Introduction
Diabetic retinopathy (DR), a specific microvascular
complication of diabetes, is the most common cause of visual
disability and blindness. The prevalence of DR increases with the
duration of diabetes (1), and
nearly 99% of patients with type 1 diabetes and 60% with type 2
have some degree of DR after 20 years (1,2).
DR can be classified into 2 stages: non-proliferative and
proliferative. The earliest visible signs of non-proliferative DR
are microaneurysms, hemorrhages, hard exudates, cotton wool spots,
intraretinal microvascular abnormalities and venous beading. The
more severe state of proliferative DR (PDR) is characterized by the
growth of new blood vessels on the surface of the retina or the
optic disc, which are prone to hemorrhaging. Finally, visual
impairment results in vitreous hemorrhage, subsequent fibrosis and
tractional retinal detachment (3,4).
Although the pathogenesis of DR has not yet been fully elucidated,
the pathogenesis of diabetes is believed to be multifactorial, with
genetic risk factors playing a fundamental role. However, several
factors, including hyperglycemia, aldose reductase, advanced
glycation end products (AGEs) and cytokines, such as vascular
endothelial growth factor (VEGF) have been implicated in the
disease pathogenesis (5).
Despite DR being a common complication of diabetes,
little is known about the underlying molecular mechanisms. In
recent years, the complex association that exists between the most
relevant contributors to the onset and progression of DR, such as
AGEs, oxidative stress, inflammation and angiogenesis have been
elucidated and analyzed, particularly via whole-genome expression
analyses using cells and animal models (6,7).
Based on published studies, several systems, pathways and processes
have been strongly implicated in DR; these include the
renin-angiotensin system, the polyol pathway, non-enzymatic
glycation, endothelial dysfunction, the maintenance of vascular
tone, extracellular matrix remodeling and angiogenesis, which is
dysregulated in diabetes and leads to the proliferation of new,
fragile retinal capillaries and culminates in PDR (8,9). A
host of genes involved in these pathways/processes have been
treated as potential candidate genes. These genes include
angiotensin I-converting enzyme (ACE), angiotensin II type 1
receptor (AGTR1), angiotensinogen (AGT), VEGF, aldose reductase
(AR2), receptor for advanced glycation end products (RAGE), glucose
transporter 1 (GLUT1), inducible nitric oxide synthase (NOS2A),
constitutive nitric oxide synthase (NOS3), transforming growth
factor-β (TGF-β), endothelin isoforms and their cellular receptors,
amongst others (10–19). However, to the best of our
knowledge, few studies to date have focused on the associated
pathways and transcription factors (TFs), or on the co-expression
patterns at the multiple pathways level.
In the present study, we employed a microarray
dataset of genome-wide gene expression profiling from the Gene
Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) (20,21), which is associated with DR. The
most well-known method of gene set enrichment analysis (GSEA) was
used to analyze the genomic data in order to uncover the regulatory
mechanisms of retinopathy (damage to the retina) caused by diabetic
complications at the multiple pathways level. GSEA is widely used
to analyze gene expression profiles, particularly to identify
pre-defined gene sets which exhibit significant differences in
expression between samples from the control and treatment groups
(22–24). The goal of GSEA is to determine
other inter esting categories (pathways) in which the constituent
genes exhibit coordinated changes in expression under the given
experimental conditions, other than in the form of sets of
differentially expressed genes (DEGs). One of the advantages of
GSEA is that it has the ability to highlight genes that are weakly
connected to the phenotype through pathway analysis, something
which may be difficult to detect using classical univariate
statistics (22).
Materials and methods
Microarray data collection and
pre-processing
We searched the GEO database(www.ncbi.nlm.nih.gov/geo/) for gene expression
profiling studies associated with DR. Data were included in our
re-analysis if they met the following conditions: i) the data were
genome-wide; ii) a comparison was conducted between DR samples and
control (CT) samples; and iii) complete microarray raw or
normalized data were available. Finally, we chose the GSE12610
dataset for our re-analysis, which was contributed by Liang et
al (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE12610).
In this dataset, a total of 5 RNA samples extracted from retinas
were examined for RNA quality and then hybridized to 2 different
GeneChip® Mouse Genome 430 2.0 arrays (technical
replicates; Affymetrix, Santa Clara, CA, USA). There were 3
biological replicates for DR (the samples from GSM315892 to
GSM315894, designated as DR-1, DR-2 and DR-3) and 2 for CT (the
samples GSM315895 and GSM315896, designated as CT-1 and CT-2).
In order to determine the influence of
pre-processing on the comparison, data pre-processing was performed
using software packages developed in version 2.6.0 of Bioconductor
and R version 2.10.1. Each Affymetrix dataset was background
adjusted, normalized, and log2 probe-set intensities were
calculated using the Robust Multichip Average (RMA) algorithm from
the affy package (25).
