Introduction
Hydrogen sulfide (H2S), a well-known
toxic gas with a characteristic smell of rotten eggs, has been
previously described as an endogenously produced labile diffusible
gasotransmitter which plays multiple roles in the cardiovascular
system in general health and also in diseases (1–6).
For example, in murine models of ischemia-induced heart failure,
endogenous and exogenous H2S clearly were shown to exert
protective effects against left ventricular structural and
functional impairment caused by ischemia-induced heart failure
(7). Wang et al reported
that H2S attenuated ventricular dysfunction and arrested
the progression of heart failure following myocardial infarction
(MI) in a rat model (8). In
addition, in relation to plasma H2S levels in patients
with coronary heart disease (CHD), a significant inverse
correlation with the severity of CHD and changes in the coronary
artery has been noted (9).
Previously, we demonstrated that exogenous H2S protects
H9c2 cardiomyocytes against chemical hypoxia- (10,11) or doxorubicin-induced (12–14) injury. The roles of H2S
in diabetes-related cardiovascular complications have attracted
considerable attention, due to the following findings. First, lower
circulating H2S concentrations have been noted in animal
models of diabetes (5,15,16) and patients with type 2 diabetes
mellitus (DM) (5,6). Second, low blood H2S
levels may be associated with the vascular inflammation observed in
diabetes since the supplementation of H2S prevents the
secretion of inflammatory factors by monocytes cultured in
high-glucose (HG) medium (5).
Third, exogenous H2S protects against the development of
HG-induced endothelial dysfunction (15). Fourth, exogenous H2S
alleviates myocardial ischemia/reperfusion (IR) injury in db/db
mice (17). Fifth, H2S
has been shown to exert protective effects against myocardial
I/R-induced damage in diabetic rats (18). Furthermore, our recent studies
demonstrated that exogenous H2S protects H9c2 cardiac
cells against HG-induced injury and inflammation (19–21). Although we have reported that
several factors, including antioxidant, anti-apoptotic and
anti-inflammatory effects, mitochondrial protection, and the
inhibition of certain intracellular signaling pathways, such as
mitogen-activated protein kinase (MAPK) (19), leptin (20) and nuclear factor-κB (NF-κB)
(21), contribute to the
protective effects of H2S against HG-induced
cardiomyocyte injury, the mechanisms responsible for these
cardioprotective effects of H2S remain unclear. Since
previous studies have indicated that H2S activates
ATP-sensitive K+ (KATP) channels in both the
heart (22,23) and vascular tissues (24) and that KATP channels
are cardioprotective (23,25–29),
we thus hypothesized that KATP channels are involved in
the protective effects which H2S exerts against
HG-induced injury in H9c2 cardiomyocytes.
KATP channels are abundant in cardiac
tissue (30). Cardiomyocytes
contain KATP channels in both the sarcolemma [surface
membrane (31)] and mitochondria
(32,33). The opening of sarcolemmal
KATP channels is associated with the shortening of
cardiac action potential, and a decrease in intracellular
Ca2+ loading and cardioprotection during ischemia
(34–36). The opening of mitochondrial
KATP channels contributes to the regulation of cardiac
mitochondrial function (37) and
cardio-protection induced by ischemic preconditioning (23,37,39). In addition, mitochondrial
KATP channels ameliorate the apoptosis induced by
oxidative stress in cardiac cells (26). Notably, previous research has
revealed that DM is associated with the dysfunction of the
cardiovascular KATP channels (40). Hyperglycemia, as well as DM, is
harmful to the vasodilation mediated by KATP channels in
human vascular smooth muscle cells (41–43). However, the roles of both
sarcolemmal KATP channels and mitochondrial
KATP channels in HG-induced cardiomyocyte injury, in
particular, in relation to the cardio-protective effects of
exogenous H2S, remain unclear.
Based on our recent studies (19–21) and other previous studies (5,6,15,17,18,22–29), we hypothesized that H2S
exerts cardioprotective effects by modulating the activation of
KATP channels in HG-treated cardiomyocytes. Therefore,
the present study was designed to examine the following points: i)
the effect of HG on the expression of cardiac KATP
channels; ii) the roles of both sarcolemmal KATP
channels and mitochondrial KATP channels in HG-induced
cardiomyocyte injury; iii) whether exogenous H2S
protects cardiomyocytes against HG-induced injury by modulating
KATP channel activity; and iv) the role of reactive
oxygen species (ROS) in the inhibitory effect of HG on the
expression of cardiac KATP channels.
Materials and methods
Materials and reagents
Anti-kir6.1 (R-14) antibody (sc-11224) was purchased
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA);
anti-cleaved caspase-3 antibody (#9662) was purchased from Cell
Signaling Technology, Inc. (Boston, MA, USA); anti-GAPDH antibody
(10494-1-AP) was purchased from Proteintech Group, Inc. (Wuhan,
China); horseradish peroxidase (HRP) conjugated secondary antibody
and the BCA Protein assay kit were obtained from KangChen Biotech
(Shanghai, China). Diazoxide (DZ), pinacidil (Pin),
5-hydroxydecanoic acid (5-HD) and glibenclamide (Gli) were all
purchased from Cayman Chemical Co. (Ann Arbor, MI, USA). Sodium
hydrosulfide (NaHS; a donor of H2S) was obtained from
Sigma-Aldrich (St. Louis, MO, USA), and was protected from sunlight
and stored at 2–4°C. The cell counting kit-8 (CCK-8) was supplied
by Dojindo Laboratories (Kumamoto, Japan).
2′,7′-Dichlorofluorescein diacetate (DCFH-DA),
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraeth-ylbenzimidazolylcarbocyanine
iodide (JC-1), Hoechst 33258 and N-acetyl-L-cysteine (NAC) were all
purchased from Sigma-Aldrich. Fetal bovine serum (FBS) and
Dulbecco's modified Eagle's medium (DMEM) medium were obtained from
Gibco-BRL (Grand Island, NY, USA). The enhanced chemiluminescence
(ECL) solution was purchased from KeyGen Biotech (Nanjing, China).
The H9c2 cardiac cells were supplied by the Sun Yat-sen University
Experimental Animal Center (Guangzhou, China).
Cell culture and treatment
The H9c2 cardiac cells, a rat cardiac myoblast cell
line, were cultured in DMEM, supplemented with 10% FBS in a
humidified atmosphere of 95% air and 5% CO2 at 37°C. The
culture medium was replaced with fresh medium every 2–3 days and
expanded to new culture plates when the cells reached approximately
80% confluence.
