1. Introduction: Overview of cell death and
its intracellular signaling pathways
Cell death is a fundamental biological process that
is required for cellular development. On the basis of its
morphological features, cell death can be grouped into three main
classes, namely, apoptosis, autophagy and necrosis (1). The deregulation of cell death is
associated with the etiology, pathogenesis and treatment of many
diseases (2–4), particularly degenerative diseases
such as cancer, Alzheimer's disease, heart disease and Parkinson's
disease (2,5). Over the past few years, increasing
evidence has indicated that cell death contributes to degenerative
disc disease (6), spinal
degenerative disease, and intervertebral disc (IVD) degeneration
(7,8). These findings have led to an
improved understanding of the etiology of these diseases as well as
providing molecular strategies for therapy. Degenerative changes in
IVDs due to aging are clinically important as these changes are
associated with back pain. Current understanding of the molecular
basis of IVD degeneration is principally focused on the regulation
of apoptotic and autophagic pathways.
Apoptosis and its signaling pathways
Apoptosis is a process of programmed cell death that
eliminates damaged or non-essential cells without causing local
inflammation from cell leakage (9). Apoptotic cells exhibit apparent
morphological changes, including cell shrinkage and plasma membrane
bubbling as well as nuclear condensation and fragmentation
(10). Triggering apoptosis
requires a group of cysteine proteases known as caspases, which may
be activated through intrinsic and extrinsic signaling pathways
(11).
The intrinsic pathway, also known as the
mitochondrial pathway, is initiated in the mitochondria (11). As shown in Fig. 1, apoptotic signals, such as DNA
damage and cytokine deprivation, activate p53, which further
initiates the intrinsic pathway by upregulating the p53 upregulated
modulator of apoptosis (Puma) and Noxa [also known as
phorbol-12-my-ristate-13-acetate-induced protein 1 (PMAIP1)]
(12). These two proteins in turn
activate pro-apoptotic proteins, such as Bax and Bak, which
eventually results in the efflux of cytochrome c (13). Cytochrome c further
interacts with the cytosolic protein apoptotic protease activating
factor-1 (Apaf-1) to recruit caspase-9 to form a complex known as
the apoptosome, thereby initiating the activation of the caspase
cascade (Fig. 1) (11). Caspase activation leads to nuclear
lamin cleavage and nuclear breakdown through caspase-3, -6 and -7
(11,13). The intrinsic pathway is regulated
by various proteins, including nuclear factor
κ-light-chain-enhancer of activated B cells (NF-κB), and B-cell
lymphoma-2 (Bcl-2) protein families. The latter is a large protein
family that contains pro-apoptotic members [Bax, Bak, Bad, Bcl-xS,
BH3 interacting domain death agonist (Bid), Bik and Bim] and
anti-apoptotic members (Hrk, Bcl-2, Bcl-xL, Bcl-W, Bfl-1 and Mcl-1)
(13–16). The anti-apoptotic Bcl-2 members
repress apoptosis by blocking the release of cytochrome c
whereas the pro-apoptotic members promote apoptosis (15). For example, the cytosolic
pro-apoptotic protein Bid is cleaved to form a truncated tBid,
which further translocates to mitochondria and oligomerizes Bak to
release cytochrome c (12). In addition, the mitochondrial
protein, second mitochondria-derived activator of caspase
(Smac/DIABLO) augments apoptosis by binding to cellular inhibitor
of apoptosis proteins (cIAPs) and reversing their grip on several
caspases including caspase-3, -6 and -7 (12).
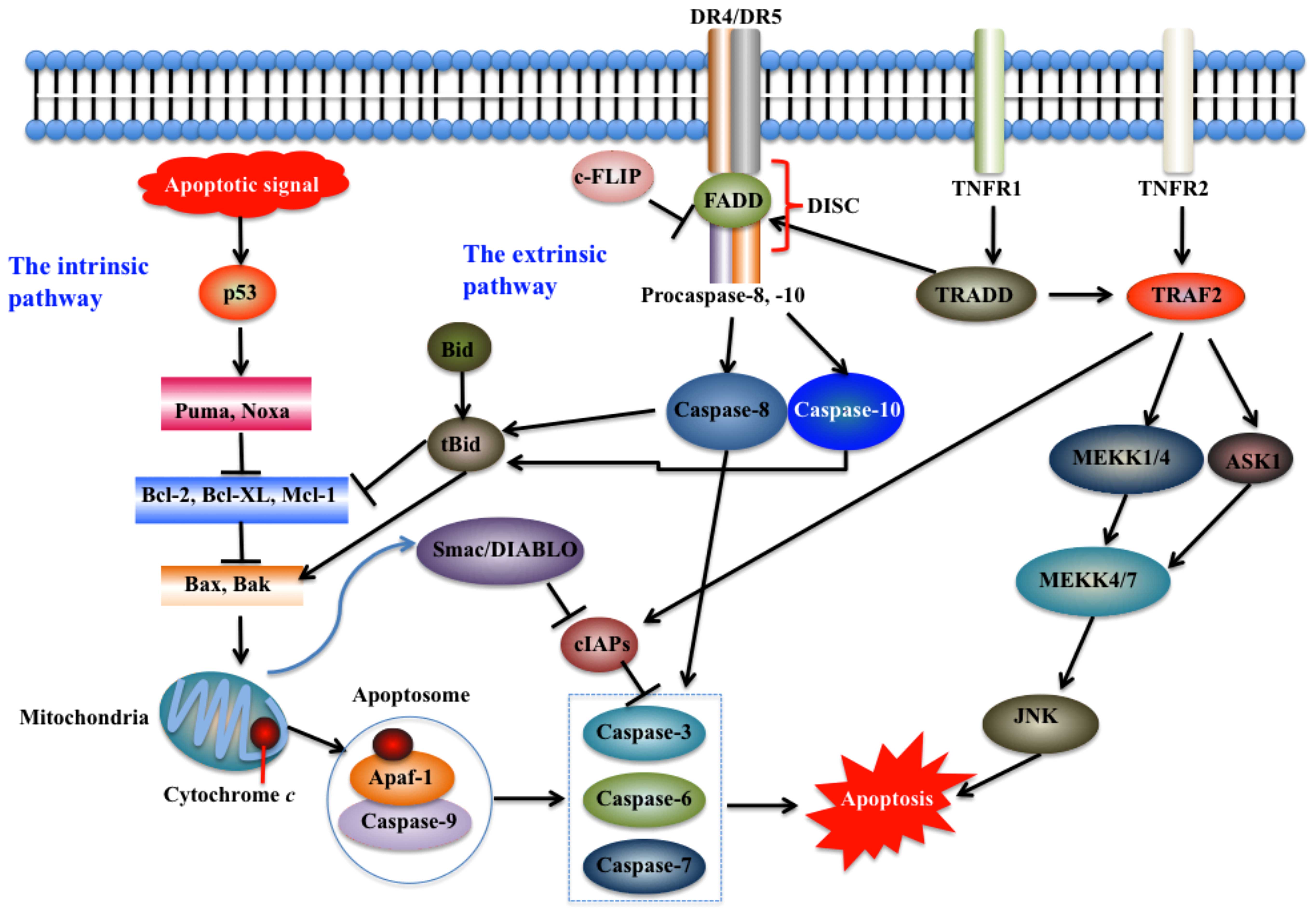 | Figure 1Intrinsic and extrinsic pathways of
apoptosis. Apoptosis pathways may be initiated in the mitochondria
(intrinsic pathway) and on the plasma membrane by death receptor
ligation (extrinsic pathway). The intrinsic pathway is initiated
intracellularly, and pro-apoptotic proteins are released from the
mitochondria to activate caspase proteases and trigger apoptosis
(12,95). The extrinsic pathway mainly
comprises two branches, namely, tumor necrosis factor (TNF)-induced
apoptosis and Fas-Fas ligand-mediated apoptosis (17). The stimulation of death receptors
(DRs) leads to receptor aggregation and recruitment of the adaptor
molecule Fas-associated death domain (FADD) and procaspase-8, which
subsequently becomes activated and initiates apoptosis by direct
cleavage of downstream effector caspases (12). JNK, c-Jun N-terminal kinase; MEKK,
mitogen-activated protein kinase kinase; Puma, p53 upregulated
modulator of apoptosis; TRADD, TNF receptor-associated death domain
protein; TRAF2, TNF receptor-associated factor 2; TNFR1, tumor
necrosis factor receptor 1; Apaf-1, apoptotic protease activating
factor-1; Bcl-2, B-cell lymphoma-2; Bid, BH3 interacting domain
death agonist; Smac/DIABLO, second mitochondria-derived activator
of caspase; cIAPs, cellular inhibitor of apoptosis proteins; DISC,
death-inducing signaling complex. |
The extrinsic pathway, also known as the cytoplasmic
pathway, is initiated by activating pro-apoptotic receptors such as
tumor necrosis factor receptor 1 (TNFR1), death receptors (DRs) and
Fas on the cell surface (17).
