmC levels are shown in Table
V. This list includes genes associated with immunity, cell
signal transduction, protein transcription and synthesis, ion
channels and transporters, and the extracellular matrix.
Of the identified candidate genes, we found that
TREX1 was hyper-hydroxymethylated in the SLE patients
compared with the normal controls. Three prime repair exonuclease 1
(TREX1) is located on chromosome 3p21.31 and is also known
as CRV, AGS1, DRN3 or HERNS. This gene encodes a
nuclear protein with 3′ exonuclease activity, which may play a role
in DNA repair and serve a proofreading function for DNA polymerase.
Mutations in this gene result in Aicardi-Goutieres syndrome,
chilblain lupus, Cree encephalitis, and other diseases of the
immune system. Alternative splicing of this gene results in
multiple transcript variants.
TREX1 plays a key role in the HIV-1 infection
process (19). This protein
degrades excess HIV-1 DNA, thereby preventing recognition by innate
immunity receptors and the type I interferon response. Rare
mutations in the TREX1 gene, the major mammalian 3′–5′
exonuclease, have been reported in sporadic SLE cases (20,21). Some of these mutations have also
been identified in a rare pediatric neurological condition
featuring an inflammatory encephalopathy known as Aicardi-Goutieres
syndrome (AGS) (22). The
mutations have also been identified in patients with several
different human diseases (23),
such as Aicardi-Goutieres syndrome 1, and account for all the
mutations in retinal vasculopathy with cerebral leukodystrophy.
These mutations include null alleles, frameshift mutations and
non-synonymous changes in the catalytic domains and the C-terminal
region. In AGS, most TREX1 mutations are autosomal recessive
and reduce exonuclease activity of the enzyme, in particular a
transition of arginine to histidine at position 114 (R114H).
Pulliero et al described mutations of the TREX1 gene
in Aicardi-Goutières syndrome 1 that increase the ability of
T-lymphocytes to inhibit the growth of neoplastic neuronal cells
and related angiogenesis (24).
In SLE, most of the mutations reported thus far are
heterozygous and are located outside of the catalytic domain in the
C-terminal region. The functional significance of these mutations
is unknown. To examine the frequency of mutations in the
TREX1 gene and their relationship with SLE, Namjou et
al (25) genotyped 40 SNPs in
the TREX1 genomic region, including previously reported rare
SNPs and more common tag SNPs that capture most of the variation in
this region. Those authors reported results indicating that
TREX1 is involved in the lupus pathogenesis and is most
likely essential for the prevention of autoimmunity. Gene Ontology
(GO) term analysis shows that TREX1 is mainly associated
with the cell process, cellular nitrogen compound metabolic
process, cell response to stress, intracellular component,
intracellular, binding, and protein binding.
We also observed that CDKN1A was
significantly hyper-hydroxymethylated and CDKN1B was
significantly hypo-hydroxymethylated in the SLE patients compared
with the normal controls. The cyclin-dependent kinase inhibitor 1A
(p21, Cip1; CDKN1A) gene is located on chromosome 6p21.2 and
is also known as P21, CIP1, SDI1, WAF1, CAP20, CDKN1, MDA-6
or p21CIP1. This gene encodes a potent cyclin-dependent
kinase inhibitor. The encoded protein binds to and inhibits the
activity of the cyclin-CDK2 or -CDK4 complexes and thus functions
as a regulator of G1 cell cycle progression. The expression of this
gene is closely regulated by the tumor-suppressor protein p53 and
mediates the p53-dependent G1 cell cycle arrest in response to a
variety of stress stimuli. This protein can interact with
proliferating cell nuclear antigen (PCNA), a DNA polymerase
accessory factor, and plays a regulatory role in DNA replication
and DNA damage repair. This protein was reported to be specifically
cleaved by CASP3-like caspases, which leads to marked activation of
CDK2 and may be instrumental in the execution of apoptosis
following caspase activation. Multiple alternatively spliced
variants have been identified for this gene.