To investigate the role of KATP channels
in HG-induced cardiomyocyte injury, the H9c2 cardiac cells were
treated with 100 µM DZ (a mitochondrial KATP
channel opener) or 50 µM Pin (a non-selective
KATP channel opener) for 30 min prior to exposure to 35
mM glucose for 24 h. To explore the protective effects of
H2S on HG-induced injury, the H9c2 cells were
conditioned with 400 µM NaHS for 30 min prior to exposure to
HG for 24 h. To further determine whether the protective effects of
NaHS were associated with the activation of KATP
channels, the cells were conditioned with 100 µM 5-HD (a
mitochondrial KATP channel blocker) or 1 mM Gli (a
non-selective KATP channel blocker) for 30 min prior to
treatment with NaHS and 35 mM glucose for 24 h. To confirm whether
there is an antagonistic interaction between ROS and
KATP channels, the H9c2 cells were treated with 500
µM NAC (a scavenger of ROS) for 60 min prior to exposure to
HG.
Cell viability assay
The H9c2 cells were seeded in 96-well plates at a
concentration of 1×104 cells/ml and incubated at 37°C.
CCK-8 assay was employed to assess the viability of the cells.
After being subjected to the above-mentioned treatments, the cells
were washed with phosphate-buffered saline (PBS), and 10 µl
CCK-8 solution at 10% dilution was added to each well, and the
plate was then incubated for approximately 2 h in an incubator. The
absorbance at 450 nm was assayed using a microplate reader
(Molecular Devices, Sunnyvale, CA, USA). The means of the optical
density (OD) of 3 wells in the indicated groups were used to
calculate the percentage of cell viability according to the
following formula: cell viability (%) = (ODtreatment
group/ODcontrol group) ×100. The experiment was
repeated 5 times.
Hoechst 33258 nuclear staining for the
assessment of apoptosis
Apoptotic cell death was assessed using the Hoechst
33258 staining method followed by photofluorography. The H9c2 cells
were plated in 35-mm dishes at a density of 1×106
cells/well. After being subjected to the indicated treatments, the
cells were harvested and fixed with paraformaldehyde in 0.1 mol/l
PBS (pH 7.4) for 10 min. The slides were then washed 5 times with
PBS. After rinsing with PBS, the nuclear DNA was stained with 5
mg/ml Hoechst 33258 for 10 min before being rinsed briefly with PBS
and then visualized under a fluorescence microscope (BX50-FLA;
Olympus, Tokyo, Japan). The viable H9c2 cells exhibited a uniform
blue fluorescence throughout the nucleus, whereas the apoptotic
cells had fragmented and condensed nuclei. The experiment was
carried out 3 times.
Measurement of intracellular ROS
levels
The determination of intracellular ROS levels was
performed by measuring the fluorescence product formed by the
oxidation of DCFH-DA, as prevoiusly described (19). Briefly, the culture medium was
removed and the cells were then washed 3 times with PBS. Following
the addition of fresh culture medium, the cells were incubated with
DCFH-DA at a final concentration of 10 µM, for 30 min at
37°C. The cells were then washed 5 times with PBS, and the relative
amount of fluorescence product was assessed using a fluorescence
microscope connected to an imaging system (BX50-FLA; Olympus). The
mean fluorescence intensity (MFI) from 5 random fields was measured
using ImageJ 1.47i software, and the MFI was used as an index for
the amount of ROS. The experiment was carried out 5 times.
Measurement of mitochondrial membrane
potential (MMP)
As previousy described (19), MMP was assessed using a
fluorescent dye, JC-1, which is a cell-permeable cationic dye that
enters the mitochondria based on a highly negative MMP. The
depolarization of MMP results in the loss of JC-1 from the
mitochondria and a decrease in intracellular green fluorescence.
The H9c2 cardiac cells were cultured on a slide with Eagle's
minimal essential medium (EMEM). After being subjected to the
indicated treatments, the slides were washed 3 times with PBS. The
H9c2 cells were incubated with 1 mg/l JC-1 at 37°C for 30 min in an
incubator and washed 3 times with PBS. JC-1 fluorescence was then
measured over the entire field of vision using a fluorescence
microscope connected to an imaging system (BX50-FLA; Olympus). The
MFI of JC-1 from 5 random fields was analyzed using ImageJ 1.47i
software and was taken as an index of the levels of MMP. The
experiment was carried out 5 times.
Western blot analysis
After being subjected to the indicated treatments,
the H9c2 cardiac cells were harvested and lysed with cell lysis
solution at 4°C for 30 min. Total proteins in the cell lysates were
quantified using a BCA protein assay kit. Loading buffer was added
to the cytosolic extracts and, after boiling for approximately 5
min, equal amounts of supernatant from each sample were
fractionated by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE). Total proteins in the gel were
transferred onto polyvinylidene difluoride (PVDF) membranes. The
membranes were blocked for approximately 90 min at room temperature
in fresh blocking buffer [0.1% Tween-20 in Tris-buffered saline
(TBS-T) containing 5% fat-free milk] and then incubated with either
anti-KATP (1:1,000 dilution), or anti-cleaved caspase-3
antibody (1:1,000 dilution) in freshly prepared TBS-T with 3%
fat-free milk overnight with slow agitation at 4°C. Following 3
washes with TBS-T, the membranes were incubated with HRP-conjugated
goat anti-rabbit secondary antibody (1:2,500 dilution; KangChen
Bio-tech) in TBS-T with 3% fat-free milk for 90 min at room
temperature. GAPDH was used as an internal control. The membranes
were then washed 3 times with TBS-T solution for 15 min. The
immunoreative signals were visualized by ECL detection. In order to
quantify protein expression, the X-ray films were scanned and
analyzed using ImageJ 1.47i software. Each experiment was repeated
3 times.
Statistical analysis
All data are presented as the means ± SEM.
Differences between groups were analyzed by one-way analysis of
variance (ANOVA) using SPSS 13.0 software (SPSS, Inc., Chicago, IL,
USA) followed by the least significant difference (LSD) post hoc
comparison test. A P-value <0.05 was considered to indicate a
statistically significant difference.