GSEA
Our GSEA of pathways and genes was performed using
the Category package in version 2.6.0 of Bioconductor (26). The goal of GSEA is to determine
whether the members of a gene set S are randomly distributed
throughout the entire reference gene list L or are primarily found
at the top or bottom. One of the advantages of GSEA is its relative
robustness in the face of noise and outliers in the data. In our
analysis, the gene sets represented by <10 genes were excluded.
The t-statistic mean of the genes was computed in each Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway. Using a
permutation test with 1,000 repetitions, the cut-off of
significance level P-values was chosen as 0.05 for the significant
pathways associated with DR. Accordingly, the significant pathways
and genes were then identified by comparing the samples with DR and
those with no DR. The following classification of identified
pathways was based on the pathway maps br08901 of BRITE Functional
Hierarchies in the KEGG database (http://www.genome.jp/kegg-bin/get_htext?br08901.keg).
The annotation of significant genes in each pathway was performed
using the biomaRt package, BioMart v 0.8 rc3 (version of 0.8
release candidate 3; http://www.biomart.org/). Next, clustering of groups
and genes was performed based on the expression of the identified
genes in each significant pathway, using the hierarchical
clustering method and Pearson’s correlation co-efficient.
Regulatory elements (REs) and TFs of
co-regulated genes
We used a web server known as the DiRE (distant
regulatory elements of co-expressed genes, http://dire.dcode.org/), which uses the Enhancer
Identification (EI) method, to predict common REs for our input
genes that have co-function in each identified, significantly
related pathway (27). It
predicts function-specific REs that consist of clusters of
specifically associated transcription factor binding sites (TFBSs),
and it also scores the association of individual TFs with the
biological function shared by the group of input genes. We selected
a random set of 5,000 genes in the genome of Mus musculus 9
(mm9) as the background genes. As a result, our predicted TFs have
two major parameters, including TF occurrence (the percentage of
candidate REs containing a conserved binding site for a particular
TF) and TF importance (the product of TF occurrence and TF weight).
From our candidate associated TFs with input gene sets, we selected
the cut-off value of TF importance as >0.05.
Results and Discussion
Identification of significant pathways
associated with DR
Compared to the approach of DEGs, the strategy of
GSEA that we used in this study is likely to be more powerful than
conventional single-gene methods in the study of complex diseases,
in which many genes make subtle contributions. According to our
GSEA of the dataset of 5 samples, achieved by comparing the DR to
the CT samples, there were 69 significant pathways associated with
DR, whose P-values were <0.05, including 10 upregulated and 59
downregulated pathways. The coregulated pathways network is
highlighted in Fig. 1 (red text
indicates upregulated pathways, and green text indicates
downregulated pathways). Furthermore, the details of significant
genes in these 69 pathways related to DR are available upon
request, as is the information on probe set ID and gene symbol.
Among these 69 pathways associated with DR, the samples were
classified and divided into DR and CT groups by clustering. For