The CDKN1A gene that encodes a cell cycle
inhibitor, p21 (WAF1/CIP1), is located in a region associated with
SLE susceptibility. Decreased cell levels of p21 are associated
with SLE (26,27). Single-nucleotide polymorphisms
(SNPs) within the promoter and the first intron of CDKN1A
are associated with SLE susceptibility. The minor allele A at
nucleotide 899 of CDKN1A is associated with increased
susceptibility to SLE and lupus nephritis and decreased cell levels
of p21.
The cyclin-dependent kinase inhibitor 1B (p27, Kip1;
CDKN1B) encodes a cyclin-dependent kinase inhibitor, which
shares a limited similarity with the CDK inhibitor
CDKN1A/p21. The encoded protein binds to and prevents the
activation of the cyclin E-CDK2 or cyclin D-CDK4
complexes and thus controls G1 cell cycle progression. The
degradation of this protein, which is triggered by its
CDK-dependent phosphorylation and subsequent ubiquitination by SCF
complexes, is required for the cellular transition from quiescence
to the proliferative state.
CDKN1B (28) may lead to defects in apoptosis or
autophagy and thus increase exposure of nuclear autoantigens to the
immune system, and its potential role in autoimmunity is supported
by numerous functional studies. CDKN1B encodes p27Kip1, a
cyclin-dependent kinase (CDK) inhibitor, which plays a critical
role in the inhibition of cell-cycle progression, especially in T
lymphocytes. p27Kip1 is essential for the induction of tolerance, a
process believed to be at the center of autoimmune diseases such as
SLE, and upregulation of p27Kip1 was found to correlate with the
induction of anergy in vitro and tolerance in vivo.
p27Kip1 is also involved in dendritic cell apoptosis, and the
potential roles of the identified susceptibility genes in SLE
etiology are noted. GO term analysis indicates that CDKN1A
and CDKN1B are strongly associated with the cell process,
intracellular component, intracellular, binding, and protein
binding.
In this study, we systematically evaluated the
genome-wide levels of 5-hmC in the DNA of SLE patients and gained
insight into the connections between key genes and 5-hmC in the
context of SLE. Our results indicate that 5-hmC is involved in the
disease state and these novel candidate genes may become potential
biomarkers or future therapeutic targets. Future investigations are
needed to clarify the roles of the identified hydroxymethylated
candidate genes in the pathogenesis of SLE.
Acknowledgments
The authors are deeply grateful to all the
volunteers. This study was supported by the Guangxi Natural Science
Foundation (no. 2012GXNSFDA053017) and by the Guangxi Key
Laboratory of Metabolic Diseases Research (no. 12-071-32).
References
|
1
|
Woodman I: Connective tissue diseases: The
MECP2/IRAK1 locus modulates SLE risk via epigenetics. Nat Rev
Rheumatol. 9:1972013.PubMed/NCBI
|
|
2
|
Baizabal-Carvallo JF, Alonso-Juarez M and
Koslowski M: Chorea in systemic lupus erythematosus. J Clin
Rheumatol. 17:69–72. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ponticelli C, Glassock RJ and Moroni G:
Induction and maintenance therapy in proliferative lupus nephritis.
J Nephrol. 23:9–16. 2010.PubMed/NCBI
|
|
4
|
Thabet Y, Cañas F, Ghedira I, Youinou P,
Mageed RA and Renaudineau Y: Altered patterns of epigenetic changes
in systemic lupus erythematosus and auto-antibody production: is
there a link? J Autoimmun. 39:154–160. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sui W, Hou X, Che W, Yang M and Dai Y: The
applied basic research of systemic lupus erythematosus based on the
biological omics. Genes Immun. 14:133–146. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tahiliani M, Koh KP, Shen Y, et al:
Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in
mammalian DNA by MLL partner TET1. Science. 324:930–935. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang Y, Pastor WA, Shen Y, Tahiliani M,
Liu DR and Rao A: The behaviour of 5-hydroxymethylcytosine in
bisulfite sequencing. PLoS One. 5:e88882010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ito S, D’Alessio AC, Taranova OV, Hong K,
Sowers LC and Zhang Y: Role of Tet proteins in 5mC to 5hmC
conversion, ES-cell self-renewal and inner cell mass specification.
