Introduction
Mitochondrial 3-hydroxy-3-methylglutaryl-CoA lyase
(HMGCL; EC 4.1.3.4) deficiency is an autosomal recessive disorder
that affects leucine catabolism and ketogenesis. The HMGCL
gene, located on chromosome 1p36.1, contains nine exons and spans
approximately 25 kb (1). In the
majority of HMGCL-deficient patients, the first hypoglycemic crisis
occurs before the age of one, while one-third of all cases may have
neonatal onset. In acute episodes, laboratory data have shown
patients with non- or hypoketotic hypoglycemia with high levels of
free fatty acids and severe metabolic acidosis with liver
dysfunction and hyperammonemia (2). In Japan, HMGCL deficiency is one of
the inborn errors of metabolism screened for in newborns by tandem
mass spectrometry. Six Japanese HMGCL-deficient patients, including
those previously reported (3)
were re-evaluated (2). Among
them, three had neonatal onset. Follow-up data showed that two
patients experienced hypoglycemic crises even after ten years of
age. Developmental delay and epilepsy were noted in two and three
patients, respectively (2).
We recently encountered two Japanese HMGCL-deficient
patients, whose inheritance patterns of single nucleotide mutations
were not consistent with transmission within their families.
HMGCL has 23 Alu elements in introns. Recombination between
Alu elements results in genomic deletions associated with a number
of human genetic disorders (4).
Hence, we hypothesized that these patients may have an intragenic
deletion by non-equal homologous recombination between Alu elements
(5–7). Large homozygous deletions can be
suspected by the absence of the deleted exons detected by PCR
amplification. However, the detection of heterozygous deletions is
difficult using routine PCR amplification of genomic DNA and direct
sequencing. Multiplex ligation-dependent probe amplification (MLPA)
has been proven to be an efficient and reliable technique for the
copy number analysis of each exon in a gene (5,8–10).
In the present study, we applied MLPA for the analysis of copy
numbers in exons of HMGCL and confirmed mutations in the two
patients with HMGCL deficiency.
Patients and methods
Patients
Patient 1 was of the female gender, born to
non-consanguineous parents, who presented with hypoglycemia at the
age of 2 days. She also experienced hypoketotic hypoglycemic crises
at the age of 6, 8 and 13 months. She was diagnosed as having HMGCL
deficiency at the age of 13 months by urine organic acid analysis,
which detected 3-hydroxymethylgluta-rate, 3-methylglutaconate and
3-hydroxy-3-methylglutarate. The patient is currenlty 13 years old.
She has epilepsy and developmental delay.
Patient 2 was of the male gender, born to
non-consanguineous parents, who presented with vomiting and
unconsciousness at the age of 3 months. He was diagnosed as having
HMGCL deficiency at the age of 3 months by urine organic acid
analysis and blood acylcarnitine analysis. He has experienced ten
or more hypoketotic hypoglycemic crises, the last of which was at
the age of 4 years. He is currently 8 years old and has achieved
normal development. A case report for this patient has been
previously published in Japanese (11).
Mutation analysis at the genomic DNA
level
The present study was approved by the Ethics
Committee of the Graduate School of Medicine, Gifu University,
Gifu, Japan. Genomic DNA was purified from peripheral blood samples
using Sepa Gene kits (Sanko Junyaku Co., Ltd., Tokyo, Japan).
Mutation screening was performed at the genomic level by PCR and
direct sequencing, using a set of primer pairs that amplify
fragments, including exons and their intron boundaries. The primer
sequences are presented in Table
I.
 | Table IAmplification primer for HMGCL
exons. |
Table I
Amplification primer for HMGCL
exons.