This pathway consists of several other proteins, including
membrane-bound Fas ligand (FasL), Fas complexes, Fas-associated
death domain (FADD), caspase-8 and -10; these proteins ultimately
activate downstream caspases and trigger apoptosis (Fig. 1) (15,17). Previous studies have demonstrated
that ligand binding induces receptor clustering and recruitment of
the adaptor protein FADD and the initiator caspases-8 and -10 as
procaspases, forming a death-inducing signaling complex (DISC)
(12). This event triggers the
activation of the apical caspases including caspase-8 and -10,
driving their autocatalytic processing and release into the
cytoplasm, where they activate the effector caspases -3, -6 and -7
(18,19) (Fig.
1). Several pathways and proteins, such as NF-κB,
Fas-associated phosphatase-1 (FAP-1), Fas-associated death
domain-like interleukin (IL)-1-converting enzyme-like inhibitory
protein (FLIP), and decoy receptors (DcR)1 (also known as TRAIL
R-3), DcR2 (also known as TRAIL R-4), and DcR3 (20,21), regulate the activation of the
extrinsic pathway.
Although the extrinsic and intrinsic pathways may
function separately, crosstalk between these two pathways has been
extensively reported. For example, the activation of the extrinsic
pathway promotes caspase-8-mediated processing of tBid, which
subsequently stimulates Bax and Bak to engage the intrinsic pathway
(Fig. 1) (12). Another well-studied crosstalk
mechanism between these two pathways regards the stimulation of the
intrinsic pathway by the tumor suppressor p53, which also
upregulates some of the pro-apoptotic receptors such as DR5 and
augments extrinsic signaling (12). In addition, a waxy lipid molecule
known as ceramide may directly interfere with the mitochondrion and
trigger the activation of the mitochondrial permeability transition
(MPT) pore, which further leads to the permeabilization of the
mitochondrial outer membrane, the release of mitochondrial
intermembrane pro-apoptotic messengers and the induction of
apoptotic cascades (22).
Autophagy and signaling pathways
Autophagy involves the degradation of unnecessary or
dysfunctional cellular components within lysosomes, and three
different forms have been described, namely, macroautophagy,
microautophagy and chaperone-mediated autophagy (23,24). Autophagy consists of several
critical steps: i) the initiation of autophagy signaling through
the unc-51 like autophagy activating kinase 1 complex (24); ii) the regulation of phagophore
formation by beclin 1/VPS34 in membranes in response to stress
signaling pathways (25); iii)
autophagy-related gene (Atg)5-Atg12 conjugation, interaction with
Atg16L, and multimerization at the phagophore (24,25); iv) microtubule-associated protein
1A/1B light chain 3 (LC3) processing and insertion into the
extending phagophore membrane (26); v) the degradation of targets and
completion of the autophagosome; and vi) the fusion of the
autophagosome with lysosomes and proteolytic degradation by
lysosomal proteases (24,27).
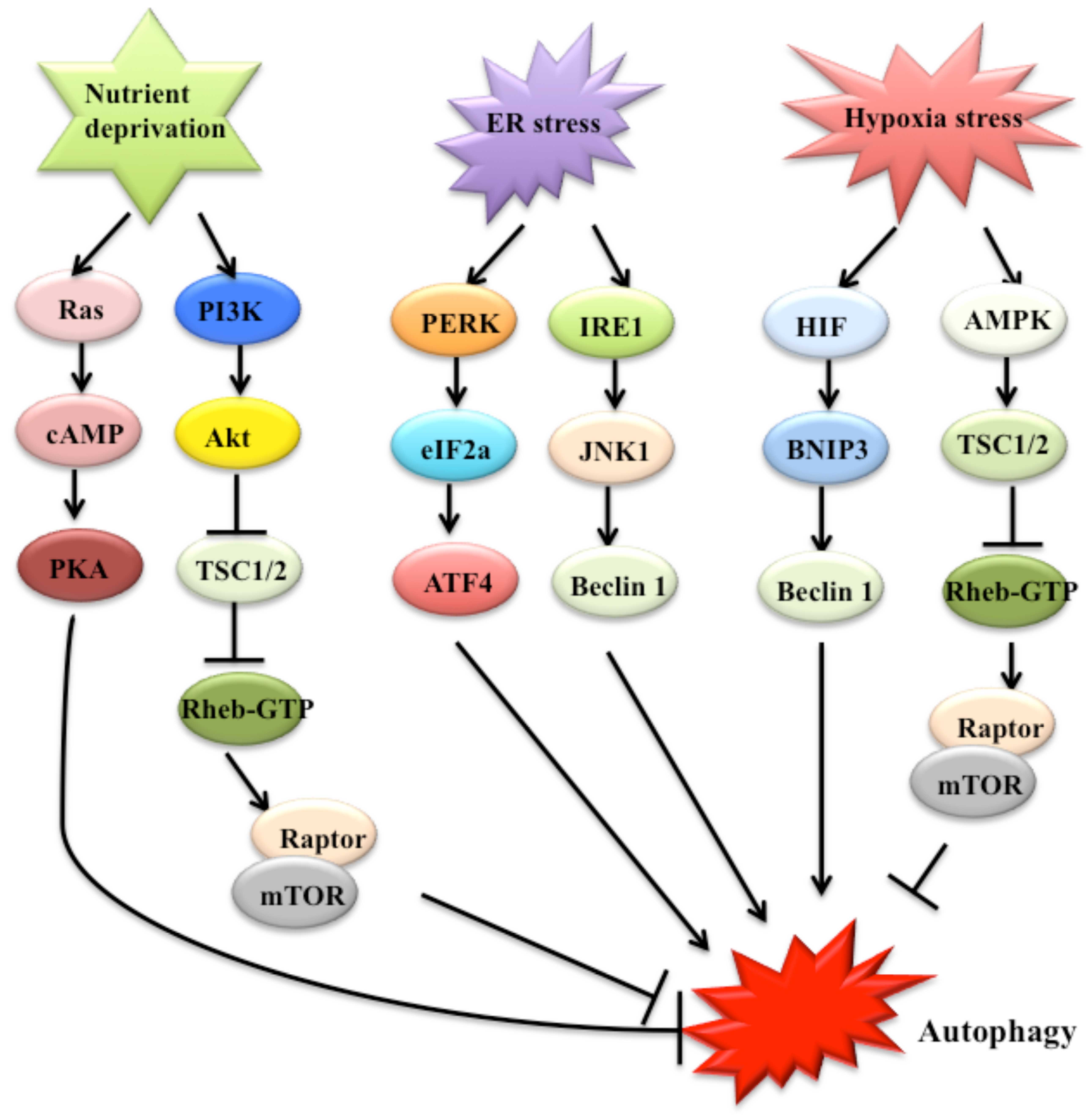
The regulation of autophagy is complicated and may
involve multiple pathways, such as nutrient deprivation, and
various stresses (23). Nutrient
deprivation may significantly induce autophagosome formation
(28). Two well-characterized
signaling cascades, including the target of rapamycin (TOR) and
Ras-cAMP-dependent protein kinase A (PKA) pathways, sense nutrient
status (Fig. 2) (23). TOR regulates nutrient sensing,
cell growth, and autophagy (24).
TOR activates downstream proteins, including Akt kinase (also known
as protein kinase B), phosphoinositide-3 kinase (PI3K) and growth
factor receptor (29).
Collectively, the Ras/cAMP-dependent PKA signaling pathway plays an
important role in glucose sensing in yeast cells and mammals. Under
nutrient-rich conditions, two Ras homologs, namely, Ras1 and Ras2,
are active and enhance cAMP generation through adenylyl cyclase in
yeast (30). Elevated cAMP binds
to bypass of cyclic-AMP requirement 1 (Bcy1) and inhibits PKA
(23). The constitutive
activation of the Ras/PKA pathway may suppress autophagy that is
induced by TOR inhibition (23).