Nature. 466:1129–1133. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yamaguchi S, Hong K, Liu R, et al:
Dynamics of 5-methylcytosine and 5-hydroxymethylcytosine during
germ cell reprogramming. Cell Res. 23:329–339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jin SG, Kadam S and Pfeifer GP:
Examination of the specificity of DNA methylation profiling
techniques towards 5-methylcytosine and 5-hydroxymethylcytosine.
Nucleic Acids Res. 38:e1252010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu SC and Zhang Y: Active DNA
demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol.
11:607–620. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Williams K, Christensen J and Helin K: DNA
methylation: TET proteins-guardians of CpG islands? EMBO Rep.
13:28–35. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song CX, Yi C and He C: Mapping recently
identified nucleotide variants in the genome and transcriptome. Nat
Biotechnol. 30:1107–1116. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stroud H, Feng S, Morey Kinney S, Pradhan
S and Jacobsen SE: 5-Hydroxymethylcytosine is associated with
enhancers and gene bodies in human embryonic stem cells. Genome
Biol. 12:R542011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu Y, Wu F, Tan L, et al: Genome-wide
regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in
mouse embryonic stem cells. Mol Cell. 42:451–464. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao Y, Chen J, Li K, et al: Replacement of
Oct4 by Tet1 during iPSC induction reveals an important role of DNA
methylation and hydroxymethylation in reprogramming. Cell Stem
Cell. 12:453–469. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thomson JP, Lempiäinen H, Hackett JA, et
al: Non-genotoxic carcinogen exposure induces defined changes in
the 5-hydroxymethylome. Genome Biol. 13:R932012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guo JU, Su Y, Zhong C, Ming GL and Song H:
Hydroxylation of 5-methylcytosine by TET1 promotes active DNA
demethylation in the adult brain. Cell. 145:423–434. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sironi M, Biasin M, Forni D, et al:
Genetic variability at the TREX1 locus is not associated with
natural resistance to HIV-1 infection. AIDS. 26:1443–1445. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hur JW, Sung YK, Shin HD, Cheong HS and
Bae SC: TREX1 polymorphisms associated with autoantibodies in
patients with systemic lupus erythematosus. Rheumatol Int.
28:783–789. 2008. View Article : Google Scholar
|
|
21
|
Lee-Kirsch MA, Gong M, Chowdhury D, et al:
Mutations in the gene encoding the 3′–5′ DNA exonuclease TREX1 are
associated with systemic lupus erythematosus. Nat Genet.
39:1065–1067. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
O’Driscoll M: TREX1 DNA exonuclease
deficiency, accumulation of single stranded DNA and complex human
genetic disorders. DNA Repair. 7:997–1003. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kavanagh D, Spitzer D, Kothari PH, et al:
New roles for the major human 3′–5′ exonuclease TREX1 in human
disease. Cell Cycle. 7:1718–1725. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pulliero A, Marengo B, Domenicotti C, et
al: Inhibition of neuroblastoma cell growth by TREX1-mutated human
lymphocytes. Oncol Rep. 27:1689–1694. 2012.PubMed/NCBI
|
|
25
|
Namjou B, Kothari PH, Kelly JA, et al:
Evaluation of the TREX1 gene in a large multi-ancestral lupus
cohort. Genes Immun. 12:270–279. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim K, Sung YK, Kang CP, Choi CB, Kang C
and Bae SC: A regulatory SNP at position -899 in CDKN1A is
associated with systemic lupus erythematosus and lupus nephritis.
Genes Immun. 10:482–486. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Miyagawa H, Yamai M, Sakaguchi D, et al:
Association of polymorphisms in complement component C3 gene with
susceptibility to systemic lupus erythematosus. Rheumatology.
47:158–164. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang W, Tang H, Zhang Y, et al:
Meta-analysis followed by replication identifies loci in or near
CDKN1B, TET3, CD80, DRAM1, and ARID5B as associated with systemic
lupus erythematosus in Asians. Am J Hum Genet. 92:41–51. 2013.
View Article : Google Scholar : PubMed/NCBI
|