| Exon | Foward primer | Sequence | Reverse primer | Sequence | Product size
(bp) |
|---|
| 1 | HL1s |
5′-GTGGAGCCAGCTTCGGAAGT-3′ | HL1as |
5′-GGGAGGGTCCAGGACTCCAACG-3′ | 324 |
| 2 | HL2s |
5′-ATGAATTCGGTCTCCCTGGGAATTG-3′ | HL2as |
5′-TAACTTGTGCAGAGGAATCACATC-3′ | 275 |
| 3 | HL3s |
5′-ATGAATTCTGCATTTTGAGGCTGTTT-3′ | HL3as |
5′-TTTGCTGCAACACAGTGCTATG-3′ | 325 |
| 4 | HL4s |
5′-ATGAATTCCTGCTCTTGGTGATGACT-3′ | HL4as |
5′-GATCACAGAGCAGTGAGTGGCA-3′ | 314 |
| 5 | HL5s |
5′-GAACCCAGGAGGTGGAGGTTGCA-3′ | HL5a |
5′-ATAAGCTTGAACGGTACAGAGGAAAGGA-3′ | 329 |
| 6 | HL6s |
5′-CTGGCACTGAATTGTACCAT-3′ | HL6as |
5′-GGGTGAATGAATGAAGTCAGGA-3′ | 336 |
| 7 | HL7s |
5′-AACTGAGTGCGTCATACCCAGA-3′ | HL7as |
5′-CAGAGCTGTACACTTCACATCTG-3′ | 473 |
| 8 | HL8s |
5′-ATGAATTCGGCAACAGACGATTGGG-3′ | HL8as |
5′-GAGCCACTGCGCCTGGCTAACC-3′ | 366 |
| 9 | HL9s |
5′-CCTGGTGTTGAGGGCATACC-3′ | HL9as |
5′-TGCCAGGAGAGACCTCTGTGTA-3′ | 300 |
Establishment of MLPA for the analysis of
HMGCL
The MLPA reaction is an efficient and reliable
technique for the analysis of exon copy numbers. We designed a pair
of MLPA probes for each HMGCL exon, using the human MLPA
probe design program (H-MAPD), as previously described (12). The MLPA probe sets for the
HMGCL exon are listed in Table II. MLPA reactions were performed
according to the manufacturer’s instructions (MRC-Holland BV,
Amsterdam, The Netherlands) using 100 ng of genomic DNA, the EK1
MLPA reagent kit and the P200-A1 Human DNA reference kit, which
includes reference probes and MLPA control fragments (MRC-Holland
BV). The PCR products were separated by capillary electrophoresis
on an ABI 3130×l genetic analyzer (Applied Biosystems, Warrington,
UK). GeneMapper v4.0 software (Applied Biosystems) was used to
analyze the separated products and to retrieve peak intensities
corresponding to each probe in the different samples. Integrated
peak areas were exported to an Excel 2003 spreadsheet. Data
generated from a combination of the HMGCL synthetic probe
mix and the P200-A1 probe mix were intra-normalized by dividing the
peak area of the amplification product of each probe by the total
area of only the reference probes in P200-A1. Secondly,
normalization was achieved by dividing this intra-normalized probe
ratio in a sample by the average intra-normalized probe ratio of
all reference samples.
| Table IIMLPA probes for the HMGCL
gene. |
Table II
MLPA probes for the HMGCL
gene.
| Exon | Product length
(base) | Primer name | Length | Probe sequence |
|---|
| 1 | 104 | MLPA-HMGCLEX1L | 50 |
GGGTTCCCTAAGGGTTGGA5016TGGACTGCCGCGGGGGATTCTGGGCCAAGAT |
| | MLPA-HMGCLEX1R | 54 |
5047GGCAGCAATGAGGAAGGCGCTTCCGCGGCGATCTAGATTGGATCTTGCTGGCAC |
| 2 | 108 | MLPA-HMGCLEX2L | 52 |
GGGTTCCCTAAGGGTTGGA9872CACCTCATCTATGGGCACTTTACCAAAGCGGGT |
| | MLPA-HMGCLEX2R | 56 |
9905GAAAATTGTGGAAGTTGGTCCCCGAGATGGACTTCTAGATTGGATCTTGCTGGCAC |
| 3 | 112 | MLPA-HMGCLEX3L | 54 |