Various extra- and intracellular stresses, such as endoplasmic
reticulum (ER) stress, hypoxia and oxidative stress, potentially
induce autophagy (23). ER stress
stimulates autophagy through the double stranded RNA-activated
protein kinase-like ER kinase-eukaryotic initiation factor-2α
(PERK-eIF2α) pathway, the inositol requiring enzyme 1 (IRE1)/c-Jun
N-terminal protein kinase (JNK)1 pathway, and Ca2+
release (23,31) (Fig.
2). The activation of eIF2α by PERK may enhance the
transcription of autophagic genes, such as ATG12 (32). Hypoxia activates autophagy through
effects that are dependent on both target genes induced by
hypoxia-inducible factor (HIF) and through HIF-independent effects
that are mediated by downstream TOR inhibition of AMP-activated
protein kinase (AMPK), regulated in development and DNA damage
responses 1 (REDD1) and tuberous sclerosis proteins 1 and 2
(TSC1/TSC2) (Fig. 2) (24,28,33). Specific targets of HIF in
autophagy include BCL2/adenovirus E1B 19 kDa protein-interacting
protein 3 (BNIP3) and BNIP3-like protein (BNIP3L), which are
noncanonical members of the Bcl-2 superfamily (Fig. 2) (25,28).
2. Degeneration of IVDs and cell death
Structure of IVDs
The IVD is a flexible joint that is localized
between adjacent spinal vertebrae (7,8).
IVDs consist of the outer endplates, the inner annulus fibrosus
(AF), and the central nucleus pulposus (NP) (34). It has been suggested that the
endplates absorb the small molecules and nutrients required for the
disc cells (35). The AF is the
tough, circular exterior of the IVD that surrounds the soft inner
NP, and the AF may prevent the NP from herniating or leaking out of
the disc by hydraulically sealing the nucleus and by evenly
distributing any pressure and force imposed on the IVD (36).
IVD degeneration and signaling pathway
regulation
As the human body ages, IVDs gradually degenerate,
which leads to degenerative disc disease in some individuals.
Various changes in the cellular phenotype and biochemical factors
occur during IVD degeneration. These changes mainly include
inflammation, matrix degradation, loss of proteoglycan in the NP,
disorganization of the concentric lamellae in the AF, spinal
instability, disc height loss and prolapse (37,38). Changes to the immune balance of
the microenvironment of the disc may cause immune cell infiltration
and attack of the NP cells (39,40).
Various intracellular signaling pathways involved in
the adaptation of IVD cells to the IVD-specific niche, pain
mediators and IVD degeneration have been extensively studied
(40). NF-κB and
mitogen-activated protein kinase (MAPK) pathways regulate
proinflammatory mediators such as TNF-α, IL-1β and IL-6 (41). The suppression of the NF-κB and
MAPK pathways controls anti-inflammatory and anticatabolic
conditions during the treatment of IVD herniation and its
associated pain (41). MAPK
activity also participates in osmoregulation, matrix production,
integrin expression and NP cell survival under HIF-1 regulation
(42). These findings suggest
that β-catenin is a fundamental factor required to maintain IVD
structure and function (43). The
Wnt pathway is also suggested to mediate the development and
progression of disc diseases (43). LiCl, an activator of the Wnt
pathway, accelerated cellular senescence in NP cells (44,45). However, β-catenin mRNA and protein
levels were decreased in NP cells following stimulation with the
PKC activator phorbol 12-myristate 13-acetate (PMA) (46). The Notch pathway is also involved
in IVD degeneration mediated by proinflammatory cytokines (47). Notch signaling is activated by
hypoxia in IVD cells, and the Notch-signaling inhibitor L685458
blocks the activity of Notch-responsive luciferase reporters and
reduces the proliferation of AF cells (48). NP cells treated with TNF-α or
IL-1β have increased levels of Notch receptors including Notch-1
and -2, the Notch ligand JAGGED2, and target genes such as HES1,
HEY1 and HEY2 (49).
Cell death and its causes in IVD
degeneration
Apoptosis and autophagy have been commonly observed
in degenerative IVDs in clinical trials, animal models and cell
culture studies. Cell death may be caused by numerous causes, such
as nutrient depletion, biotic and abiotic stress as well as viral
infection (49). The cells
located at the center of the IVD only acquire nutrients through
fluid flow or diffusion through the vertebral endplates and the AF
(50). Consequently, nutrients
and oxygen tension within the disc are significantly reduced as a
result of the long distance from the vasculature to the center of
the NP (50). Therefore, the
metabolism in disc cells is partly anaerobic, which leads to high
lactic acid concentrations and low pH conditions (50). With disc degeneration, the
increased loss of NP proteoglycans reduces the hydrodynamic
transfer of axial stress to the outer AF (50). Concurrently, the integrity of the
AF is affected by radial fissures. Endplates undergo ossification,
which further reduces the nutritional supply to the disc (51). Consequently, changes to the
microenvironment, nutrient depletion and stress result in cell
death during IVD degeneration.
3. Apoptosis and IVD degeneration
Apoptosis has been demonstrated to participate in
IVD degeneration for many years. Apoptosis was initially identified
using the terminal deoxynucleotidyl transferase-mediated dUTP
nick-end labeling (TUNEL) assay; the IVDs from patients were found
to have considerably more TUNEL positive cells compared with the
healthy control discs (52).
Thereafter, numerous studies determined that NP and AF cells
undergo apoptosis in degenerative discs through complicated
mechanisms.
Apoptosis in NP cells
The excessive apoptosis of NP cells, which produce
cartilage-specific extracellular matrix (ECM) components, is an
evident cellular and biochemical change which occurs during IVD
degeneration (53,54). The dynamic balance between ECM
synthesis and degradation is disrupted during IVD degeneration,
which results in a gradual loss of disc ECM, structural failure and
biomechanical changes (54). Both
intrinsic and extrinsic pathways of apoptosis play critical roles
in NP cell degeneration.
Intrinsic pathway and the degeneration of
NP cells
Emerging evidence suggests that the intrinsic
pathway of apoptosis participates in NP cell degeneration mainly by
regulating the levels of Bcl-2, caspase-3, collagen and aggrecan.
Bcl-2 inhibits the intrinsic pathway of apoptosis in various cell
systems, including factor-dependent lymphohematopoietic and neural
cells (55). Bcl-2 regulates
apoptosis by controlling mitochondrial membrane permeability and
inhibiting caspase activity either by preventing the release of
cytochrome c from the mitochondria and/or by binding to
Apaf-1 (55). Notably, Bcl-2
significantly prevents apoptosis during IVD degeneration mainly by
inhibiting caspase-3 activity (Fig.
3). It has been demonstrated that NP cells overexpressing
Bcl-2 under conditions of serum starvation exhibit reduced
apoptosis, decreased mRNA levels of caspase-3 and increased mRNA
levels of type II collagen and aggrecan (56). Bcl-2 has been found to bind to
nucleotide-binding domain and leucine-rich repeat containing
protein 1 (NLRP1) and suppress its activation, thereby inhibiting
the release of IL-1β, a pro-inflammatory cytokine that is processed
to its active form by caspase-1 (57).