Results
NaHS attenuates the HG-induced decrease
in protein expression of KATP channels in H9c2 cardiac
cells
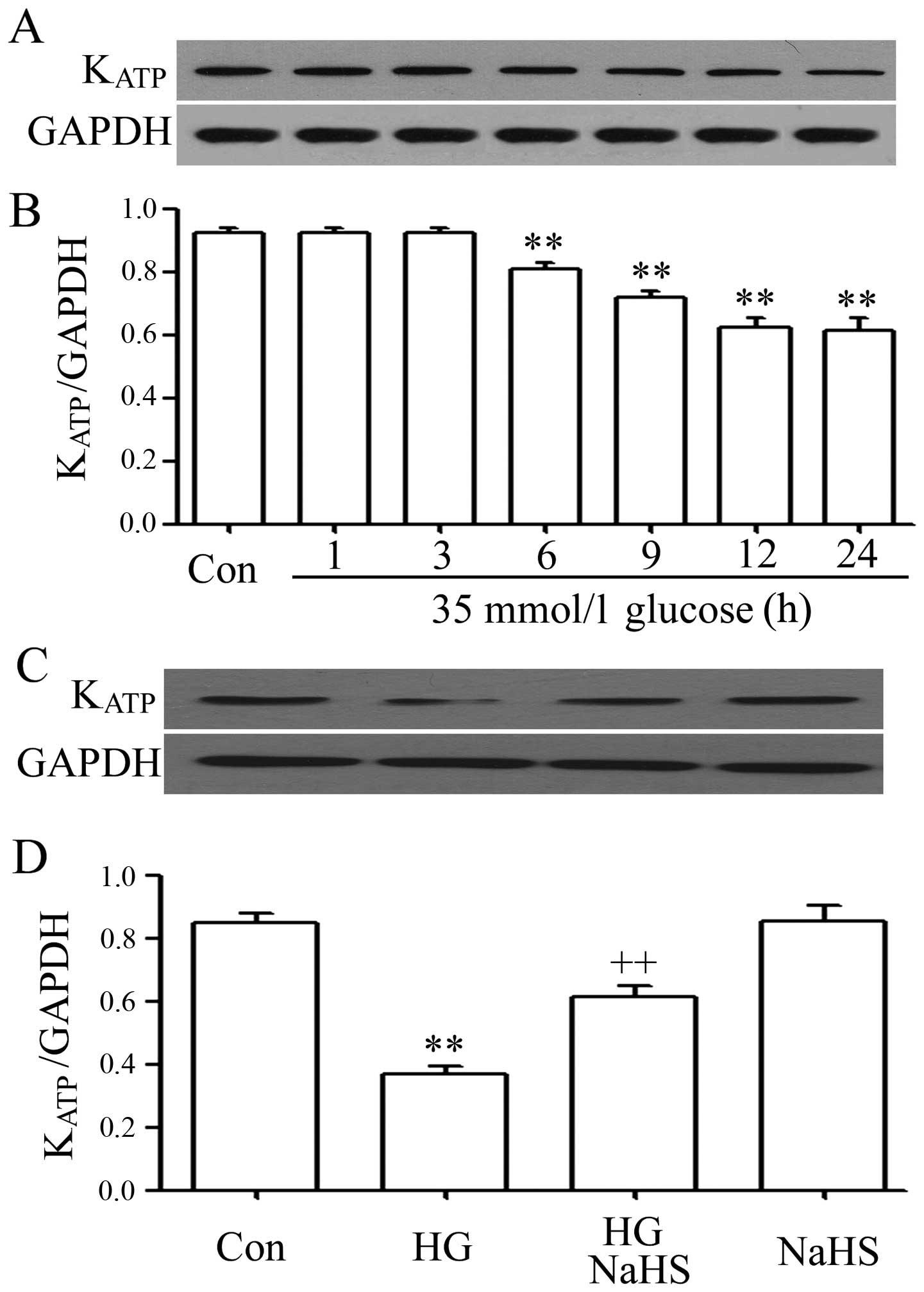
In order to investigate the influence of HG (35 mM
glucose) on the protein expression of KATP channels in
H9c2 cardiac cells, a time-response experiment to determine the
protein expression levels of KATP channels was
performed. As shown in Fig. 1A and
B, the cells were exposed to HG for 1, 3, 6, 9, 12 and 24 h,
respectively. Following exposure to HG for 6 h, the protein
expression level of KATP channels began to decrease, and
the maximum decrease in expression levels was observed after the
cells were exposed to HG for 12 and 24 h. Based on these results,
the expression levels of KATP channels were detected at
12 h following exposure to HG in the subsequent experiments.
It is important to note that the decrease in the
KATP channel levels was ameliorated by treatment with
400 µM NaHS (a donor of H2S) for 30 min prior to
exposure to HG for 12 h (Fig. 1C and
D). However, the basal expression level of KATP
channels was not markedly altered by treatment with 400 µM
NaHS alone for 30 min. These data indicate that exogenous
H2S alleviates the decrease in the protein expression
levels of KATP channels induced by HG in H9c2 cardiac
cells.
KATP channels are involved in
the protective effects which H2S exerts against
HG-induced cytotoxicity to H9c2 cardiac cells
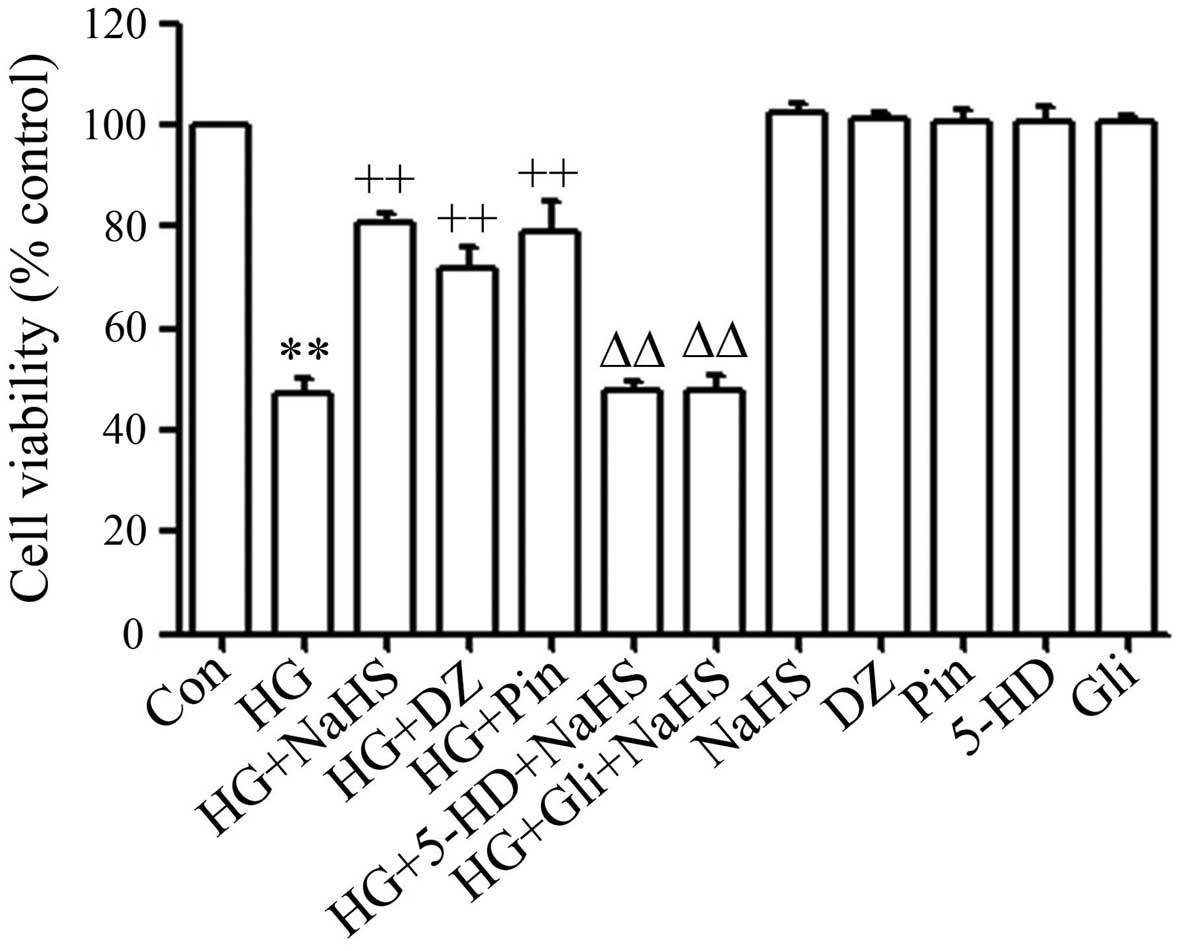
Consistent with our recent studies (19–21), treatment of the cells with 400
µM NaHS for 30 min prior to exposure to HG for 24 h
significantly inhibited HG-induced cytotoxicity, leading to an
increase in cell viability (Fig.