example, based on the expression of 86 significant genes whose
significance level P-value was <0.05 in the downregulated ErbB
signaling pathway, which may be clustered into 7 groups of gene
sets (Fig. 2; group A-G), 5
samples were clustered into 2 groups, with DR-1, DR-2 and DR-3 in
one group and CT-1 and CT-2 in the other group. Similarly, in the
downregulated mammalian target of rapamycin (mTOR) pathway, 5
samples were also grouped, as CT-1 and CT-2 in the CT group and
DR-1, DR-2 and DR-3 in the DR group (Fig. 3). The 52 genes involved in the
mTOR signaling pathway may also be clustered into 4 groups of gene
sets (Fig. 3, group A-D).
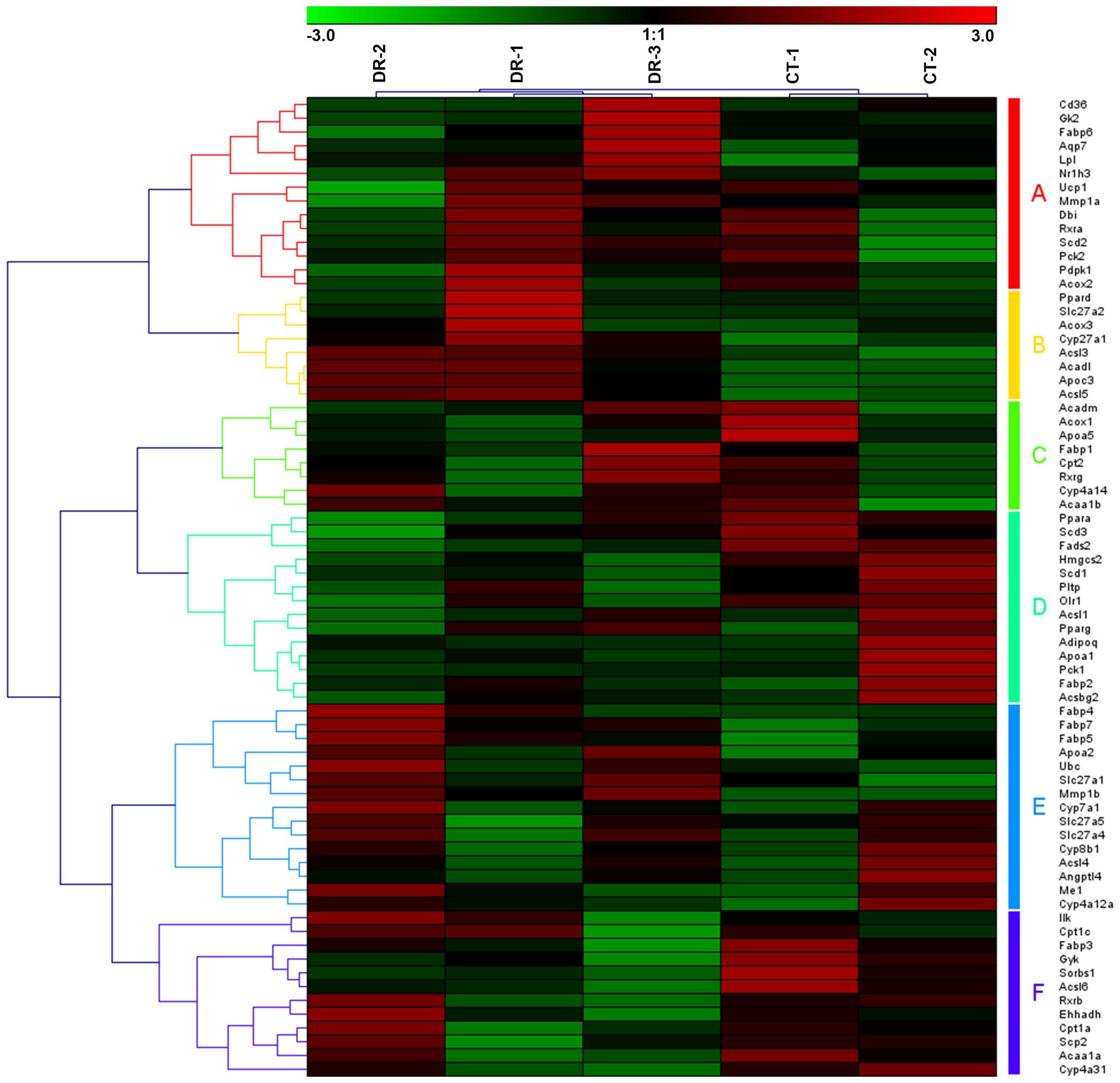
Moreover, in the upregulated peroxisome proliferator-activated
receptor (PPAR) pathway, 5 samples were also clustered into 2
groups, with CT-1 and CT-2 in one group and DR-1, DR-2 and DR-3 in
the other group (Fig. 4).
Furthermore, 71 genes were involved in the PPAR signaling pathway
associated with DR, which may be clustered into 6 groups of gene
sets (Fig. 4, group A–F).
Moreover, based on the KEGG pathway maps in the KEGG database
(http://www.genome.jp/kegg/), the 69
significant pathways could be mapped into 6 functional classes:
cellular processes, environmental information processing, genetic
information processing, human diseases, metabolism and organismal
systems. The details of the pathways involved in each class are
described in Tables ITable IITable IIITable IV–V.
 | Table ISignificant pathways associated with
DR in the functional class of cellular processes. |
Table I
Significant pathways associated with
DR in the functional class of cellular processes.
| Pathways | Map B | No. of genes | No. of TFs |
|---|
| 04142: Lysosome | Transport and
catabolism | 119 | 28 |
| 04144:
Endocytosis | Transport and
catabolism | 199 | 22 |
| 04145:
Phagosome | Transport and
catabolism | 140 | 22 |
| 04530: Tight
junction | Cell
communication | 127 | 34 |
| 04810: Regulation
of actin cytoskeleton | Cell motility | 206 | 26 |
| 05142: Chagas
disease (American trypanosomiasis) | | 99 | 28 |
| Table IISignificant pathways associated with
DR in the functional class of environmental and genetic information
processing. |
Table II
Significant pathways associated with
DR in the functional class of environmental and genetic information
processing.
| Pathways | Map B | No. of genes | No. of TFs |
|---|
| 02010: ABC
transporters | Membrane
transport | 44 | 25 |
| 04512: ECM-receptor
interaction | Signaling molecules
and interaction | 83 | 34 |
| 04012: ErbB
signaling pathway | Signal
transduction | 86 | 40 |
| 04150: mTOR
signaling pathway | Signal
transduction | 52 | 30 |
| 04370: VEGF
signaling pathway | Signal
transduction | 74 | 25 |
| 04310: Wnt
signaling pathway | Signal
transduction | 145 | 30 |
| 04070:
Phosphatidylinositol signaling system | Signal
transduction | 73 | 26 |
| 04130: SNARE
interactions in vesicular transport | Folding, sorting
and degradation | 35 | 6 |
| 03050:
Proteasome | Folding, sorting
and degradation | 44 | 20 |
| 03440: Homologous
recombination | Replication and
repair | 27 | 17 |
| 03030: DNA
replication | Replication and
repair | 35 | 23 |
| 00970:
Aminoacyl-tRNA biosynthesis | Translation | 41 | 7 |
| 03010:
Ribosomea | Translation | 53 | 19 |
| 03020: RNA
polymerasea | Transcription | 24 | 29 |
| Table IIISignificant pathways associated with
DR in the functional class of human diseases. |
Table III
Significant pathways associated with
DR in the functional class of human diseases.