GGGTTCCCTAAGGGTTGGA12925GAAGCAGGACTCTCTGTTATAGAAACCACCAGCTT |
| | MLPA-HMGCLEX3R | 58 |
12960TGTGTCTCCTAAGTGGGTTCCCCAGGTGAGCCCTATCTAGATTGGATCTTGCTGGCAC |
| 4 | 116 | MLPA-HMGCLEX4L | 56 |
GGGTTCCCTAAGGGTTGGA13730TTCCTGGCATCAACTACCCAGTCCTGACCCCAAATTT |
| | MLPA-HMGCLEX4R | 60 |
13767GAAAGGCTTCGAGGCAGCGGTAAGAGGATAGCTTGTTTCTAGATTGGATCTTGCTGGCAC |
| 5 | 120 | MLPA-HMGCLEX5L | 58 |
GGGTTCCCTAAGGGTTGGA16193TTGTTCCATAGAGGAGAGTTTTCAGAGGTTTGACGCAAT |
| | MLPA-HMGCLEX5R | 62 |
16232CCTGAAGGCAGCGCAGTCAGCCAATATTTCTGTGCGGGGTCTAGATTGGATCTTGCTGGCAC |
| 6 | 124 | MLPA-HMGCLEX6L | 60 |
GGGTTCCCTAAGGGTTGGA19636TCATTCCTCCCCTGTCTTCCCACAGGTACGTCTCCTGTGCT |
| | MLPA-HMGCLEX6R | 64 |
19677CTTGGCTGCCCTTATGAAGGGAAGATCTCCCCAGCTAAAGTTCTAGATTGGATCTTGCTGGCAC |
| 7 | 128 | MLPA-HMGCLEX7L | 62 |
GGGTTCCCTAAGGGTTGGA22152TACTCAATGGGCTGCTACGAGATCTCCCTGGGGGACACCATTG |
| | MLPA-HMGCLEX7R | 66 |
22195GTGTGGGCACCCCAGGGATCATGAAAGACATGCTATCTGCTGTTCTAGATTGGATCTTGCTGGCAC |
| 8 | 132 | MLPA-HMGCLEX8L | 64 |
GGGTTCCCTAAGGGTTGGA25930TCTAGATGGGAGTGAGTGTCGTGGACTCTTCTGTGGCAGGACTTG |
| | MLPA-HMGCLEX8R | 68 |
25975GAGGCTGTCCCTACGCACAGGGGGCATCAGGAAACTTGGCCACAGTCTAGATTGGATCTTGCTGGCAC |
| 9 | 136 | MLPA-HMGCLEX9L | 66 |
GGGTTCCCTAAGGGTTGGA27975CTCAGGCTACCTGTAAACTCTGAGCCCCTTGCCCACCTGAAGCCCTG |
| | MLPA-HMGCLEX9R | 70 |
28022GGGATGATGTGGAAATAGGGGCACACACAGATGATTCATGGATGGGGTCTAGATTGGATCTTGCTGGCAC |
Deletion breakpoint characterization
The region surrounding the deletion from intron 1 to
intron 4 in patient 1 was amplified using three primer pairs as
follows: fragment A: sense primer (In1s1,
5′-ACGAACGGTGGTAAAGAGGCAACAG-3′) located at position g.6421–6445 in
intron 1 and antisense primer (Ex5as,
5′-TTGGCTGACTGCGCTGCCTTCAGGA-3′) located at position g.16255–16231
in exon 5; fragment B: sense primer (In1s3,
5′-GTGATGATTCCAGGAGGTCAGA GGA-3′) located at position g.8701–8725
in intron 1 and antisense primer Ex5as; and fragment C: sense
primer In1s3 and antisense primer (In4as1:
5′-GAGAGGCATAGGACAGATTCTCC-3′) located at position g.15110–15088 in
intron 4 (GenBank accession no. NG_013061).
PCR was carried out for 40 cycles at 94°C for 1 min,
64°C for 2 min, 72°C for 2 min followed by a 5-min extension at
72°C using Takara r-Taq (Takara Shuzo Co., Ltd., Shiga, Japan) and
a Takara PCR thermal cycler. After subcloning into the pGEM-T Easy
vector (Promega, Madison, WI, USA), the fragments were
sequenced.
Comparative genomic hybridization (CGH)
and single nucleotide polymorphism (SNP) microarray analysis
Whole genomic copy number analysis was performed for
patient 2 using an Agilent SurePrint G3 Hmn CGH + SNP 180K
Microarray kit (Agilent Technologies, Santa Clara, CA, USA),
according to the manufacturer’s instructions. Genomic DNA extracted
from peripheral blood was used as a template. Data were extracted
using Feature Extraction version 9 (Agilent Technologies) and the
results were visualized using Agilent Genomic Workbench version 6.5
(Agilent Technologies).