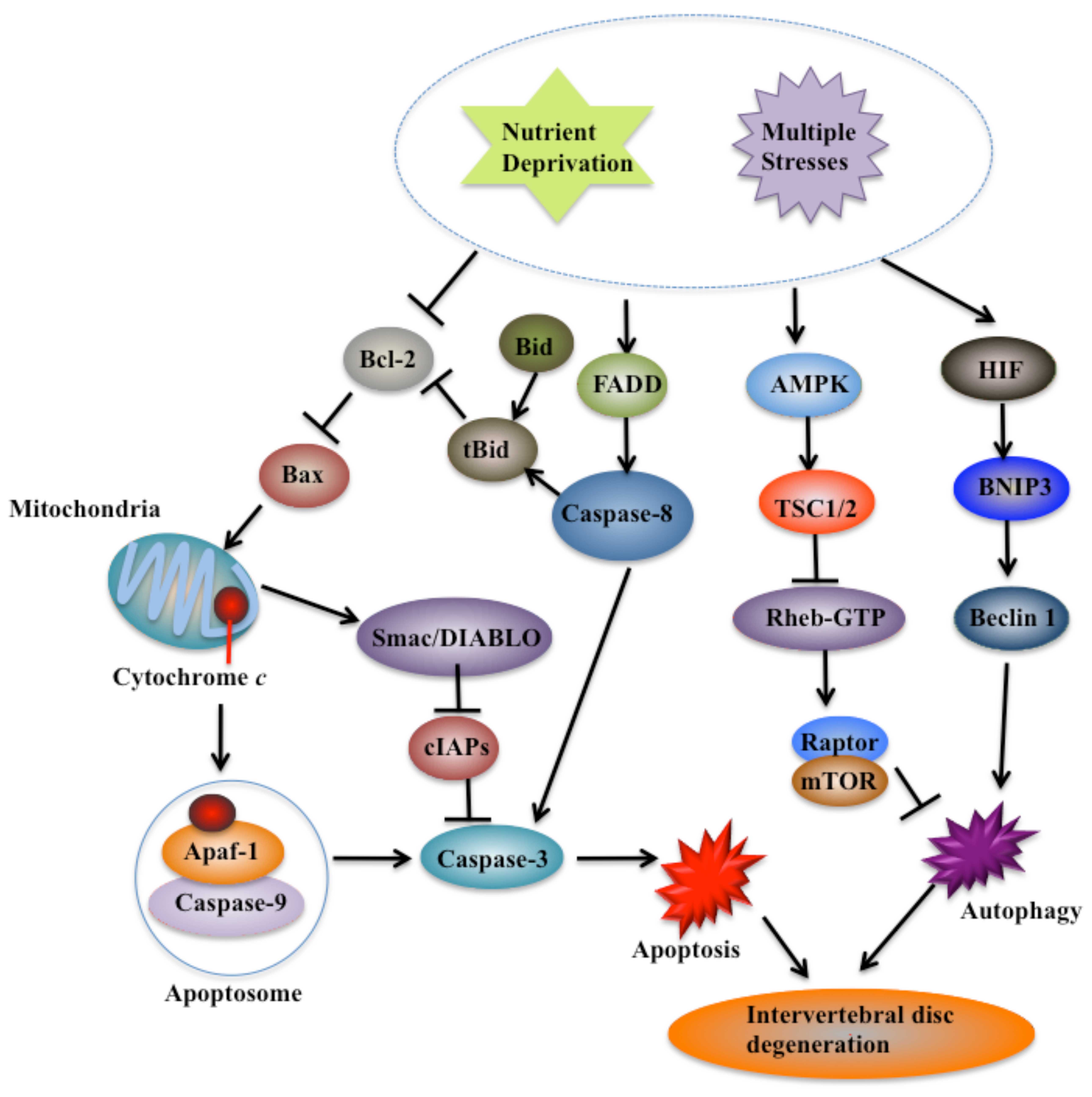 | Figure 3Principal cell death pathways
involved in intervertebral disc (IVD) degeneration. With aging,
IVDs suffer nutrient deprivation and multiple stresses, and undergo
cell death, which eventually results in degeneration. Cells undergo
apoptosis through both intrinsic and extrinsic pathways. The
leakage of cytochrome c from the mitochondria, the
activation of crosstalk between caspase-8 and BH3 interacting
domain death agonist (Bid)-tBid, the important downstream molecules
of caspase-9 such as inhibitor of apoptosis protein (IAP), second
mitochondria-derived activator of caspase (Smac/DIABLO) and the
upstream regulators of mitochondrial maintenance such as Bax and
B-cell lymphoma-2 (Bcl-2), play critical roles in IVD degeneration
(65,66). Two main autophagic pathways,
including signaling via AMP-activated protein kinase (AMPK) to
inhibit mTOR activity and the HIF-BINP3-beclin-1 pathway, are also
induced during IVD degeneration. BINP3, BCL2/adenovirus E1B 19 kDa
protein-interacting protein 3; TSC1/2, tuberous sclerosis proteins
1 and 2; HIF, hypoxia-inducible factor. |
Numerous studies have shown that oxidative stress
leads to the apoptosis of NP cells during IVD degeneration. NP
cells treated with IL-1β exhibited elevated production of nitric
oxide (NO) and decreased levels of proteoglycan, which triggered
apoptosis (54); apoptosis is
increased in NP cells treated with H2O2 and
the mRNA levels of aggrecan and type II collagen are decreased
(58). Notably, the deleterious
effects of either H2O2 or IL-1β may be
efficiently prevented by glutathione (58), a powerful antioxidant that
protects NP cells from apoptosis. Pyrroloquinoline quinone (PQQ), a
redox cofactor for bacterial dehydrogenases, potentially scavenges
reactive oxygen species (ROS) and inhibits apoptosis (59). PQQ protects rat NP cells against
H2O2-induced apoptosis by inhibiting the
intrinsic pathway (59). In the
presence of PQQ, ECM production is maintained despite being in an
apoptotic environment (59). In
addition, the pre-treatment of cells with PQQ increases
Bcl-2 expression, inhibits cytochrome c release, and
decreases Bax expression and caspase-3 cleavage (59). These results indicate that
glutathione and PQQ are possible therapeutic options for the
management of disc degeneration.
Sirtuin-1 (SIRT1), an NAD(+)-dependent
deacetylase, has been suggested to reduce apoptosis in NP cells by
enhancing the expression of many cartilage-specific ECM genes, such
as type II collagen (COL2A1) and aggrecan (54,60). Recent studies have also shown that
SIRT1 protects human NP cells from apoptosis by activating the Akt
anti-apoptotic signaling pathway (54). In addition, degenerative NP cells
obtained from patients have decreased numbers of autophagosomes and
low LC3 and beclin 1 levels (6).
These findings suggest that autophagy plays an important role in
IVD degeneration and that SIRT1 protects the degenerative NP cells
in humans against apoptosis by promoting autophagy.
Extrinsic pathway and the degeneration of
NP cells
NP cells undergo apoptosis through the extrinsic
pathway during IVD degeneration by regulating the levels of Fas and
FasL, thereby affecting caspase activities. The expression levels
of FasL and Fas were elevated in a co-culture system
of human NP cells and human microvascular endothelial (HMEC-1)
cells (61). FasL expression in
human NP cells prevents angiogenesis in the IVD by inducing
Fas-mediated apoptosis with the activation of downstream FADD and
caspase-3 (61). The NP is
derived from the notochord, a rod-like structure of mesodermal
origin (62). Notochordal cells
protect NP cells from matrix protein degradation and apoptosis
induced by IL-1β and FasL, and this apoptotic process is inhibited
by notochordal cell-conditioned medium by suppressing activated
caspase-9 and -3 (63). Bid,
cytochrome c and activated caspases-9 and -3 were robustly
detected in herniated NP tissues (63). Apoptotic signaling downstream of
activated caspase-9 involves complex interactions between mediators
of Smac/DIABLO and X-linked inhibitor of apoptosis protein (XIAP)
that control activated caspase-3 signaling (Fig. 3) (64). These findings suggest that
Smac/DIABLO and XIAP play a role in the degeneration of NP cells.
However, this aspect remains to be clarified. The strong expression
of Fas and FasL and the TUNEL-positive staining of a
few NP cells in human herniated lumbar IVD tissues indicated the
involvement of the DR pathway in IVD degeneration (64). Similar results were also observed
in the IVD tissues obtained from patients with scoliosis (65). Recently, a member of a disintegrin
and metalloproteinase with thrombospondin motifs (ADAMTS) family,
ADAMTS-7, was found to have markedly elevated levels in both human
and rat degenerative NP tissues compared with those in normal
controls (66). The findings of
this study suggest that IL-17A may induce ADAMTS-7
expression through TNF-α, which may form a molecular axis in human
NP cells (66).
Apoptosis is regulated by miRNAs, which are key
post-transcriptional regulators that target the 3′-untranslated
regions of the genes that they repress. Aberrant expression
profiles of miRNAs are considered as one of the etiologies of IVD
degeneration (67). A study
examining the miRNA expression profiles revealed that 29 miRNAs are
differentially expressed and that miR-155 is significantly
downregulated in degenerative NP cells (58). Additional evidence indicated that
miR-155 promotes Fas-mediated apoptosis by targeting FADD and
caspase-3 (67). Some studies
examined the expression of several other miRNAs, such as miR-10b in
degenerative NP cells; however, the upstream regulation of miRNAs
and their interactions with cytokines remain elusive (67). In addition, miR-27a was recently
found to regulate apoptosis in NP cells by targeting PI3K (69). The elevated expression of
miR-21 was found in human degenerative NP tissues, and
functional analysis revealed that the overexpression of
miR-21 increases Akt phosphorylation by targeting
phosphatase and tensin homolog (PTEN) (70).