| Pathways | Map B | No. of genes | No. of TFs |
|---|
| 04950: Maturity
onset diabetes of the young | Endocrine and
metabolic diseases | 25 | 25 |
| 04940: Type I
diabetes mellitus | Endocrine and
metabolic diseases | 40 | 30 |
| 05330: Allograft
rejection | Immune
diseases | 33 | 22 |
| 05416: Viral
myocarditis | Cardiovascular
diseases | 65 | 21 |
| 05410: Hypertrophic
cardiomyopathy (HCM) | Cardiovascular
diseases | 81 | 32 |
| 05412:
Arrhythmogenic right ventricular cardiomyopathy (ARVC) | Cardiovascular
diseases | 73 | 43 |
| 05100: Bacterial
invasion of epithelial cells | Infectious
diseases: bacterial | 66 | 29 |
| 05140:
Leishmaniasis | Infectious
diseases: parasitic | 62 | 20 |
| 05143: African
trypanosomiasis | Infectious
diseases: parasitic | 30 | 19 |
| 05146:
Amoebiasis | Infectious
diseases: parasitic | 106 | 26 |
| 05145:
Toxoplasmosis | Infectious
diseases: parasitic | 123 | 19 |
| 05211: Renal cell
carcinoma | Cancers | 70 | 30 |
| 05212: Pancreatic
cancer | Cancers | 69 | 25 |
| Table IVSignificant pathways associated with
DR in the functional class of metabolism. |
Table IV
Significant pathways associated with
DR in the functional class of metabolism.
| Pathways | Map B | No. of genes | No. of TFs |
|---|
| 00010:
Glycolysis/gluconeogenesis | Carbohydrate
metabolism | 57 | 14 |
| 00500: Starch and
sucrose metabolism | Carbohydrate
metabolism | 32 | 12 |
| 00520: Amino sugar
and nucleotide sugar metabolism | Carbohydrate
metabolism | 48 | 20 |
| 00620: Pyruvate
metabolism | Carbohydrate
metabolism | 39 | 22 |
| 00030: Pentose
phosphate pathway | Carbohydrate
metabolism | 25 | 14 |
| 00630: Glyoxylate
and dicarboxylate metabolism | Carbohydrate
metabolism | 18 | 30 |
| 00250: Alanine,
aspartate and glutamate metabolism | Amino acid
metabolism | 32 | 22 |
| 00290: Valine,
leucine and isoleucine biosynthesis | Amino acid
metabolism | 11 | 30 |
| 00310: Lysine
degradation | Amino acid
metabolism | 43 | 26 |
| 00230: Purine
metabolism | Nucleotide
metabolism | 152 | 24 |
| 00590: Arachidonic
acid metabolism | Lipid
metabolism | 77 | 21 |
| 00591: Linoleic
acid metabolism | Lipid
metabolism | 40 | 13 |
| 00100: Steroid
biosynthesisa | Lipid
metabolism | 18 | 17 |
| 00670: One carbon
pool by folate | Metabolism of
cofactors and vitamins | 17 | 26 |
| 00790: Folate
biosynthesisa | Metabolism of
cofactors and vitamins | 11 | 28 |
| 00900: Terpenoid
backbone biosynthesisa | Metabolism of
terpenoids and polyketides | 13 | 22 |
| 00604:
Glycosphingolipid biosynthesis-ganglio seriesa | Glycan biosynthesis
and metabolism | 15 | 19 |
| 00534:
Glycosaminoglycan biosynthesis-heparan sulfate | Glycan biosynthesis
and metabolism | 24 | 25 |
| Table VSignificant pathways associated with
DR in the functional class of organismal systems. |
Table V
Significant pathways associated with
DR in the functional class of organismal systems.