Results
Mutation analysis
Mutations were screened at the genomic level using
PCR amplification followed by direct sequencing. Patient 1 had a
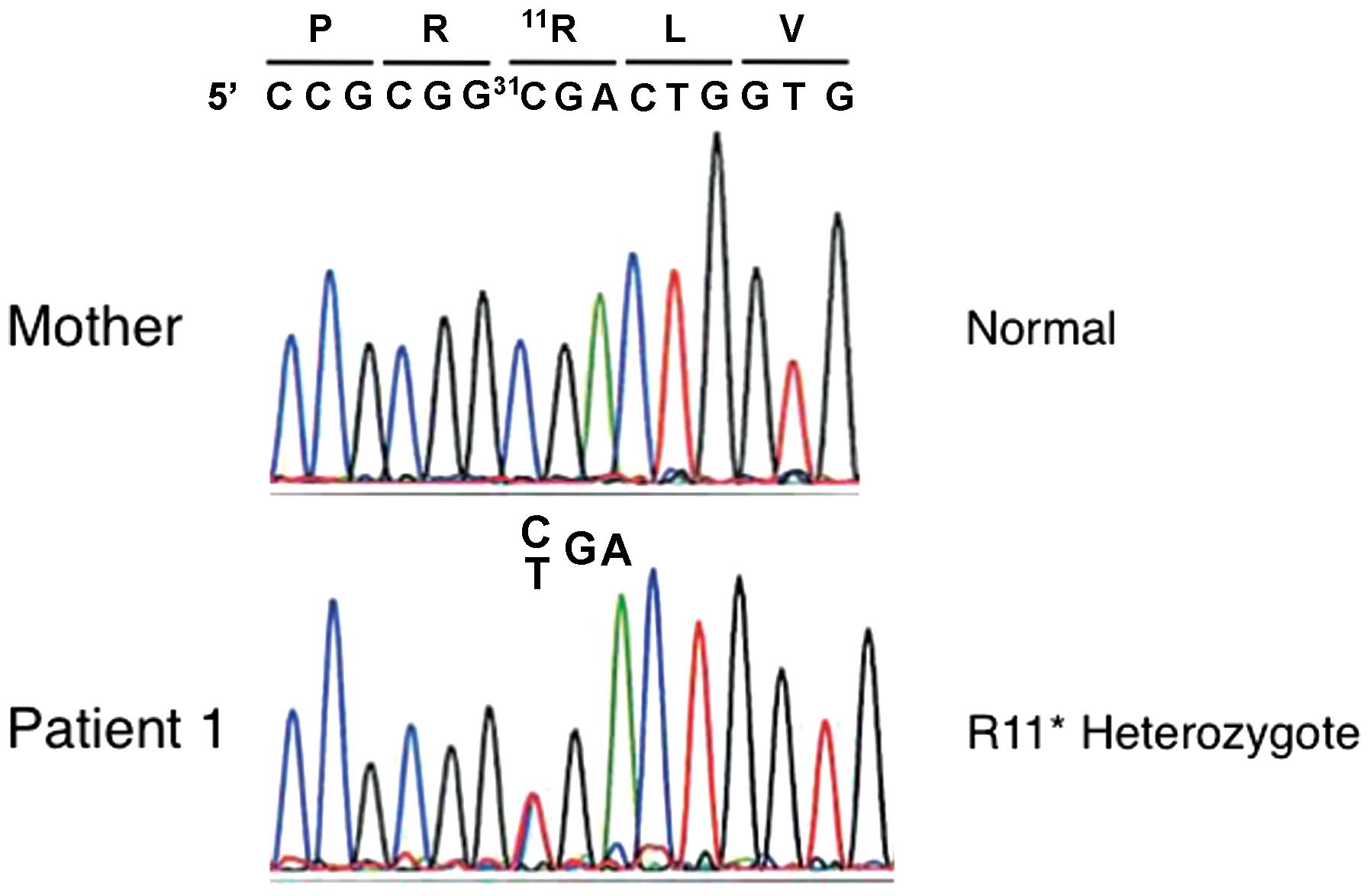
heterozygous c. 31C>T (p.R11*) mutation in exon 1,
and her mother did not have this mutation (Fig. 1). No mutation in the maternal
allele was identified. The DNA of the father was not available.
Patient 2 had a homozygous c.242G>A (p.W81*) mutation
in exon 3. Genomic analyses of his patients revealed that the
father was heterozygous for the p.W81* mutation;
however, the mother did not have the p.W81* mutation
(Fig. 2). Hence, we hypothesized
the following: i) the maternal allele may have a large deletion not
including exon 1 in patient 1; and ii) the maternal allele in
patient 2 may have a deletion including exon 3.
MLPA analysis of HMGCL
We successfully performed MLPA for HMGCL.
Similar patterns of amplification were obtained in three controls,
enabling copy number to be evaluated in the patient samples
(Fig. 3). In patient 1 and his
mother, only one copy of exons 2–4 was present, whereas two copies
of all other exons were present, suggesting a large deletion
including exons 2–4 in the maternal allele (Fig. 3). Patient 2 and his parents all
had two copies of all HMGCL exons (Fig. 3). This means that both copies of
exon 3 in patient 2 were from the father. We, therefore, suspected
that patient 2 has a paternal uniparental disomy of the
HMGCL region.