2). To explore the role of KATP channels in
HG-induced cytotoxicity, the cells were treated with 100 µM
DZ (a mitochondrial KATP channel opener) or 50 µM
Pin (a non-selective KATP channel opener) for 30 min
prior to exposure to HG. As shown in Fig. 2, pre-treatment of the H9c2 cardiac
cells with DZ or Pin considerably reduced HG-induced cytotoxicity,
as evidenced by an increase in cell viability. To further
investigate the role which KATP channels play in the
protective effects exerted by H2S against HG-induced
cytotoxicity, the cells were treated with 100 µM 5-HD (a
mitochondrial KATP channel blocker) or 1 mM Gli (a
non-selective KATP channel blocker) for 30 min prior to
treatment with NaHS and exposure to HG. It was demonstrated that
the blockade of KATP channels with 5-HD or Gli markedly
reduced the protective effects of NaHS against HG-induced
cytotoxicity, resulting in a decrease in cell viability (Fig. 2). Alone, 5-HD and Gli did not
significantly alter cell viability. These data suggest that
KATP channels mediate the protective effects of
H2S against cytotoxicity to H9c2 cardiac cells induced
by HG.
KATP channels are involved in
the protective effects which H2S exerts against
HG-induced apoptosis in H9c2 cardiac cells
In agreement with our recent studies (19–21), exposure of the cells to HG for 24
h markedly increased the number of apoptotic cells (Fig. 3A, panel b), leading to an increase
in the percentage of apoptotic cells (Fig. 3A, panel m). The increased number
of apoptotic cells was decreased by pre-treatment with NaHS, Pin or
DZ (Fig. 3A, panels c, d and e).
Similarly, exposure of the cells to 35 mM glucose for 24 h markedly
enhanced the expression level of cleaved caspase-3 (Fig. 3B). However, the increased
expression level of cleaved caspase-3 was attenuated by treatment
with 400 µM NaHS or 50 µM Pin or 100 µM DZ for
30 min prior to exposure to HG for 24 h (Fig. 3B, panels a to d). Furthermore,
treatment of the H9c2 cardiac cells with 100 µM 5-HD
(Fig. 3A, panel f and B, panels c
and d) or 1 mM Gli (Fig. 3A,
panel g and B, panels c and d) for 30 min prior to treatment with
NaHS and exposure to HG blocked the above-mentioned anti-apoptotic
effects of NaHS, as evidenced by the increase in the percentage of
apoptotic cells (Fig. 3A, panels
f and g) and the increase in cleaved caspase-3 expression (Fig. 3B, panels c and d). Alone, NaHS,
Pin, DZ, 5-HD and Gli did not significantly affect the percentage
of apoptotic cells or the basal expression level of cleaved
caspase-3.
KATP channels are implicated
in the protective effects exerted by H2S against
HG-induced oxidative stress in H9c2 cardiac cells
In agreement with our recent findings (19–21), treatment of the cells with 35 mM
glucose for 24 h significantly increased the intracellular
production of ROS (Fig. 4B and
M). The increased ROS production was ameliorated by treatment
with 400 µM NaHS for 30 min prior to exposure to HG for 24 h
(Fig. 4C and M). Similarly,
treatment with 50 µM Pin (Fig.
4D and M) or 100 µM DZ (Fig. 4E and M) for 30 min prior to
exposure to HG for 24 h also attenuated the generation of ROS. To
confirm the role played by KATP channels in the
protective effects of NaHS against HG-induced oxidative stress, the
cells were treated with 100 µM 5-HD or 1 mM Gli for 30 min
prior to exposure to NaHS and HG. Our data indicated that
pre-treatment with 5-HD (Fig. 4F and
M) or Gli (Fig. 4G and M)
blocked the inhibitory effects which NaHS exerted on the generation
of ROS induced by HG, suggesting that the KATP channels
contribute to the protective effects which H2S exerts
against the HG-induced overproduction of ROS.
KATP channels are linked to
the protective effects of H2S against HG-induced
mitochondrial insults in H9c2 cardiac cells
Consistent with our recent studies (19–21), treatment of the cells with 35 mM
glucose for 24 h markedly induced mitochondrial damage, as
evidenced by the loss of MMP (Fig.
5A, panel b and 5M). In addition, we noted that the HG-induced
decrease in MMP was reversed by treatment of the cells with 400
µM NaHS for 30 min prior to exposure to HG (Fig. 5A, panel c and 5M). Notably,
treatment of the cells with 50 µM Pin (Fig. 5A, panel d and 5M) or 100 µM
DZ (Fig. 5A, panel e and 5M) for
30 min prior to exposure to HG also blocked the HG-induced loss of
MMP. Subsequently, we further explored the role of KATP
channels in the protective effects of NaHS against the dissipation
of MMP induced by HG. Our findings revealed that treatment with 100
µM 5-HD or 1 mM Gli for 30 min prior to treatment with NaHS
and exposure to HG considerably reduced the inhibitory effects of
NaHS on the HG-induced dissipation of MMP (Fig. 5A, panels f and g and 5M). These
results demonstrate that KATP channels play a critical
role in the protective effects of H2S against the
HG-induced mitochondrial damage.