| Pathways | Map B | No. of genes | No. of TFs |
|---|
| 03320: PPAR
signaling pathwaya | Endocrine
system | 71 | 25 |
| 04910: Insulin
signaling pathway | Endocrine
system | 128 | 27 |
| 04916:
Melanogenesis | Endocrine
system | 95 | 40 |
| 04920:
Adipocytokine signaling pathway | Endocrine
system | 67 | 19 |
| 04320:
Dorso-ventral axis formation | Development | 22 | 24 |
| 04360: Axon
guidance | Development | 129 | 28 |
| 04380: Osteoclast
differentiation | Development | 112 | 34 |
| 04270: Vascular
smooth muscle contraction | Circulatory
system | 110 | 32 |
| 04960:
Aldosterone-regulated sodium reabsorption | Excretory
system | 44 | 33 |
| 04962:
Vasopressin-regulated water reabsorption | Excretory
system | 43 | 27 |
| 04970: Salivary
secretion | Digestive
system | 70 | 41 |
| 04972: Pancreatic
secretion | Digestive
system | 99 | 44 |
| 04720: Long-term
potentiation | Nervous system | 63 | 27 |
| 04730: Long-term
depression | Nervous system | 68 | 22 |
| 04660: T cell
receptor signaling pathway | Immune system | 106 | 33 |
| 04664: Fc epsilon
RI signaling pathway | Immune system | 77 | 28 |
| 04666: Fc gamma
R-mediated phagocytosis | Immune system | 86 | 39 |
| 04670: Leukocyte
transendothelial migration | Immune system | 115 | 26 |
In the functional class of cellular processes, there
were 6 significantly downregulated pathways related to DR (Table I). These pathways were involved in
cell communication, cell motility, and transport and catabolism. Of
these pathways, the tight junction one was the most significant,
and was classified as belonging to the functional group of cell
communication. As is well known, the breakdown of the blood-retinal
barrier (BRB) is one of the most important characteristics of DR
and is largely attributed to the disruption of endothelial tight
junction. X-box binding protein 1 (XBP1), which is a major
transcription factor activated by ER stress, plays an important
role in maintaining endothelial barrier function (28). The activation of XBP1 protects
against ER stress-induced tight junction damage.
There were 7 significantly downregulated pathways in
the functional class of environmental information processing, 5
significantly downregulated pathways in the functional class of
genetic information processing, and only 2 significantly
upregulated pathways in the functional class of genetic information
processing related to DR (Table
II). The ATP-binding cassette (ABC) transporters pathway was
associated with the function of membrane transport, and the
extracellular matrix (ECM)-receptor interaction pathway was
associated with the function of the signaling of molecules and
interaction. The environmental information processing pathways,
such as the ErbB, mTOR, VEGF and Wnt signaling pathways, and the
phosphatidylinositol signaling system were related to the functions
of signal transduction. The genetic information processing
pathways, such as soluble N-ethylmaleimide-sensitive attachment
protein receptor (SNARE) interactions in vesicular transport and
proteasome were related to the functions of folding, sorting and
degradation. The genetic information processing pathways of
homologous recombination and DNA replication were related to the
functions of replication and repair, and. Tthe genetic information
processing pathways of aminoacyl-tRNA biosynthesis and ribosome
were related to the functions of translation, and the RNA
polymerase pathway was transcription-related. Of these
aforementioned pathways, the ErbB and mTOR signaling pathways were
the most significant in this class (the functional class of
environmental and genetic information processing). The epidermal
growth factor receptor (EGFR), also known as ErbB1/human epidermal
growth factor receptor 1 (HER1), is a member of the ErbB family of
receptor tyrosine kinases which also includes ErbB2 (Neu, HER2),
ErbB3 (HER3) and ErbB4 (HER4). It was recently observed that
hyperglycemia perturbs the EGFR-PI3K-AKT and extracellular
signal-regulated kinase (ERK) signaling pathways in normal and
healing corneas and that increased levels of cellular apoptosis and
decreased cell proliferation may be contributing factors in the
impairment of corneal epithelial wound healing in diabetic corneas
(29,30). Signaling through the mTOR pathway
plays a major role in smooth muscle and endothelial cell
proliferation in response to hypoxia (31). There have been signs that the
inhibition of the PI3K/Akt/mTOR pathway may have beneficial
therapeutic effects in the management of PDR, which stems from
findings that indicate that growth factors known to play major
roles in the induction of angiogenesis depend on PI3K/Akt/mTOR for
prolonging the cell survival signals that are operating in
pathological angiogenesis (32).
Thirteen significantly associated pathways were
classified and assigned to the functional class of human diseases,
including 2 upregulated endocrine and metabolic diseases related
pathways, 1 upregulated immune diseases related pathways, 3
downregulated cardiovascular diseases related pathways, 1
downregulated infectious diseases: bacterial related pathways, 4
downregulated infectious diseases: parasitic related pathways and 2
downregulated cancer related pathways (Table III). Of these, the pathway of
type I diabetes mellitus was one of the most significant endocrine
and metabolic diseases-related pathways. The prevalence of DR
increases with the duration of diabetes. After 20 years of
diabetes, nearly all patients with type I diabetes and >60% of
patients with type II diabetes have some degree of retinopathy
(33).
In the functional class of metabolism, there were 14
downregulated and 4 upregulated significant pathways associated
with DR (Table IV). These were
involved in 7 different types of metabolism, including carbohydrate
metabolism, amino acid metabolism, nucleotide metabolism, lipid
metabolism, metabolism of co-factors and vitamins, metabolism of
terpenoids and polyketides, and glycan biosynthesis and metabolism.
Of these, the glycolysis/gluconeogenesis pathway was one of the
most significant pathways, which was classified and assigned to the
functional group of carbohydrate metabolism. Protein kinase C (PKC)
is a serine/threonine kinase, which is involved in signal
transduction events with regard to specific hormonal, neuronal and
growth factor stimuli. Hyperglycaemia leads to an increase in
glucose flux through the glycolysis pathway, which in turn
increases the de novo synthesis of diacylglycerol (DAG), the
key activator of PKC in physiology (34). PKC is a molecule which plays an
important role in the regulation of numerous physiological
processes, whose upregulation contributes to the pathogenesis of
DR.