MAPK and the degeneration of NP
cells
The MAPK family members are crucial for the
maintenance of cell development. Three subfamilies of MAPKs have
been identified: extracellular signal-regulated kinases (ERKs),
JNKs and p38-MAPKs (71). ERKs
are important for cell survival, and JNKs and p38-MAPKs are
involved in both the intrinsic and extrinsic pathways of apoptosis
(71). The p38-MAPK, JNK1/2 and
ERK1/2 signaling pathways exist in NP cells and are required for
cell growth, differentiation, and apoptosis (71). A highly osmotic microenvironment
may be established during IVD degeneration. Mimicking
high-osmolality conditions in vitro activated the p38-MAPK,
JNK1/2 and ERK1/2 signaling pathways in rabbit NP cells (72). Furthermore, activated p38-MAPKs
and JNK1/2 may induce cell apoptosis whereas activated ERK1/2
promotes cell survival (72).
Recently, β1 integrin was found to inhibit apoptosis induced by
cyclic stretch in AF cells through the ERK1/2 MAPK pathway, and
this process correlates with the activation of caspase-3 (73).
Apoptosis in AF cells
AF cells play an important role in providing the
structural properties of the disc, and the apoptosis of AF cells
contributes to IVD degeneration through both the intrinsic and
extrinsic pathways.
Intrinsic pathway and the degeneration of
AF cells
Studies of TUNEL-positive staining in rat AF tissues
as well as rabbit models of overload-induced IVD degeneration
showed that many cells release anti-cytochrome c; however,
no anti-FasL-positive cells were identified in these tissues
(74,75). These results imply that AF cells
undergo apoptosis through the intrinsic pathway under mechanical
conditions of overload. In addition, cell proliferation was
inhibited after subjecting rabbit AF cells to pressure for 24 or 36
h; this result was associated with increased apoptosis and
caspase-9 activity (75). The
activity of caspase-9 is suggested to be proportional to the
apoptotic index of rabbit AF cells cultured in silicon elastic
membranes; however, no detectable change in caspase-8 activity was
observed in these cells (76).
Notably, only caspase-9 inhibitor was capable of suppressing the
apoptosis of AF cells induced by cyclic stretch (76). Moreover, it was demonstrated that
cyclic stretch-induced apoptosis is partially mediated by ER stress
through NO production (77). The
cyclic stretch of AF cells caused NO overproduction, the
upregulation of ER stress markers (CHOP, GRP78 and caspase-12),
mitochondrial depolarization and caspase-9 activation (77). The specific inhibitors of
caspase-12 (Z-ATAD-FMK) and caspase-9 (Z-LEHD-FMK) partially
suppressed apoptosis (77).
Electroacupuncture (EA) inhibited the apoptosis of
AF cells by suppressing the intrinsic pathway in a rat model of IVD
degeneration (78). Treatment
with EA reduced the number of TUNEL-positive stained cells whereas
it increased the number of Bcl-2-positive cells as revealed by
immunohistochemical staining (78). Moreover, EA treatment
significantly inhibits the activation of caspase-9 and -3, and
enhances the mRNA and protein levels of Crk and ERK2 (78).
Extrinsic pathway and the degeneration of
AF cells
An in vitro study examined the involvement of
the extrinsic pathway in IVD degeneration and demonstrated that
rabbit AF cells undergo increased apoptosis under conditions of
serum deprivation (76). This
process is associated with the increased activity of caspase-3 and
-8; however, there was no substantial increase in cytochrome
c protein levels in the cytosolic fraction (76). The inductive effect of serum
deprivation on apoptosis may be reduced by caspase-8 inhibitor but
not by caspase-9 inhibitor (76).
Apoptosis in human IVD cells subjected to acute trauma force may be
simultaneously and interdependently mediated by extrinsic and
intrinsic pathways (79).
Apoptosis in endplate cells
Apoptosis evidently occurs in the cartilaginous
endplate cells during IVD degeneration, which results in a marked
decrease in cell density (80).
The number of TUNEL-positive cells in the cartilaginous endplate
increases with age and with the destruction of the cartilaginous
endplate following apoptosis (81).
Mechanical stress induced the apoptosis of endplate
chondrocytes in organ-cultured mouse IVDs (82). Apoptosis occurred after subjecting
the cells to a static mechanical load (82). MAPK inhibitors increase the
occurrence of apoptosis, suggesting that MAPKs counteract
mechanical stress-induced apoptosis (82). In rat endplate chondrocytes, the
increased phosphorylation of JNK, ERK1/2 and p38-MAPK; increased
cytochrome c release; and activated caspase-9 and -3
indicate the occurrence of static mechanical stress-induced
apoptosis through the MAPK and intrinsic signaling pathways
(82). Treatment with inhibitors
of JNK (SP600125), p38-MAPK (SB203580) and ERK (PD98059) prior to
mechanical stimulation reversed both the static load-induced
apoptosis of chondrocytes and the activation of JNK, p38 MAPK and
ERK (82). Collectively, these
findings demonstrate that mechanical stress induces apoptosis in
rat cervical endplate chondrocytes through the MAPK-mediated
mitochondrial apoptotic pathway.
Low levels of fetal bovine serum may induce the
apoptosis of rat endplate cells, and serum deprivation leads to the
elevated expression of caspase-9, -3, poly(ADP-ribose) polymerase,
cytochrome c and Bax (83). The caspase-9 inhibitor Z-LEHD-FMK
significantly suppressed serum deprivation-induced apoptosis
(80). In addition, the
activation of acid-sensing ion channel 1a (ASIC1a) in endplate
chondrocytes may trigger Ca2+-dependent protease
activity and signaling, leading to the apoptosis of endplate
chondrocytes in IVDs (84).
4. Autophagy and IVD degeneration
Autophagy consists of multiple processes that are
highly regulated by Atg proteins and LC3 (23). During autophagy, the cytosolic
microtubule-associated protein LC3-I is converted to LC3-II through
lipidation, and LC3-II is translocated to the autophagosomal
membrane (85). Thus, the
conversion of LC3-I to LC3-II and the accumulation of LC3 are
widely used as autophagy markers (23). Beclin 1 is a BH3 member of the
Bcl-2 gene family that drives autophagy in mammalian cells
(86). Various studies have
demonstrated that autophagy occurs in both NP and AF cells. For
example, rat NP and AF cells cultured in high glucose
concentrations demonstrated the increased expression of beclin 1,
LC3 and Atg3, 5, 7 and 12 (85).
Autophagy in NP cells
Rat NP cells exposed to compression undergo
ROS-mediated autophagy, which leads to cell degeneration (87). Compression increases the levels of
beclin 1 and the processing of LC3B-I to LC3B-II, which is a major
step in autophagosome formation (87). The autophagy inhibitor
3-methyladenine (3-MA) attenuates the formation of LC3B and beclin
1 (87). Moreover, glucosamine,
an amino sugar and a precursor in the synthesis of glycosylated
proteins and lipids, is capable of protecting NP cells and inducing
autophagy through the mTOR-dependent pathway (88). Glucosamine activates autophagy in
a dose-dependent manner within 24 h and inhibits the
phosphorylation of mTOR and p70S6K (88). Autophagy in IL-1β- or
H2O2-treated cells is increased by
glucosamine (88). Glucosamine
attenuates the reduction in aggrecan levels and prevents the
apoptosis of NP cells induced by IL-1β, whereas 3-MA partially
reverses these effects (88).
H2O2 increases the lysosomal membrane
permeability in NP cells and subsequently induces apoptosis through
the mitochondrial pathway (88).
Moreover, H2O2 stimulates an early autophagic
response through the ERK/mTOR signaling pathway (88). The inhibition of autophagy
significantly decreases the rate of apoptosis in the cells
disrupted with H2O2 (89). These results suggest that
controlling the autophagy response in NP cells under oxidative
stress enhances cell survival and probably delays disc
degeneration.