Determination of breakpoints in patient
1
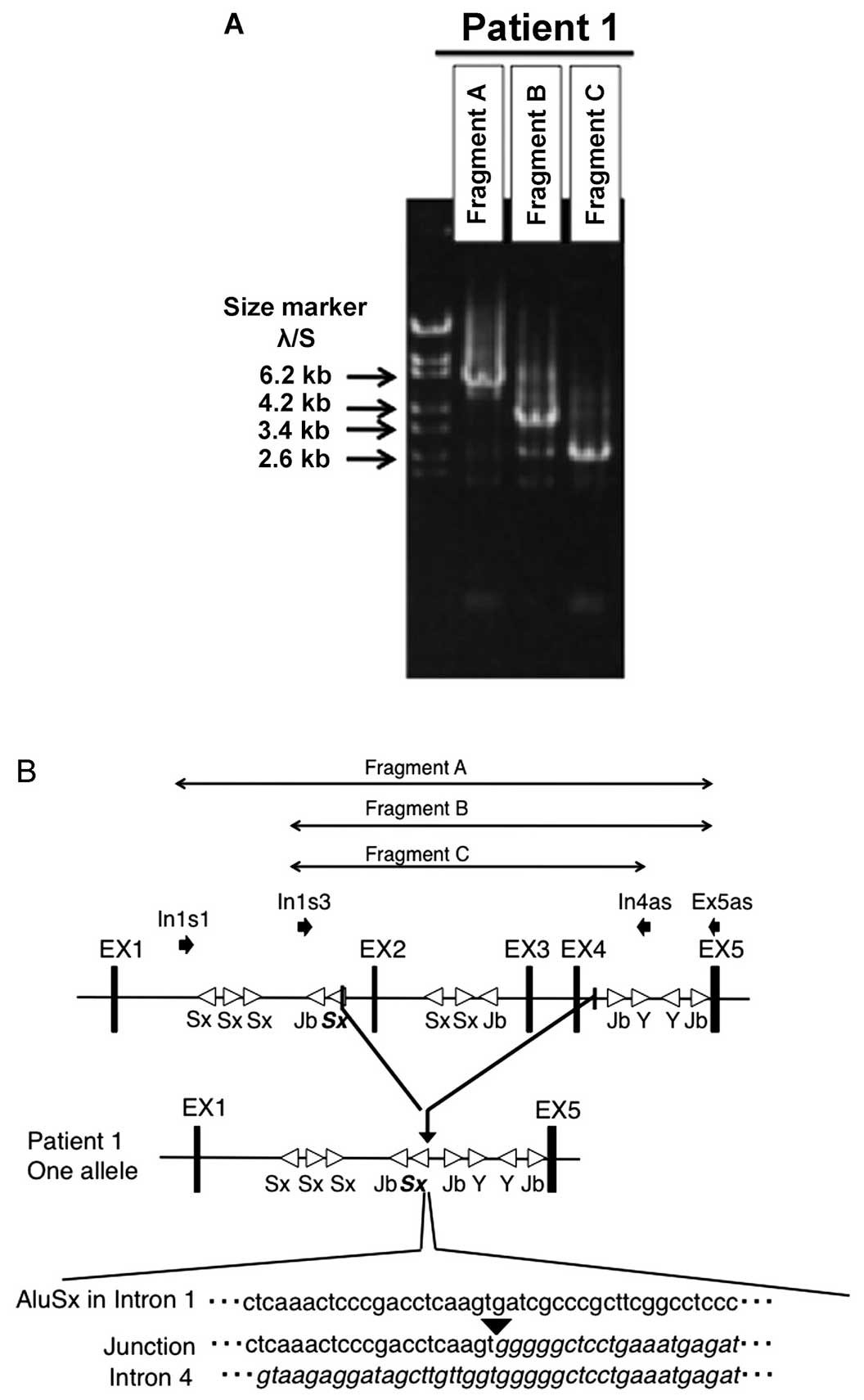
Long-range PCR amplification using DNA from patient
1 yielded fragments that included an approximate 4-kb deletion
(Fig. 4A). A large deletion from
g.9326 to g.13806 (4481 bp; NG_013061) was identified. We confirmed
that one breakpoint was within an Alu element in intron 1 and the
other was in a non-Alu element in intron 4 (Fig. 4B).