In the last functional class of organismal systems,
there was 1 significantly upregulated and 17 significantly
downregulated pathways associated with DR (Table V). These were involved in the
endocrine system, development, and the circulatory, excretory,
digestive, nervous and immune systems. Of these, the PPAR signaling
pathway was one of the most associated pathways, and was classified
and assigned to the functional group of the endocrine system. PPARs
are ligand-activated TFs (members of the nuclear receptor family)
which offer promising targets for the development of novel,
efficient treatments for several metabolic disorders. An indication
suggesting that antidiabetic thiazolidinediones and adipogenic
prostanoids are ligands of one of the PPARs reveals a novel
signaling pathway that directly links these compounds to processes
involved in glucose homeostasis and lipid metabolism, including
adipocyte differentiation (35).
Candidate TF selection associated with
DR
To predict TFs potentially involved in the
regulation of DR, we performed an analysis of TFBSs and predicted
TFs using the significant genes in each identified pathway. Based
on the cut-off value of TF importance, we identified the candidate
TFs related to DR with potential target genes which are
co-regulated in each of the above 69 pathways. The details are
available upon request. As a result, 2 protein families, PPARs and
SMADs, including members PPARα, PPARγ, PPAR_DR1, SMAD, SMAD3 and
SMAD4, were predicted as candidate TFs in the majority of the
identified pathways, particularly the downregulated pathways. PPARs
are ligand-activated nuclear TFs that control gene expression by
binding to specific response elements (PPREs) within promoters.
They play a critical physiological role as lipid sensors and
regulators of lipid metabolism (36). More potent synthetic PPAR ligands,
including the fibrates and thiazolidinediones, have proven
effective in the treatment of dyslipidemia and diabetes (35). The powerful therapeutic ability of
PPARα and PPARγ agonists to favorably influence systemic lipid
levels, glucose homeostasis, and the risk of atherosclerosis (in
the case of PPARα activation in humans) have been demonstrated
(37). PPARγ plays a vitally
important role in the pathogenesis of DR by inhibiting retinal
leukostasis and leakage in response to diabetes (38). Fenofibrate, a PPARα agonist, has
demonstrated robust protective effects against DR in diabetic
patients (39). Our data also
support the hypothesis that PPARα and PPARγ may be important
therapeutic targets for the management of DR.
The TGF-β signal is predominantly transduced by a
family of TFs, the Smad proteins (40). After binding the TGF-β ligand from
outside the cell surface, the type II receptor activates the type I
receptor kinase, and this is followed by the phosphorylation of
receptor-regulated Smads (R-Smads), Smad2 and Smad3. After
associating with a common-partner Smad (co-Smad), or Smad4, the
Smad complex translocates to the nucleus where it regulates the
expression of target genes (40,41). TGF-β is a multifunctional cytokine
with a number of biological effects, such as cell growth,
differentiation and immunomodulation (42). A recent study found that the
expression of TGF-β and Smad4 was increased in the retinal
neovascular membrane of mice with oxygen-induced retinopathy
(43). A previous study also
indicated that increased Smad2/3 phosphorylation levels and
increased TGF-β signaling do in fact occur in the retinal vessels
of diabetic rats, and the concordant attenuation of such signaling
by drugs which protect the vessels from the effects of diabetes
through different mechanisms suggests that the increased signaling
contributes to the vascular pathology (44).
In conclusion, we applied GSEA to re-analyze the
published microarray datasets of DR and performed candidate TF
selection. Finally, we identified 10 upregulated pathways,
including the type I diabetes mellitus and PPAR signaling pathways,
as well as 59 downregulated pathways, including the ErbB signaling
pathway and the mTOR signaling pathway. Furthermore, co-expression
networks of related pathways were constructed using the significant
core genes and TFs, such as PPARG and SMAD4. These may be helpful
to systematically understand the molecular mechanisms of diabetic
retinopathy in genome-wide.
Acknowledgments
We acknowledge financial support from the Scientific
Research Foundation and Academic and Technology Leaders
Introduction Project, the 211 Project of Anhui University (grant
nos. 10117700023 and 02303203-32030081), the Student Research
Training Program of Anhui University (grant no. J18520131), the
Natural Science Foundation Project of Anhui (grant nos.
1508085MH189 and 1508085QC63) and the Education Revitalization
Project of Anhui: Stem Cell and Translational Medicine (grant no.
Y05201374).
References
|
1
|
Klein R, Klein BE, Moss SE and
Cruickshanks KJ: The Wisconsin Epidemiologic Study of Diabetic
Retinopathy: XVII. The 14-year incidence and progression of
diabetic retinopathy and associated risk factors in type 1
diabetes. Ophthalmology. 105:1801–1815. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wong TY, Klein R, Islam FM, Cotch MF,
Folsom AR, Klein BE, Sharrett AR and Shea S: Diabetic retinopathy
in a multi-ethnic cohort in the United States. Am J Ophthalmol.