Hypoxia facilitates NP cell survival under
conditions of serum deprivation by downregulating excessive
autophagy through restricting the generation of ROS (90). Appropriate autophagic activity
enhances the survival of NP cells under conditions of serum
deprivation, whereas excessive autophagy triggers the apoptosis of
NP cells (90). Hypoxia
facilitates the survival of NP cells in serum deprivation by
downregulating excessive autophagy (90). Hypoxia downregulates the
autophagic activity of NP cells by restricting the production of
ROS and inactivating the AMPK/mTOR signaling pathway, and possibly
through a pathway involving HIF-1α (90). Nutrient starvation may also induce
NP cell autophagy by increasing the ratios of LC3-II/LC3-I and
beclin-1/β-actin, and by producing autophagosomes (90). Treatment with 3-MA may suppress
autophagosome formation (90).
Autophagy in AF cells
Under conditions of serum deprivation, autophagy was
detected in rat AF cells by transmission electron microscopy
(91). IL-1β may dose-dependently
enhance the autophagy-induction effect of serum deprivation
(91). However, IL-1β alone fails
to induce autophagy in AF cells cultured under conditions of serum
starvation (91). The suppression
of autophagy by 3-MA treatment increases the apoptosis of cells
(91). Serum supplementation also
partially reverses the incidence of autophagy without affecting the
incidence of apoptosis in the same cells. IL-1β dose-dependently
upregulates the serum deprivation-induced autophagy of AF cells
(91). Autophagy may act as a
protective mechanism against apoptosis in AF cells and IVD
degeneration.
Autophagy in endplate cells
The IVD obtains nutrients by diffusion from blood
vessels through the cartilaginous endplate (92). Thus, endplate calcification may
result in disc degeneration by decreasing nutrient diffusion and
failing to maintain cellular activity and homeostasis, which leads
to apoptosis (92). Autophagic
activity may correlate with IVD development and degeneration. For
example, a previous study demonstrated that autophagy protects the
endplate cells from calcification induced by intermittent cyclic
mechanical tension (93), and the
expression of the autophagy related-genes LC3 and beclin
1 significantly decreases as endplate chondrocyte activity
decreases during aging (94).
5. Conclusion and future aspects
Cell death is closely associated with the pathology
of IVD degeneration. Different types of cells undergo cell death
through different signaling pathways in response to various stimuli
(Fig. 3). On the basis of current
studies, different IVD cell types undergo apoptosis largely through
the caspase-9 pathway which is clearly associated with cytochrome
c leakage (Fig. 3). In
addition, the activation of crosstalk between caspase-8 and
Bid-tBid, the important downstream molecules of caspase-9 such as
the inhibitor of apoptosis protein (IAP) and Smac/DIABLO as well as
the upstream regulators of mitochondrial maintenance such as Bax
and Bcl-2, also play critical roles in IVD degeneration (Fig. 3). Moreover, emerging evidence also
suggests that several autophagic pathways, such as the AMPK/mTOR
signaling pathway and the HIF-BINP3-beclin 1 pathway, are also
induced by multiple stresses (Fig.
3).
Over the past few years, significant progress has
been achieved to enhance our understanding of the molecular
mechanisms that involve apoptosis and autophagy in IVD
degeneration. However, the underlying mechanisms remain
incompletely understood. With regard to the apoptotic pathways,
future studies should build on current knowledge in order to
identify all the key factors of the intrinsic and extrinsic
pathways in the different IVD cells, as well as to establish their
crosstalk. With regard to the autophagic pathways, current evidence
provides either an indication of the factors involved or incomplete
signaling pathways. Thus, it is necessary to exert considerable
efforts in order to identify the factors that are specifically
involved in autophagy during IVD degeneration. Importantly, greater
efforts are necessary in order to develop clinical treatments that
potentially retard or prevent IVD degeneration in the future.
Acknowledgments
We apologize to all authors whose contributions
were not cited due to space limitations.
References
|
1
|
Kroemer G, Galluzzi L, Vandenabeele P,
Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS,
Golstein P, Green DR, et al: Nomenclature Committee on Cell Death
2009: Classification of cell death: recommendations of the
Nomenclature Committee on Cell Death 2009. Cell Death Differ.
16:3–11. 2009. View Article : Google Scholar :
|
|
2
|
Gorman AM: Neuronal cell death in
neurodegenerative diseases: recurring themes around protein
handling. J Cell Mol Med. 12:2263–2280. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xu J, Wang D and Ma W: Cell death in human
health and disease. BioMed Res Int. 2014:2430172014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang C and Zhang F: Iron homeostasis and
tumorigenesis: molecular mechanisms and therapeutic opportunities.
Protein Cell. 6:88–100. 2015. View Article : Google Scholar :
|
|
5
|
McIlwain DR, Berger T and Mak TW: Caspase
functions in cell death and disease. Cold Spring Harb Perspect
Biol. 7:72015. View Article : Google Scholar
|
|
6
|
Jiang W, Zhang X, Hao J, Shen J, Fang J,
Dong W, Wang D, Zhang X, Shui W, Luo Y, et al: SIRT1 protects
against apoptosis by promoting autophagy in degenerative human disc
nucleus pulposus cells. Sci Rep. 4:74562014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ding F, Shao ZW and Xiong LM: Cell death
in intervertebral disc degeneration. Apoptosis. 18:777–785. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao CQ, Jiang LS and Dai LY: Programmed
cell death in intervertebral disc degeneration. Apoptosis.
11:2079–2088. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Taylor RC, Cullen SP and Martin SJ:
Apoptosis: controlled demolition at the cellular level. Nat Rev Mol
Cell Biol. 9:231–241. 2008. View Article : Google Scholar
|
|
10
|
Ashkenazi A and Salvesen G: Regulated cell
death: signaling and mechanisms. Annu Rev Cell Dev Biol.
30:337–356. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Elmore S: Apoptosis: a review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ashkenazi A: Directing cancer cells to
self-destruct with pro-apoptotic receptor agonists. Nat Rev Drug
Discov. 7:1001–1012. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tait SW and Green DR: Mitochondria and
cell death: outer membrane permeabilization and beyond. Nat Rev Mol
Cell Biol. 11:621–632. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gogvadze V, Orrenius S and Zhivotovsky B:
Multiple pathways of cytochrome c release from mitochondria in
apoptosis. Biochim Biophys Acta. 1757:639–647. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ghobrial IM, Witzig TE and Adjei AA:
Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin.
55:178–194. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kalimuthu S and Se-Kwon K: Cell survival
and apoptosis signaling as therapeutic target for cancer: marine
bioactive compounds. Int J Mol Sci. 14:2334–2354. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stanzer S, Janesch B, Resel M, Augustin T,
Samonigg H and Bauernhofer T: The role of activation-induced cell
death in the higher onset of spontaneous apoptosis of NK cell
subsets in patients with metastatic epithelial cancer. Cell
Immunol. 261:99–104. 2010. View Article : Google Scholar
|
|
18
|
Zhang C and Zhang F: The multifunctions of
WD40 proteins in genome integrity and cell cycle progression. J
Genomics. 3:40–50. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Graumann K, Hippe D, Gross U and Lüder CG:
Mammalian apoptotic signalling pathways: multiple targets of
protozoan parasites to activate or deactivate host cell death.
Microbes Infect. 11:1079–1087. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Plati J, Bucur O and Khosravi-Far R:
Apoptotic cell signaling in cancer progression and therapy. Integr
Biol Camb. 3:279–296. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Klener P Jr, Andera L, Klener P, Necas E
and Zivný J: Cell death signalling pathways in the pathogenesis and
therapy of haematologic malignancies: overview of apoptotic
pathways. Folia Biol (Praha). 52:34–44. 2006.
|
|
23
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: a
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yamamoto A and Yue Z: Autophagy and its
normal and pathogenic states in the brain. Annu Rev Neurosci.
37:55–78. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Glick D, Barth S and Macleod KF:
Autophagy: cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Matsushita M, Suzuki NN, Obara K, Fujioka
Y, Ohsumi Y and Inagaki F: Structure of Atg5.Atg16, a complex
essential for autophagy. J Biol Chem. 282:6763–6772. 2007.
View Article : Google Scholar
|
|
28
|
Pattingre S, Espert L, Biard-Piechaczyk M
and Codogno P: Regulation of macroautophagy by mTOR and Beclin 1
complexes. Biochimie. 90:313–323. 2008. View Article : Google Scholar
|
|
29
|
Russell RC, Yuan HX and Guan KL: Autophagy
regulation by nutrient signaling. Cell Res. 24:42–57. 2014.