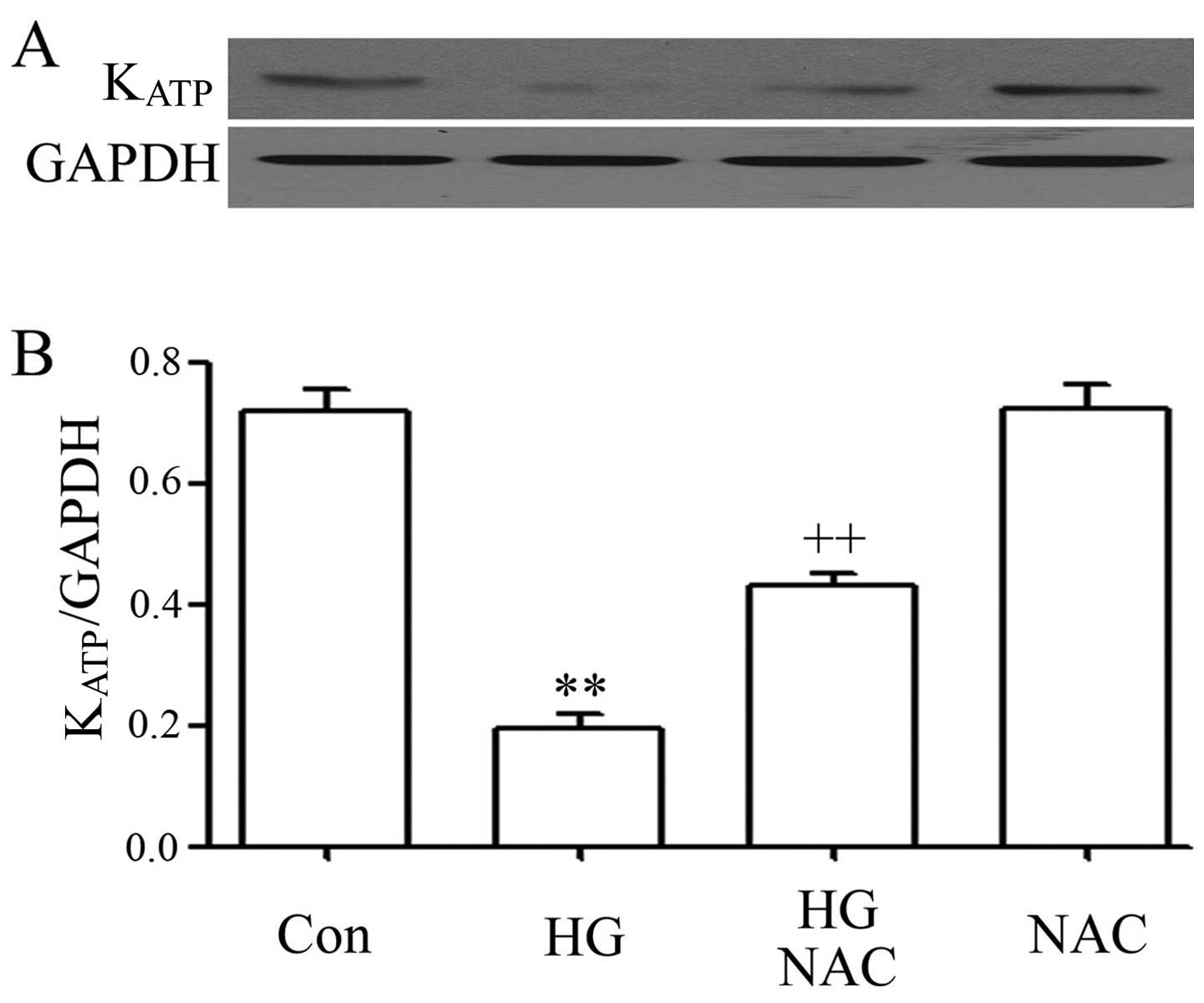
ROS scavenger blocks the HG-induced
downregulation of the expression of KATP channels in
H9c2 cardiac cells
Since hyperglycemia, as well as DM, has been
reported to impair KATP channels in human vascular
smooth muscle cells via ROS (41–43), we investigated the role of ROS in
the HG-induced decrease in KATP channel expression in
the H9c2 cardiac cells. As shown in Fig. 6, treatment of the cells with 500
µM NAC (a scavenger of ROS and NAC) for 60 min prior to
exposure to 35 mM glucose for 12 h blocked the inhibitory effects
of HG on the expression levels of KATP channels. Alone,
NAC did not significantly alter the basal expression level of
KATP channels. These results suggest that ROS
participates in the HG-induced downregulation of the expression of
KATP channels.
Discussion
Cardiac KATP channels are key sensors
and effectors of the metabolic status of cardiomyocytes, and their
roles in HG-induced cardiomyocyte injury and in the
cardioprotective effects of H2S are noteworthy. The
major findings of this study can be summarized as follows: i) HG
markedly downregulated the expression levels of cardiac
KATP channels; ii) exogenous H2S attenuated
the inhibitory effects of HG on the expression of cardiac
KATP channels; iii) the KATP channel openers,
DZ (a mitochondrial KATP channel opener) and Pin (a
non-selective KATP channel openner), ameliorated the
HG-induced cardiomyocyte injury, including cytotoxicity, apoptosis,
ROS generation and the dissipation of MMP; iv) the KATP
channel antagonists, namely 5-HD (a mitochondrial KATP
channel antagonist) and Gli (a non-selective KATP
channel antagonist), blocked the cardio-protective effects of
exogenous H2S against the HG-induced cardiomyocyte
injury; v) the ROS scavenger, NAC, reduced the inhibition of
cardiac KATP channel expression induced by HG. These
results strongly suggest that the impairment of KATP
channels is implicated in HG-induced cardiomyocyte injury and that
the activation of KATP channels is linked to the
protective effects which exogenous H2S exerts against
HG-induced cardiomyocyte injury.
KATP channels have the unique ability to
regulate membrane excitability in response to changes in the
energetic status of cells (31,44,45). This ability serves to decrease
myocardial energy consumption and vulnerability to stress (44,45). Previous research has indicated
that the ability of cardiac KATP channels to affect
cellular excitability and function depends on their abundance at
the membrane surface (46,47).
For example, an increase in cardiac sarcolemmal KATP
channels enhances the speed and degree of shortening of action
potentials and reduces cardiac energy consumption in response to
escalating workloads (46).
Several studies have indicated a correlation between dysfunction in
KATP channel gating and insulin secretory disorders
(40,48), such as neonatal diabetes (48). Furthermore, in human vascular
smooth muscle cells, HG impairs vasorelaxation by inhibiting the
activity of KATP channels (41–43). However, our knowledge of the roles
of KATP channels in HG-induced cardiac cells remains
incomplete.