141:446–455. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ng DP: Human genetics of diabetic
retinopathy: Ccurrent perspectives. J Ophthalmol.
2010:1725932010.
|
|
4
|
Mohamed Q, Gillies MC and Wong TY:
Management of diabetic retinopathy: Aa systematic review. JAMA.
298:902–916. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Todd JA, Walker NM, Cooper JD, Smyth DJ,
Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, et
al: Genetics of Type 1 Diabetes in Finland; Wellcome Trust Case
Control Consortium: Robust associations of four new chromosome
regions from genome-wide analyses of type 1 diabetes. Nat Genet.
39:857–864. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brucklacher RM, Patel KM, VanGuilder HD,
Bixler GV, Barber AJ, Antonetti DA, Lin CM, LaNoue KF, Gardner TW,
Bronson SK and Freeman WM: Whole genome assessment of the retinal
response to diabetes reveals a progressive neurovascular
inflammatory response. BMC Med Genomics. 1:262008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Costa V, Gallo MA, Letizia F, Aprile M,
Casamassimi A and Ciccodicola A: PPARG: Gene expression regulation
and next-generation sequencing for unsolved issues. PPAR Res.
2010:4091682010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lorenzi M and Gerhardinger C: Early
cellular and molecular changes induced by diabetes in the retina.
Diabetologia. 44:791–804. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Warpeha KM and Chakravarthy U: Molecular
genetics of microvascular disease in diabetic retinopathy. Eye
(Lond). 17:305–311. 2003. View Article : Google Scholar
|
|
10
|
Ray D, Mishra M, Ralph S, Read I, Davies R
and Brenchley P: Association of the VEGF gene with proliferative
diabetic retinopathy but not proteinuria in diabetes. Diabetes.
53:861–864. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kumaramanickavel G, Ramprasad VL, Sripriya
S, Upadyay NK, Paul PG and Sharma T: Association of Gly82Ser
polymorphism in the RAGE gene with diabetic retinopathy in type II
diabetic Asian Indian patients. J Diabetes Complications.
16:391–394. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Awata T, Inoue K, Kurihara S, Ohkubo T,
Watanabe M, Inukai K, Inoue I and Katayama S: A common polymorphism
in the 5′-untranslated region of the VEGF gene is associated with
diabetic retinopathy in type 2 diabetes. Diabetes. 51:1635–1639.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Y, Ng MC, Lee SC, So WY, Tong PC,
Cockram CS, Critchley JA and Chan JC: Phenotypic heterogeneity and
associations of two aldose reductase gene polymorphisms with
nephropathy and retinopathy in type 2 diabetes. Diabetes Care.
26:2410–2415. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Suganthalakshmi B, Anand R, Kim R,
Mahalakshmi R, Karthikprakash S, Namperumalsamy P and Sundaresan P:
Association of VEGF and eNOS gene polymorphisms in type 2 diabetic
retinopathy. Mol Vis. 12:336–341. 2006.PubMed/NCBI
|
|
15
|
Beránek M, Kanková K, Benes P,
Izakovicová-Hollá L, Znojil V, Hájek D, Vlková E and Vácha J:
Polymorphism R25P in the gene encoding transforming growth
factor-beta (TGF-beta1) is a newly identified risk factor for
proliferative diabetic retinopathy. Am J Med Genet. 109:278–283.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lam HC, Lee JK, Lu CC, Chu CH, Chuang MJ
and Wang MC: Role of endothelin in diabetic retinopathy. Curr Vasc
Pharmacol. 1:243–250. 2003. View Article : Google Scholar
|
|
17
|
Santos KG, Tschiedel B, Schneider J, Souto
K and Roisenberg I: Diabetic retinopathy in Euro-Brazilian type 2
diabetic patients: relationship with polymorphisms in the aldose
reductase, the plasminogen activator inhibitor-1 and the
methylenetetrahydrofolate reductase genes. Diabetes Res Clin Pract.
61:133–136. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Amano S, Yamagishi S, Koda Y, Tsuneoka M,
Soejima M, Okamoto T, Inagaki Y, Yamada K and Kimura H:
Polymorphisms of sorbitol dehydrogenase (SDH) gene and
susceptibility to diabetic retinopathy. Med Hypotheses. 60:550–551.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yoshioka K, Yoshida T, Takakura Y, Umekawa
T, Kogure A, Toda H and Yoshikawa T: Relation between polymorphisms
G1704T and G82S of rage gene and diabetic retinopathy in Japanese
type 2 diabetic patients. Intern Med. 44:417–421. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Edgar R, Domrachev M and Lash AE: Gene
Expression Omnibus: NCBI gene expression and hybridization array
data repository. Nucleic Acids Res. 30:207–210. 2002. View Article : Google Scholar :
|
|
21
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Aarchive for functional
genomics data set-update. Nucleic Acids Res. 41:D991–D995. 2013.