View Article : Google Scholar :
|
|
30
|
Richardson CJ, Schalm SS and Blenis J:
PI3-kinase and TOR: PIKTORing cell growth. Semin Cell Dev Biol.
15:147–159. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
D'Souza CA and Heitman J: Conserved cAMP
signaling cascades regulate fungal development and virulence. FEMS
Microbiol Rev. 25:349–364. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shen S, Zhang Y, Wang Z, Liu R and Gong X:
Bufalin induces the interplay between apoptosis and autophagy in
glioma cells through endoplasmic reticulum stress. Int J Biol Sci.
10:212–224. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang C, Liu G and Huang M: Ribonucleotide
reductase metallocofactor: Assembly, maintenance and inhibition.
Front Biol (Beijing). 9:104–113. 2014. View Article : Google Scholar
|
|
34
|
Le Maitre CL, Binch AL, Thorpe AA and
Hughes SP: Degeneration of the intervertebral disc with new
approaches for treating low back pain. J Neurosurg Sci. 59:47–61.
2015.
|
|
35
|
Bron JL, Helder MN, Meisel HJ, Van Royen
BJ and Smit TH: Repair, regenerative and supportive therapies of
the annulus fibrosus: achievements and challenges. Eur Spine J.
18:301–313. 2009. View Article : Google Scholar
|
|
36
|
Moore RJ: The vertebral endplate: disc
degeneration, disc regeneration. Eur Spine J. 15(Suppl 3):
S333–S337. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sivan SS, Hayes AJ, Wachtel E, Caterson B,
Merkher Y, Maroudas A, Brown S and Roberts S: Biochemical
composition and turnover of the extracellular matrix of the normal
and degenerate intervertebral disc. Eur Spine J. 23(Suppl 3):
S344–S353. 2014. View Article : Google Scholar
|
|
38
|
Iatridis JC, Nicoll SB, Michalek AJ,
Walter BA and Gupta MS: Role of biomechanics in intervertebral disc
degeneration and regenerative therapies: what needs repairing in
the disc and what are promising biomaterials for its repair? Spine
J. 13:243–262. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Urban JP and Roberts S: Degeneration of
the intervertebral disc. Arthritis Res Ther. 5:120–130. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu ZH, Sun Z, Wang HQ, Ge J, Jiang TS,
Chen YF, Ma Y, Wang C, Hu S, Samartzis D and Luo ZJ: FasL
expression on human nucleus pulposus cells contributes to the
immune privilege of intervertebral disc by interacting with
immunocytes. Int J Med Sci. 10:1053–1060. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hiyama A, Sakai D and Mochida J: Cell
signaling pathways related to pain receptors in the degenerated
disk. Global Spine J. 3:165–174. 2013. View Article : Google Scholar :
|
|
42
|
Risbud MV, Schipani E and Shapiro IM:
Hypoxic regulation of nucleus pulposus cell survival: from niche to
notch. Am J Pathol. 176:1577–1583. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kondo N, Yuasa T, Shimono K, Tung W, Okabe
T, Yasuhara R, Pacifici M, Zhang Y, Iwamoto M and Enomoto-Iwamoto
M: Intervertebral disc development is regulated by Wnt/β-catenin
signaling. Spine. 36:E513–E518. 2011. View Article : Google Scholar
|
|
44
|
Hiyama A, Sakai D, Risbud MV, Tanaka M,
Arai F, Abe K and Mochida J: Enhancement of intervertebral disc
cell senescence by WNT/β-catenin signaling-induced matrix
metalloproteinase expression. Arthritis Rheum. 62:3036–3047. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Arai F, Hiyama A, Sakai D, Yokoyama K and
Mochida J: The expression and role of non-canonical (PKC) signaling
in nucleus pulposus cell metabolism. J Orthop Res. 30:1478–1485.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Risbud MV and Shapiro IM: Role of
cytokines in intervertebral disc degeneration: pain and disc
content. Nat Rev Rheumatol. 10:44–56. 2014. View Article : Google Scholar :
|
|
47
|
Hiyama A, Skubutyte R, Markova D, Anderson
DG, Yadla S, Sakai D, Mochida J, Albert TJ, Shapiro IM and Risbud
MV: Hypoxia activates the Notch signaling pathway in cells of the
intervertebral disc: implications in degenerative disc disease.
Arthritis Rheum. 63:1355–1364. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang H, Tian Y, Wang J, Phillips KL, Binch
AL, Dunn S, Cross A, Chiverton N, Zheng Z, Shapiro IM, et al:
Inflammatory cytokines induce NOTCH signaling in nucleus pulposus
cells: implications in intervertebral disc degeneration. J Biol
Chem. 288:16761–16774. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu Y and Levine B: Autosis and autophagic
cell death: the dark side of autophagy. Cell Death Differ.
22:367–376. 2015. View Article : Google Scholar :
|
|
50
|
Sakai D and Grad S: Advancing the cellular
and molecular therapy for intervertebral disc disease. Adv Drug
Deliv Rev. 84:159–171. 2015. View Article : Google Scholar
|
|
51
|
Wang HQ and Samartzis D: Clarifying the
nomenclature of intervertebral disc degeneration and displacement:
from bench to bedside. Int J Clin Exp Pathol. 7:1293–1298.
2014.PubMed/NCBI
|
|
52
|
Gruber HE and Hanley EN Jr: Analysis of
aging and degeneration of the human intervertebral disc. Comparison
of surgical specimens with normal controls. Spine. 23:751–757.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gruber HE and Hanley EN Jr: Biologic
strategies for the therapy of intervertebral disc degeneration.
Expert Opin Biol Ther. 3:1209–1214. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang D, Hu Z, Hao J, He B, Gan Q, Zhong X,
Zhang X, Shen J, Fang J and Jiang W: SIRT1 inhibits apoptosis of
degenerative human disc nucleus pulposus cells through activation
of Akt pathway. Age (Dordr). 35:1741–1753. 2013. View Article : Google Scholar
|
|
55
|
Cory S, Huang DC and Adams JM: The Bcl-2
family: roles in cell survival and oncogenesis. Oncogene.
22:8590–8607. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sudo H and Minami A: Regulation of
apoptosis in nucleus pulposus cells by optimized exogenous Bcl-2
overexpression. J Orthop Res. 28:1608–1613. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Gabelloni ML, Sabbione F, Jancic C, Fuxman
Bass J, Keitelman I, Iula L, Oleastro M, Geffner JR and Trevani AS:
NADPH oxidase derived reactive oxygen species are involved in human
neutrophil IL-1β secretion but not in inflammasome activation. Eur
J Immunol. 43:3324–3335. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yang D, Wang D, Shimer A, Shen FH, Li X
and Yang X: Glutathione protects human nucleus pulposus cells from
cell apoptosis and inhibition of matrix synthesis. Connect Tissue
Res. 55:132–139. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yang L, Rong Z, Zeng M, Cao Y, Gong X, Lin
L, Chen Y, Cao W, Zhu L and Dong W: Pyrroloquinoline quinone
protects nucleus pulposus cells from hydrogen peroxide-induced
apoptosis by inhibiting the mitochondria-mediated pathway. Eur
Spine J. 24:1702–1710. 2015. View Article : Google Scholar
|
|
60
|
Dvir-Ginzberg M, Gagarina V, Lee EJ and
Hall DJ: Regulation of cartilage-specific gene expression in human
chondrocytes by SirT1 and nicotinamide phosphoribosyltransferase. J
Biol Chem. 283:36300–36310. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sun Z, Wan ZY, Guo YS, Wang HQ and Luo ZJ:
FasL on human nucleus pulposus cells prevents angiogenesis in the
disc by inducing Fas-mediated apoptosis of vascular endothelial
cells. Int J Clin Exp Pathol. 6:2376–2385. 2013.PubMed/NCBI
|
|
62
|
Risbud MV and Shapiro IM: Notochordal
cells in the adult intervertebral disc: new perspective on an old
question. Crit Rev Eukaryot Gene Expr. 21:29–41. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Erwin WM, Islam D, Inman RD, Fehlings MG
and Tsui FW: Notochordal cells protect nucleus pulposus cells from
degradation and apoptosis: implications for the mechanisms of
intervertebral disc degeneration. Arthritis Res Ther. 13:R2152011.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Park JB, Kim KW, Han CW and Chang H:
Expression of Fas receptor on disc cells in herniated lumbar disc
tissue. Spine. 26:142–146. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Chen B, Fellenberg J, Wang H, Carstens C
and Richter W: Occurrence and regional distribution of apoptosis in
scoliotic discs. Spine. 30:519–524. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wang SS, Zhang W, Zhang YQ, Zhao Y, Liu Y,
Li JK, Zhang HX, Cheng L and Nie L: IL-17A enhances ADAMTS-7
expression through regulation of TNF-α in human nucleus pulposus
cells. J Mol Histol. 46:475–483. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wang HQ, Yu XD, Liu ZH, Cheng X, Samartzis
D, Jia LT, Wu SX, Huang J, Chen J and Luo ZJ: Deregulated miR-155
promotes Fas-mediated apoptosis in human intervertebral disc
degeneration by targeting FADD and caspase-3. J Pathol.