CGH and SNP arrays in patient 2
To confirm the copy number and SNP haplotype of the
HMGCL region, microarray analysis using CGH + SNP array was
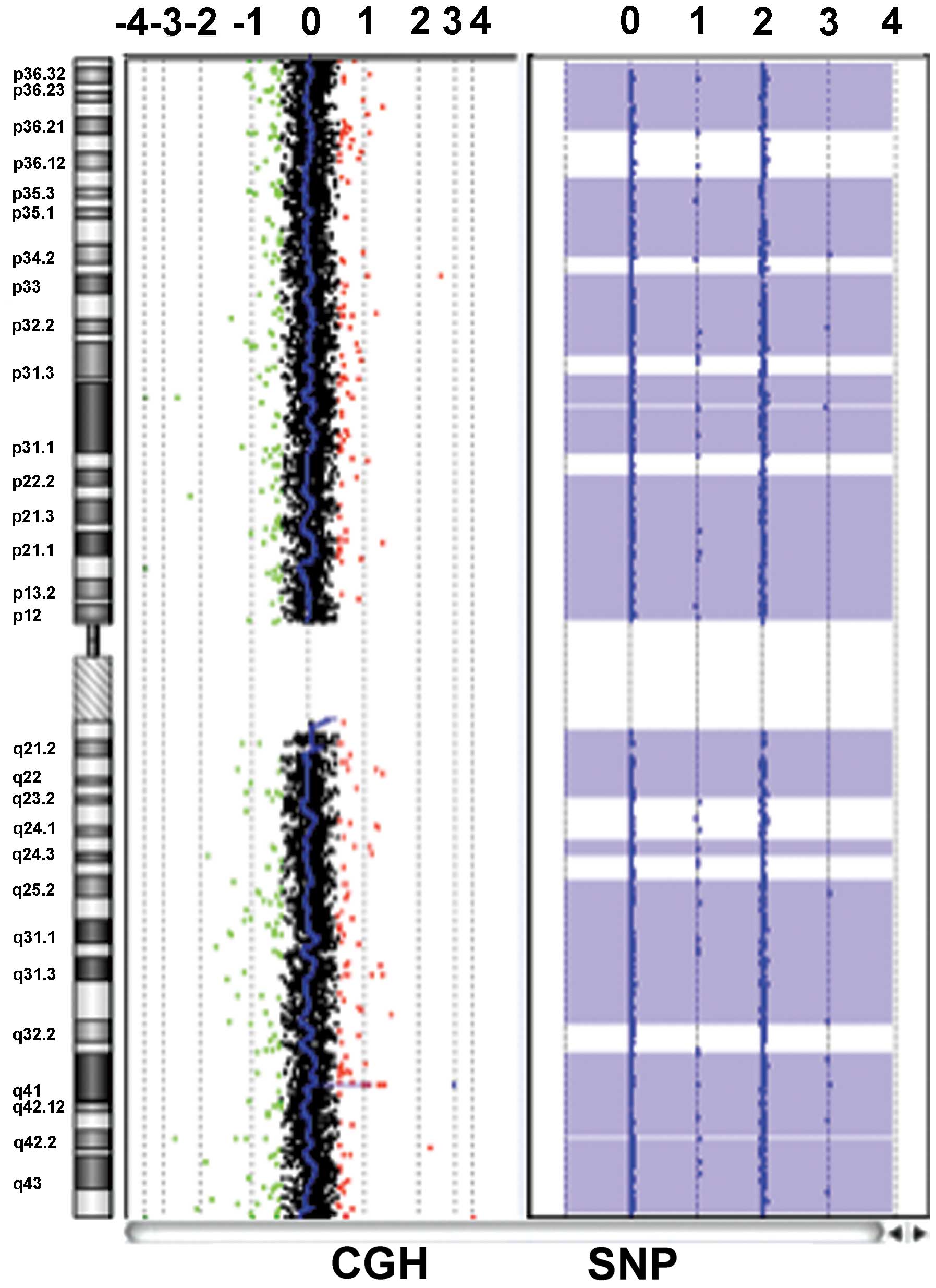
performed for patient 2. In the CGH array, no copy number
aberration was detected in the whole of chromosome 1 including the
HMGCL region, which confirmed the homozygous status of the
HMGCL mutation (Fig.
5).
Furthermore, a loss of heterozygosity (LOH) for
almost all of chromosome 1, where HMGCL is located, was
revealed. This indicated paternal uniparental disomy of chromosome
1. In conclusion, a homozygous mutation in HMGCL was
determined to be caused by non-Mendelian inheritance due to
paternal uniparental isodisomy of the whole chromosome 1, including
the 1p36.1 region.
Discussion
In the present study, we investigated the molecular
genetic basis of two Japanese HMGCL-deficient patients using
standard Sanger sequencing. Although we identified a mutation in
each patient, inheritance patterns were not consistent in their
families. Human HMGCL is an Alu element-rich gene, having 23
Alu elements within 23 kb. We first hypothesized that a
heterozygous intragene deletion caused by non-equal homologous
recombination between Alu elements was a possible cause of
HMGCL mutation in these patients. Therefore, we used the
MLPA method to detect the copy numbers of each exon in
HMGCL.