View Article : Google Scholar
|
|
22
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES, et al: Gene set enrichment analysis: a
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
He K, Chen Z, Ma Y and Pan Y:
Identification of high-copper-responsive target pathways in Atp7b
knockout mouse liver by GSEA on microarray data sets. Mamm Genome.
22:703–713. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao H, Wang Q, Bai C, He K and Pan Y: A
cross-study gene set enrichment analysis identifies critical
pathways in endometriosis. Reprod Biol Endocrinol. 7:942009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: affy - analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chiaretti S, Li X, Gentleman R, Vitale A,
Vignetti M, Mandelli F, Ritz J and Foa R: Gene expression profile
of adult T-cell acute lymphocytic leukemia identifies distinct
subsets of patients with different response to therapy and
survival. Blood. 103:2771–2778. 2004. View Article : Google Scholar
|
|
27
|
Gotea V and Ovcharenko I: DiRE:
Iidentifying distant regulatory elements of co-expressed genes.
Nucleic Acids Res. 36:W133–W139. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ma JH, Li J, Wang JJ and Zhang SX: X-box
binding protein 1 (XBP1) is a crucial regulator of endothelial
tight junction and protects the blood-retinal barrier in diabetic
retinopathy. Invest Ophthalmol Vis Sci. 55:22532014.
|
|
29
|
Akhtar S, Almubrad T, Bron AJ, Yousif MH,
Benter IF and Akhtar S: Role of epidermal growth factor receptor
(EGFR) in corneal remodelling in diabetes. Acta Ophthalmol.
87:881–889. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
McClintock JL and Ceresa BP: Transforming
growth factor-{alpha} enhances corneal epithelial cell migration by
promoting EGFR recycling. Invest Ophthalmol Vis Sci. 51:3455–3461.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Humar R, Kiefer FN, Berns H, Resink TJ and
Battegay EJ: Hypoxia enhances vascular cell proliferation and
angiogenesis in vitro via rapamycin (mTOR)-dependent signaling.
FASEB J. 16:771–780. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cai J, Ahmad S, Jiang WG, Huang J, Kontos
CD, Boulton M and Ahmed A: Activation of vascular endothelial
growth factor receptor-1 sustains angiogenesis and Bcl-2 expression
via the phosphatidylinositol 3-kinase pathway in endothelial cells.
Diabetes. 52:2959–2968. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Klein R, Lee KE, Gangnon RE and Klein BE:
The 25-year incidence of visual impairment in type 1 diabetes
mellitus the wisconsin epidemiologic study of diabetic retinopathy.
Ophthalmology. 117:63–70. 2010. View Article : Google Scholar
|
|
34
|
Wang QJ: PKD at the crossroads of DAG and
PKC signaling. Trends Pharmacol Sci. 27:317–323. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lemberger T, Desvergne B and Wahli W:
Peroxisome proliferator-activated receptors: a nuclear receptor
signaling pathway in lipid physiology. Annu Rev Cell Dev Biol.
12:335–363. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hihi AK, Michalik L and Wahli W: PPARs:
Transcriptional effectors of fatty acids and their derivatives.
Cell Mol Life Sci. 59:790–798. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Berger J and Moller DE: The mechanisms of
action of PPARs. Annu Rev Med. 53:409–435. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Song MK, Roufogalis BD and Huang TH:
Modulation of diabetic retinopathy pathophysiology by natural
medicines through PPAR-γ-related pharmacology. Br J Pharmacol.
165:4–19. 2012. View Article : Google Scholar :
|
|
39
|
Chen Y, Hu Y, Lin M, Jenkins AJ, Keech AC,
Mott R, Lyons TJ and Ma JX: Therapeutic effects of PPARα agonists
on diabetic retinopathy in type 1 diabetes models. Diabetes.
62:261–272. 2013. View Article : Google Scholar
|
|
40
|
Massagué J and Wotton D: Transcriptional
control by the TGF-β/Smad signaling system. EMBO J. 19:1745–1754.
2000. View Article : Google Scholar
|
|
41
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-β family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kim ES, Kim MS and Moon A:
TGF-beta-induced upregulation of MMP-2 and MMP-9 depends on p38
MAPK, but not ERK signaling in MCF10A human breast epithelial
cells. Int J Oncol. 25:1375–1382. 2004.PubMed/NCBI
|
|
43
|
Yingchuan F, Chuntao L, Hui C and Jianbin
H: Increased expression of TGF-β1 and Smad 4 on oxygen-induced
retinopathy in neonatal mice. Retinal Degenerative Diseases.
Springer-Verlag; New York, NY: pp. 71–77. 2010, View Article : Google Scholar
|
|
44
|
Gerhardinger C, Dagher Z, Sebastiani P,
Park YS and Lorenzi M: The transforming growth factor-β pathway is
a common target of drugs that prevent experimental diabetic
retinopathy. Diabetes. 58:1659–1667. 2009. View Article : Google Scholar : PubMed/NCBI
|