225:232–242. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhao B, Yu Q, Li H, Guo X and He X:
Characterization of microRNA expression profiles in patients with
intervertebral disc degeneration. Int J Mol Med. 33:43–50.
2014.
|
|
69
|
Liu G, Cao P, Chen H, Yuan W, Wang J and
Tang X: MiR-27a regulates apoptosis in nucleus pulposus cells by
targeting PI3K. PLoS One. 8:e752512013. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Liu H, Huang X, Liu X, Xiao S, Zhang Y,
Xiang T, Shen X, Wang G and Sheng B: miR-21 promotes human nucleus
pulposus cell proliferation through PTEN/AKT signaling. Int J Mol
Sci. 15:4007–4018. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Wada T and Penninger JM: Mitogen-activated
protein kinases in apoptosis regulation. Oncogene. 23:2838–2849.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Dong ZH, Wang DC, Liu TT, Li FH, Liu RL,
Wei JW and Zhou CL: The roles of MAPKs in rabbit nucleus pulposus
cell apoptosis induced by high osmolality. Eur Rev Med Pharmacol
Sci. 18:2835–2845. 2014.PubMed/NCBI
|
|
73
|
Zhang K, Ding W, Sun W, Sun XJ, Xie YZ,
Zhao CQ and Zhao J: Beta1 integrin inhibits apoptosis induced by
cyclic stretch in annulus fibrosus cells via ERK1/2 MAPK pathway.
Apoptosis. Oct 14–2015.Epub ahead of print.
|
|
74
|
Yurube T, Hirata H, Kakutani K, Maeno K,
Takada T, Zhang Z, Takayama K, Matsushita T, Kuroda R, Kurosaka M
and Nishida K: Notochordal cell disappearance and modes of
apoptotic cell death in a rat tail static compression-induced disc
degeneration model. Arthritis Res Ther. 16:R312014. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Xie M, Yang S, Win HL, Xiong L, Huang J
and Zhou J: Rabbit annulus fibrosus cell apoptosis induced by
mechanical overload via a mitochondrial apoptotic pathway. J
Huazhong Univ Sci Technolog Med Sci. 30:379–384. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Rannou F, Lee TS, Zhou RH, Chin J, Lotz
JC, Mayoux-Benhamou MA, Barbet JP, Chevrot A and Shyy JY:
Intervertebral disc degeneration: the role of the mitochondrial
pathway in annulus fibrosus cell apoptosis induced by overload. Am
J Pathol. 164:915–924. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zhang YH, Zhao CQ, Jiang LS and Dai LY:
Cyclic stretch-induced apoptosis in rat annulus fibrosus cells is
mediated in part by endoplasmic reticulum stress through nitric
oxide production. Eur Spine J. 20:1233–1243. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Liao J, Ke M, Xu T and Lin L:
Electroacupuncture inhibits apoptosis in annulus fibrosis cells
through suppression of the mitochondria-dependent pathway in a rat
model of cervical intervertebral disc degradation. Genet Mol Biol.
35:686–692. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Alkhatib B, Rosenzweig DH, Krock E,
Roughley PJ, Beckman L, Steffen T, Weber MH, Ouellet JA and Haglund
L: Acute mechanical injury of the human intervertebral disc: link
to degeneration and pain. Eur Cell Mater. 28:98–111.
2014.PubMed/NCBI
|
|
80
|
Zhao CQ, Wang LM, Jiang LS and Dai LY: The
cell biology of intervertebral disc aging and degeneration. Ageing
Res Rev. 6:247–261. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Ariga K, Miyamoto S, Nakase T, Okuda S,
Meng W, Yonenobu K and Yoshikawa H: The relationship between
apoptosis of endplate chondrocytes and aging and degeneration of
the intervertebral disc. Spine. 26:2414–2420. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Ariga K, Yonenobu K, Nakase T, Hosono N,
Okuda S, Meng W, Tamura Y and Yoshikawa H: Mechanical
stress-induced apoptosis of endplate chondrocytes in organ-cultured
mouse intervertebral discs: an ex vivo study. Spine. 28:1528–1533.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Li D, Zhu B, Ding L, Lu W, Xu G and Wu J:
Role of the mitochondrial pathway in serum deprivation-induced
apoptosis of rat endplate cells. Biochem Biophys Res Commun.
452:354–360. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Li X, Wu FR, Xu RS, Hu W, Jiang DL, Ji C,
Chen FH and Yuan FL: Acid-sensing ion channel 1a-mediated calcium
influx regulates apoptosis of endplate chondrocytes in
intervertebral discs. Expert Opin Ther Targets. 18:1–14. 2014.
View Article : Google Scholar
|
|
85
|
Kong CG, Park JB, Kim MS and Park EY: High
glucose accelerates autophagy in adult rat intervertebral disc
cells. Asian Spine J. 8:543–548. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Kang R, Zeh HJ, Lotze MT and Tang D: The
Beclin 1 network regulates autophagy and apoptosis. Cell Death
Differ. 18:571–580. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Ma KG, Shao ZW, Yang SH, Wang J, Wang BC,
Xiong LM, Wu Q and Chen SF: Autophagy is activated in
compression-induced cell degeneration and is mediated by reactive
oxygen species in nucleus pulposus cells exposed to compression.
Osteoarthritis Cartilage. 21:2030–2038. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Jiang L, Jin Y, Wang H, Jiang Y and Dong
J: Glucosamine protects nucleus pulposus cells and induces
autophagy via the mTOR-dependent pathway. J Orthop Res.
32:1532–1542. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Chen JW, Ni BB, Li B, Yang YH, Jiang SD
and Jiang LS: The responses of autophagy and apoptosis to oxidative
stress in nucleus pulposus cells: implications for disc
degeneration. Cell Physiol Biochem. 34:1175–1189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Chen JW, Ni BB, Zheng XF, Li B, Jiang SD
and Jiang LS: Hypoxia facilitates the survival of nucleus pulposus
cells in serum deprivation by down-regulating excessive autophagy
through restricting ROS generation. Int J Biochem Cell Biol.
59:1–10. 2015. View Article : Google Scholar
|
|
91
|
Shen C, Yan J, Jiang LS and Dai LY:
Autophagy in rat annulus fibrosus cells: evidence and possible
implications. Arthritis Res Ther. 13:R1322011. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Jackson AR, Huang CY and Gu WY: Effect of
endplate calcification and mechanical deformation on the
distribution of glucose in intervertebral disc: a 3D finite element
study. Comput Methods Biomech Biomed Engin. 14:195–204. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Xu HG, Yu YF, Zheng Q, Zhang W, Wang CD,
Zhao XY, Tong WX, Wang H, Liu P and Zhang XL: Autophagy protects
end plate chondrocytes from intermittent cyclic mechanical tension
induced calcification. Bone. 66:232–239. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Yu YF, Xu HG, Wang H, Zhang W, Xiong SL
and Zhang M: Change of autophagy in endplate chondrocytes of rats
during aging process. Zhonghua Yi Xue Za Zhi. 93:3632–3635. 2013.In
Chinese.
|
|
95
|
Fadeel B and Orrenius S: Apoptosis: a
basic biological phenomenon with wide-ranging implications in human
disease. J Intern Med. 258:479–517. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Xu Y, Xia X and Pan H: Active autophagy in
the tumor microenvironment: a novel mechanism for cancer
metastasis. Oncol Lett. 5:411–416. 2013.PubMed/NCBI
|