Patient 1 had a heterozygous p.R11*
mutation from the father, but a mutation from the mother was not
identified by conventional sequence analysis. MLPA revealed that
patient 1 and her mother had only one copy of exons 2–4, which was
consistent with our hypothesis. However, this large deletion that
included exons 2–4 was not caused by non-equal Alu-mediated
homologous recombination. One breakpoint was within an Alu element
in intron 1 and the other was in a non-Alu element in intron 4.
There were no homologous sequences around the breakpoint. The
genesis of this deletion is unknown.
Patient 2 had an apparent homozygous
p.W81* mutation and his father was heterozygous for this
mutation; however, his mother did not have this mutation. MLPA
revealed that patient 2 had two copies of each HMGCL exon.
Hence, our new hypothesis was that LOH of the HMGCL region
may be the molecular basis for HMGCL deficiency in this patient. We
successfully identified a paternal uniparental isodisomy of almost
the entire chromosome 1 by microarray analysis using a CGH + SNP
array. To the best of our knowledge, this is the first case of
HMGCL deficiency shown to be caused by uniparental disomy.
There are two types of uniparental disomy,
heterodisomy and isodisomy. In heterodisomy, a pair of
non-identical chromosomes is inherited from one parent and in
isodisomy a single chromosome from one parent is duplicated.
Isodisomy is potentially dangerous as it may lead to the
duplication of lethal recessive genes, while heterodisomy is
essentially benign. Uniparental disomy in humans is mainly caused
by meiotic non-disjunction events followed by i) gamete
complementation; ii) trisomy rescue; iii) monosomy duplication; and
iv) somatic crossing over (13).
Our present results from HMGCL gene analysis, MLPA and CGH +
SNP arrays indicate that patient 2 had a monosomic duplication of
paternal chromosome 1. Partial and whole uniparental disomy of
chromosome 1 has been reported in autism, fumarase deficiency,
Stargardt disease, Pelizaeus- Merzbacher-like disease, leptin
receptor deficiency, rhizomelic chondrodysplasia punctata type 2
and CD45-deficient severe combined immunodeficiency (14–20). This study demonstrates the
advantage of using MLPA method for the analysis and identification
of such heterozygous gene alterations. The identification of such
mutations may facilitate mutation analysis in newly diagnosed
patients.
Acknowledgments
We are grateful to the patients for their
participation in this study. The present study was supported in
part by a Grant-in-Aid for Scientific Research from the Ministry of
Education, Culture, Sports, Science and Technology of Japan
(26114708, 24591505) and Health and Labor Science Research Grants
for Research on Intractable Diseases from the Ministry of Health,
Labor and Welfare of Japan to T.F.
Abbreviations:
|
HMGCL
|
3-hydroxy-3-methylglutaryl-CoA
lyase
|
|
MLPA
|
multiplex ligation-dependent probe
amplification
|
|
CGH
|
comparative genomic hybridization
|
|
SNP
|
single nucleotide polymorphism
|
|
LOH
|
loss of heterozygosity
|
References
|
1
|
Wang SP, Robert MF, Gibson KM, Wanders RJ
and Mitchell GA: 3-Hydroxy-3-methylglutaryl CoA lyase (HL): mouse
and human HL gene (HMGCL) cloning and detection of large gene
deletions in two unrelated HL-deficient patients. Genomics.
33:99–104. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fukao T, Mitchell G, Sass JO, Hori T, Orii
K and Aoyama Y: Ketone body metabolism and its defects. J Inherit
Metab Dis. 37:541–551. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Muroi J, Yorifuji T, Uematsu A, Shigematsu
Y, Onigata K, Maruyama H, Nobutoki T, Kitamura A and Nakahata T:
Molecular and clinical analysis of Japanese patients with
3-hydroxy-3-methylglutaryl CoA lyase (HL) deficiency. Hum Genet.
107:320–326. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sen SK, Han K, Wang J, Lee J, Wang H,
Callinan PA, Dyer M, Cordaux R, Liang P and Batzer MA: Human
genomic deletions mediated by recombination between Alu elements.