In order to explore this issue, in the present
study, we first investigated the effects of HG on the expression
levels of KATP channels in H9c2 cardiac cells. Our data
demonstrated that the exposure of the cells to HG markedly reduced
the expression levels of KATP channels. This reduced
expression level of KATP channels indirectively suggests
a decrease in their presence in the HG-treated cardiac cells. Since
a previous study indicated that a reduction in the number of
sarcolemmal KATP channels slows cardiac action potential
during shortening under hypoxia (47),. Thus, we hypothesized that the
inhibition of cardiac KATP channels is a critical
mechanism which underlies HG-induced cardiomyocyte injury. To
confirm this hypothesis, we observed the influence of
KATP channel activation on HG-induced injury. In
agreement with our recent studies (19–21), the findings of this study
demonstrated that treatment of the H9c2 cardiac cells with HG
induced considerable injuries, including a decrease in cell
viability, an increase in apoptotic cells, cleaved caspase-3
expression and ROS generation, as well as the loss of MMP. However,
treatment of the H9c2 cardiac cells with DZ (a mitochondrial
KATP channel opener) or Pin (a non-selective
KATP channel opener) markedly attenuated the
above-mentioned HG-induced injuries, as evidenced by an increase in
cell viability, and a decrease in the number of apoptotic cells,
decreased cleaved caspase-3 expression and ROS generation, as well
as the loss of MMP. The above-mentioned results suggest that HG
impairs the function of cardiac KATP channels, which
contributes to HG-induced injuries. In support of our results are
experimental studies which demonstrate that hyperglycemia damages
the functionality of the human ether-ago-go-related gene (HERG)
K+ channels, reduces the transient outward K+
current, enhances the intracellular concentration of
Ca2+, and impairs the excitation-contraction coupling in
the heart (49,50). It has also been noted that the
opening of the mitochondrial KATP channels plays an
important role in the regulation of cardiac mitochondrial function
(37) and attenuates the
oxidative stress-induced apoptosis of cardiac cells.
Of note, sulfonylurea drugs have been shown to
stimulate insulin secretion by closing KATP channels
(51,52). Despite the fact that their target
is not completely clear, these drugs have been used to treat type 2
DM since the 1950s and they are still used today. Thus, the
findings of this study drive us to explore the effects of
sulfonylurea drugs on HG-induced cardiomyocyte injury in future
experiments.
Another important result of this study relates to
the role of the activation of KATP channels in the
cardioprotective effects of exogenous H2S against
HG-induced cardiac injuries. Previously, the protective effects of
H2S on DM-related cardiovascular insults have received
much attention. Exogenous H2S attenuates I/R-induced
injury in db/db mice (17) and
diabetic rats (18). Our recent
studies have also examined the protective effects of exogenous
H2S against HG-induced injury and inflammation in H9c2
cardiac cells (19–21). The mechanisms responsible for
these cardioprotective effects of H2S are associated
with the inhibition of the MAPK (19), leptin (20) and NF-κB (21) pathways. However, the protective
mechanisms of H2S are complex, and other factors are
likely involved in these cardioprotective effects of
H2S. Since the KATP channels in the heart
(22,23) have been reported to be activated
by H2S, this study further investigated that roles
KATP channels play in the protective effects which
H2S exerts against HG-induced cardiomyocyte injury. In
agreement with our recent studies, the findings of this study
demonstrated that exogenous H2S exerted protective
effects against HG-induced cardiomyocyte injuries, as indicated by
an increase in cell viability, and a decrease in the number of
apoptotic cells, decreased cleaved caspase-3 expression and ROS
generation, as well as the loss of MMP. Notably, our results
demonstrated that exogenous H2S markedly reduced the
downregulation of KATP channel expression by HG and that
both 5-HD (a mitochondrial KATP channel blocker) and Gli
(a non-selective KATP channel blocker) blocked the
cardioprotective effects of H2S mentioned above. These
results suggest that KATP channels, in particular
mitochondrial KATP channels, play a critical role in the
cardioprotective effects of H2S. Similarly, Zhao et
al demonstrated the protective effects which H2S
exerts against arrhythmia by opening mitochondrial KATP
channels (24). However, Bian
et al (23) revealed that
sarcolemmal KATP channels, but not mitochondrial
KATP channels, mediate the cardioprotective effects of
H2S in isolated cardiac myocytes exposed to simulated
ischemia solution. Therefore, the roles of subtypes of
KATP channels involved in the cardioprotective effects
of H2S in the present study differ from the ones
reported by Bian et al (23), although we did not use a
sarcolemmal KATP channels blocker or MCC-134 that opens
sarcolemmal KATP channels and blocks minochondrial
KATP channels. One possible explanation for this
difference in the results may lie in the use of different
experimental models.
In the present study, we also investigated the
interaction between ROS and cardiac KATP channel
activation. As aforementioned, both DZ and Pin ameliorated
HG-induced ROS generation, suggesting that KATP channel
activity had an inhibitory effect on ROS generation. A previous
study reporting that KATP channel opener (DZ) reduces
mitochondrial ROS production at reoxygenation (53) supports our results. On the other
hand, we noted that NAC, a ROS scavenger, blocked the decrease in
the expression of KATP channel protein induced by HG.
Collectively, these results suggest that there is an interaction
between ROS and KATP channels in HG-treated cardiac
cells. The elucidation of the molecular mechanism underlying this
interaction may be significant for the treatment and prevention of
DM-related cardiovascular complications.
In conclusion, the present study provides novel
evidence that the impairment of KATP channels is
associated with HG-induced multiple cardiac injuries, including
cytotoxicity, apoptosis, oxidative stress and mitochondrial damage.
Therefore, the inhibition of cardiac KATP channels by
insulin secretagogues should be considered to increase cardiac
risk. Importantly, the present study has demonstrated that
exogenous H2S protects cardiomyocytes against HG-induced
injuries by activating KATP channels. Based on the
results of the present study and previous studies (5,6,15,16), we suggest that low levels of
endogenous H2S and the impairment of KATP
channels are an important pathophysiological mechanism underlying
huperglycemia-induced cardiovascular complications. Therefore,
supplementation with H2S and modulation of the
H2S-KATP channel pathway should be considered
a potential approach to attenuating hyperglycemia-induced
cardiomyocyte injury.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (no. 81270296) and the
Guangdong Province Finance Science and Technical Fund (no.
2014Sc107).
References
|
1
|
Wang R: Two's company, three's a crowd:
can H2S be the third endogenous gaseous transmitter?
FASEB J. 16:1792–1798. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fiorucci S, Distrutti E, Cirino G and
Wallace JL: The emerging roles of hydrogen sulfide in the
gastrointestinal tract and liver. Gastroenterology. 131:259–271.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Szabo C: Hydrogen sulphide and its
therapeutic potential. Nat Rev Drug Discov. 6:917–935. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li L and Moore PK: Putative biological
roles of hydrogen sulfide in health and disease: a breath of not so
fresh air? Trends Pharmacol Sci. 29:84–90. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jain SK, Bull R, Rains JL, Bass PF, Levine
SN, Reddy S, McVie R and Bocchini JA Jr: Low levels of hydrogen
sulfide in the blood of diabetes patients and
streptozotocin-treated rats causes vascular inflammation? Antioxid
Redox Signal. 12:1333–1337. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Whiteman M, Gooding KM, Whatmore JL, Ball
CI, Mawson D, Skinner K, Tooke JE and Shore AC: Adiposity is a
major determinant of plasma levels of the novel vasodilator
hydrogen sulphide. Diabetologia. 53:1722–1726. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Calvert JW, Elston M, Nicholson CK,
Gundewar S, Jha S, Elrod JW, Ramachandran A and Lefer DJ: Genetic
and pharmacologic hydrogen sulfide therapy attenuates
ischemia-induced heart failure in mice. Circulation. 122:11–19.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang X, Wang Q, Guo W and Zhu YZ: Hydrogen
sulfide attenuates cardiac dysfunction in a rat model of heart
failure: a mechanism through cardiac mitochondrial protection.
Biosci Rep. 31:87–98. 2011. View Article : Google Scholar
|
|
9
|
Zhang Y, Tang ZH, Ren Z, Qu SL, Liu MH,
Liu LS and Jiang ZS: Hydrogen sulfide, the next potent preventive
and therapeutic agent in aging and age-associated diseases. Mol
Cell Biol. 33:1104–1113. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen SL, Yang CT, Yang ZL, Guo RX, Meng
JL, Cui Y, Lan AP, Chen PX and Feng JQ: Hydrogen sulphide protects
H9c2 cells against chemical hypoxia-induced injury. Clin Exp
Pharmacol Physiol. 37:316–321. 2010. View Article : Google Scholar
|
|
11
|
Yang Z, Yang C, Xiao L, Liao X, Lan A,
Wang X, Guo R, Chen P, Hu C and Feng J: Novel insights into the
role of HSP90 in cytoprotection of H2S against chemical
hypoxia-induced injury in H9c2 cardiac myocytes. Int J Mol Med.
28:397–403. 2011.PubMed/NCBI
|
|
12
|
Wang XY, Yang CT, Zheng DD, Mo LQ, Lan AP,
Yang ZL, Hu F, Chen PX, Liao XX and Feng JQ: Hydrogen sulfide
protects H9c2 cells against doxorubicin-induced cardiotoxicity
through inhibition of endoplasmic reticulum stress. Mol Cell
Biochem. 363:419–426. 2012. View Article : Google Scholar
|
|
13
|
Guo R, Lin J, Xu W, Shen N, Mo L, Zhang C
and Feng J: Hydrogen sulfide attenuates doxorubicin-induced
cardiotoxicity by inhibition of the p38 MAPK pathway in H9c2 cells.
Int J Mol Med. 31:644–650. 2013.PubMed/NCBI
|
|
14
|
Guo R, Wu K, Chen J, Mo L, Hua X, Zheng D,
Chen P, Chen G, Xu W and Feng J: Exogenous hydrogen sulfide
protects against doxorubicin-induced inflammation and cytotoxicity
by inhibiting p38MAPK/NF-κB pathway in H9c2 cardiac cells. Cell
Physiol Biochem. 32:1668–1680. 2013.
|
|
15
|
Suzuki K, Olah G, Modis K, Coletta C, Kulp
G, Gero D, Szoleczky P, Chang T, Zhou Z, Wu L, et al: Hydrogen
sulfide replacement therapy protects the vascular endothelium in
hyperglycemia by preserving mitochondrial function. Proc Natl Acad
Sci USA. 108:13829–13834. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ahmad FU, Sattar MA, Rathore HA, Abdullah
MH, Tan S, Abdullah NA and Johns EJ: Exogenous hydrogen sulfide
(H2S) reduces blood pressure and prevents the
progression of diabetic nephropathy in spontaneously hypertensive
rats. Ren Fail. 34:203–210. 2012. View Article : Google Scholar
|
|
17
|
Peake BF, Nicholson CK, Lambert JP, Hood
RL, Amin H, Amin S and Calvert JW: Hydrogen sulfide preconditions
the db/db diabetic mouse heart against ischemia-reperfusion injury
by activating Nrf2 signaling in an Erk-dependent manner. Am J
Physiol Heart Circ Physiol. 304:H1215–H1224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gao Y, Yao X, Zhang Y, Li W, Kang K, Sun L
and Sun X: The protective role of hydrogen sulfide in myocardial
ischemia-reperfusion-induced injury in diabetic rats. Int J
Cardiol. 152:177–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu W, Wu W, Chen J, Guo R, Lin J, Liao X
and Feng J: Exogenous hydrogen sulfide protects H9c2 cardiac cells
against high glucose-induced injury by inhibiting the activities of
the p38 MAPK and ERK1/2 pathways. Int J Mol Med. 32:917–925.
2013.PubMed/NCBI
|
|
20
|
Zhuang XD, Hu X, Long M, Dong XB, Liu DH
and Liao XX: Exogenous hydrogen sulfide alleviates high
glucose-induced cardiotoxicity via inhibition of leptin signaling
in H9c2 cells. Mol Cell Biochem. 391:147–155. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu W, Chen J, Lin J, Liu D, Mo L, Pan W,
Feng J, Wu W and Zheng D: Exogenous H2S protects H9c2
cardiac cells against high glucose-induced injury and inflammation
by inhibiting the activation of the NF-κB and IL-1β pathways. Int J
Mol Med. 35:177–186. 2015.
|
|
22
|
Geng B, Yang J, Qi Y, Zhao J, Pang Y, Du J
and Tang C: H2S generated by heart in rat and its
effects on cardiac function. Biochem Biophys Res Commun.
313:362–368. 2004. View Article : Google Scholar
|
|
23
|
Bian JS, Yong QC, Pan TT, Feng ZN, Ali MY,
Zhou S and Moore PK: Role of hydrogen sulfide in the
cardioprotection caused by ischemic preconditioning in the rat
heart and cardiac myocytes. J Pharmacol Exp Ther. 316:670–678.
2006. View Article : Google Scholar
|
|
24
|
Zhao W, Zhang J, Lu Y and Wang R: The
vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP)
channel opener. EMBO J. 20:6008–6016. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Eisen A, Fisman EZ, Rubenfire M, Freimark
D, McKechnie R, Tenenbaum A, Motro M and Adler Y: Ischemic
preconditioning: nearly two decades of research. A comprehensive
review. Atherosclerosis. 172:201–210. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Akao M, Ohler A, O'Rourke B and Marban E:
Mitochondrial ATP-sensitive potassium channels inhibit apoptosis
induced by oxidative stress in cardiac cells. Circ Res.