Am J Hum Genet. 79:41–53. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fukao T, Aoyama Y, Murase K, Hori T,
Harijan RK, Wierenga RK, Boneh A and Kondo N: Development of MLPA
for human ACAT1 gene and identification of a heterozygous
Alu-mediated deletion of exons 3 and 4 in a patient with
mitochondrial acetoacetyl-CoA thiolase (T2) deficiency. Mol Genet
Metab. 110:184–187. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fukao T, Zhang G, Rolland MO, Zabot MT,
Guffon N, Aoki Y and Kondo N: Identification of an Alu-mediated
tandem duplication of exons 8 and 9 in a patient with mitochondrial
acetoacetyl-CoA thiolase (T2) deficiency. Mol Genet Metab.
92:375–378. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang G, Fukao T, Sakurai S, Yamada K,
Michael Gibson K and Kondo N: Identification of Alu-mediated, large
deletion-spanning exons 2–4 in a patient with mitochondrial
acetoacetyl-CoA thiolase deficiency. Mol Genet Metab. 89:222–226.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gatta V1, Scarciolla O, Gaspari AR, Palka
C, De Angelis MV, Di Muzio A, Guanciali-Franchi P, Calabrese G,
Uncini A and Stuppia L: Identification of deletions and
duplications of the DMD gene in affected males and carrier females
by multiple ligation probe amplification (MLPA). Hum Genet.
117:92–98. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Janssen B, Hartmann C, Scholz V, Jauch A
and Zschocke J: MLPA analysis for the detection of deletions,
duplications and complex rearrangements in the dystrophin gene:
potential and pitfalls. Neurogenetics. 6:29–35. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lalic T, Vossen RH, Coffa J, Schouten JP,
Guc-Scekic M, Radivojevic D, Djurisic M, Breuning MH, White SJ and
den Dunnen JT: Deletion and duplication screening in the DMD gene
using MLPA. Eur J Hum Genet. 13:1231–1234. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tomoko T, Takanori S, Kazumi O, et al: A
case of 3-Hydroxy-3-methylglutaryl CoA lyase deficiency. J Jpn
Pediatr Soc. 112:1249–1254. 2008.In Japanese.
|
|
12
|
Zhi J and Hatchwell E: Human MLPA Probe
Design (H-MAPD): a probe design tool for both electrophoresis-based
and bead-coupled human multiplex ligation-dependent probe
amplification assays. BMC Genomics. 9:4072008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ledbetter DH and Engel E: Uniparental
disomy in humans: development of an imprinting map and its
implications for prenatal diagnosis. Hum Mol Genet. 4:Spec No.
1757–1764. 1995.PubMed/NCBI
|
|
14
|
Riveiro-Alvarez R, Valverde D,
Lorda-Sanchez I, Trujillo-Tiebas MJ, Cantalapiedra D, Vallespin E,
Aguirre-Lamban J, Ramos C and Ayuso C: Partial paternal uniparental
disomy (UPD) of chromosome 1 in a patient with Stargardt disease.
Mol Vis. 13:96–101. 2007.PubMed/NCBI
|
|
15
|
Shimojima K, Tanaka R, Shimada S, Sangu N,
Nakayama J, Iwasaki N and Yamamoto T: A novel homozygous mutation
of GJC2 derived from maternal uniparental disomy in a female
patient with Pelizaeus-Merzbacher-like disease. J Neurol Sci.
330:123–126. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wassink TH, Losh M, Frantz RS, Vieland VJ,
Goedken R, Piven J and Sheffield VC: A case of autism and
uniparental disomy of chromosome 1. Hum Genet. 117:200–206. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zeng WQ, Gao H, Brueton L, Hutchin T, Gray
G, Chakrapani A, Olpin S and Shih VE: Fumarase deficiency caused by
homozygous P131R mutation and paternal partial isodisomy of
chromosome 1. Am J Med Genet A. 140:1004–1009. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nimmo G, Monsonego S, Descartes M,
Franklin J, Steinberg S and Braverman N: Rhizomelic
chrondrodysplasia punctata type 2 resulting from paternal isodisomy
of chromosome 1. Am J Med Genet A. 152A:1812–1817. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roberts JL1, Buckley RH, Luo B, Pei J,
Lapidus A, Peri S, Wei Q, Shin J, Parrott RE and Dunbrack RL Jr:
CD45-deficient severe combined immunodeficiency caused by
uniparental disomy. Proc Natl Acad Sci USA. 109:10456–10461. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Le Beyec J, Cugnet-Anceau C, Pépin D,
Alili R, Cotillard A, Lacorte JM, Basdevant A and Laville Mand
Clément K: Homozygous leptin receptor mutation due to uniparental
disomy of chromosome 1: response to bariatric surgery. J Clin
Endocrinol Metab. 98:E397–E402. 2013. View Article : Google Scholar : PubMed/NCBI
|