88:1267–1275. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yamada S, Kane GC, Behfar A, Liu XK, Dyer
RB, Faustino RS, Miki T, Seino S and Terzic A: Protection conferred
by myocardial ATP-sensitive K+ channels in pressure
overload-induced congestive heart failure revealed in KCNJ11
Kir6.2-null mutant. J Physiol. 577:1053–1065. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zingman LV, Alekseev AE, Hodgson-Zingman
DM and Terzic A: ATP-sensitive potassium channels: Metabolic
sensing and cardioprotection. J Appl Physiol. 103:1888–1893. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sierra A, Zhu Z, Sapay N, Sharotri V,
Kline CF, Luczak ED, Subbotina E, Sivaprasadarao A, Snyder PM,
Mohler PJ, et al: Regulation of cardiac ATP-sensitive potassium
channel surface expression by calcium/calmodulin-dependent protein
kinase II. J Biol Chem. 288:1568–1581. 2013. View Article : Google Scholar :
|
|
30
|
Nichols CG and Lederer WJ: Adenosine
triphosphate-sensitive potassium channels in the cardiovascular
system. Am J Physiol. 261:H1675–H1686. 1991.PubMed/NCBI
|
|
31
|
Noma A: ATP-regulated K+
channels in cardiac muscle. Nature. 305:147–148. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Garlid KD, Paucek P, Yarov-Yarovoy V, Sun
X and Schindler PA: The mitochondrial KATP channel as a receptor
for potassium channel openers. Biol Chem. 271:8796–8799. 1996.
View Article : Google Scholar
|
|
33
|
Liu Y, Sato T, O'Rourke B and Marban E:
Mitochondrial ATP-dependent potassium channels: novel effectors of
cardioprotection? Circulation. 97:2463–2469. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Findlay I: Interactive regulation of the
ATP-sensitive potassium channel of cardiac muscle. J Cardiovasc
Pharmacol. 24:S6–S11. 1994.PubMed/NCBI
|
|
35
|
López JR, Ghanbari RA and Terzic A: A KATP
channel opener protects cardiomyocytes from Ca2 waves: a
laser confocal microscopy study. Am J Physiol. 270:H1384–H1389.
1996.
|
|
36
|
Grover GJ: Pharmacology of ATP-sensitive
potassium channel (KATP) openers in models of myocardial ischemia
and reperfusion. Can J Physiol Pharmacol. 75:309–315. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Holmuhamedov EL, Jovanović S, Dzeja PP,
Jovanović A and Terzic A: Mitochondrial ATP-sensitive K+
channels modulate cardiac mitochondrial function. Am J Physiol.
275:H1567–H1576. 1998.PubMed/NCBI
|
|
38
|
Yellon DM and Downey JM: Preconditioning
the myocardium: from cellular physiology to clinical cardiology.
Physiol Rev. 83:1113–1151. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gross GJ and Peart JN: KATP channels and
myocardial preconditioning: an update. Am J Physiol Heart Circ
Physiol. 285:H921–H930. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Weintraub NL: Impaired hypoxic coronary
vasodilation and ATP-sensitive potassium channel function: a
manifestation of diabetic microangiopathy in humans? Circ Res.
92:127–129. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Miura H, Wachtel RE, Loberiza FR Jr, Saito
T, Miura M, Nicolosi AC and Gutterman DD: Diabetes mellitus impairs
vasodilation to hypoxia in human coronary arterioles: reduced
activity of ATP-sensitive potassium channels. Circ Res. 92:151–158.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kinoshita H, Azma T, Nakahata K, Iranami
H, Kimoto Y, Dojo M, Yuge O and Hatano Y: Inhibitory effect of high
concentration of glucose on relaxations to activation of
ATP-sensitive K+ channels in human omental artery.
Arterioscler Thromb Vasc Biol. 24:2290–2295. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kinoshita H, Azma T, Iranami H, Nakahata
K, Kimoto Y, Dojo M, Yuge O and Hatano Y: Synthetic peroxisome
proliferator-activated receptor-gamma agonists restore impaired
vasorelaxation via ATP-sensitive K+ channels by high
glucose. J Pharmacol Exp Ther. 318:312–318. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Seino S and Miki T: Physiological and
pathophysiological roles of ATP-sensitive K+ channels.
Prog Biophys Mol Biol. 81:133–176. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Flagg TP, Enkvetchakul D, Koster JC and
Nichols CG: Muscle KATP channels: recent insights to
energy sensing and myoprotection. Physiol Rev. 90:799–829. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zingman LV, Zhu Z, Sierra A, Stepniak E,
Burnett CM, Maksymov G, Anderson ME, Coetzee WA and Hodgson-Zingman
DM: Exercise-induced expression of cardiac ATP-sensitive potassium
channels promotes action potential shortening and energy
conservation. J Mol Cell Cardiol. 51:72–81. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhu Z, Burnett CM, Maksymov G, Stepniak E,
Sierra A, Subbotina E, Anderson ME, Coetzee WA, Hodgson-Zingman DM
and Zingman LV: Reduction in number of sarcolemmal KATP channels
slows cardiac action potential duration shortening under hypoxia.
Biochem Biophys Res Commun. 415:637–641. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Pearson ER, Flechtner I, Njølstad PR,
Malecki MT, Flanagan SE, Larkin B, Ashcroft FM, Klimes I, Codner E,
Iotova V, et al: Switching from insulin to oral sulfonylureas in
patients with diabetes due to Kir6.2 mutations. N Engl J Med.
355:467–477. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang Y, Han H, Wang J, Wang H, Yang B and
Wang Z: Impairment of human ether-à-go-go-related gene (HERG)
K+ channel function by hypoglycemia and hyperglycemia.
Similar phenotypes but different mechanisms. J Biol Chem.
278:10417–10426. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Smogorzewski M, Galfayan V and Massry SG:
High glucose concentration causes a rise in [Ca2+]i of
cardiac myocytes. Kidney Int. 53:1237–1243. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gribble FM and Reimann F: Sulphonylurea
action revisited: the post-cloning era. Diabetologia. 46:875–891.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Trube G, Rorsman P and Ohno-Shosaku T:
Opposite effects of tolbutamide and diazoxide on the ATP-dependent
K+ channel in mouse pancreatic beta-cells. Pflugers
Arch. 407:493–499. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ozcan C, Bienengraeber M, Dzeja PP and
Terzic A: Potassium channel openers protect cardiac mitochondria by
attenuating oxidant stress at reoxygenation. Am J Physiol Heart
Circ Physiol. 282:H531–H539. 2002. View Article : Google Scholar : PubMed/NCBI
|