Introduction
Atherosclerosis is a chronic inflammatory disease
and is associated with high mortality and disability when the
atheroma ruptures (1).
Atherosclerosis is likely to be initiated by the activation of
endothelial cells with the expression of adhesion molecules,
including intercellular adhesion molecule-1 (ICAM-1) and vascular
cell adhesion molecule-1 (VCAM-1). These adhesion molecules in turn
enable the adhesion of mononuclear leukocytes, such as monocytes,
to endothelial cells and also their transmigration into the intima,
which further leads to a cascade of inflammatory reactions
(2–6). Therefore, targeting monocyte
adhesion to the endothelium is considered a novel treatment
strategy for atherosclerosis.
A number of inflammatory signaling pathways are
involved in the initiation and progression of atherosclerosis. The
nuclear factor-κB (NF-κB) pathway, which can be activated by a
number of inflammatory stimuli, such as tumor necrosis factor-α
(TNF-α) and interleukin (IL)-1β, is the most important signaling
pathway in atherosclerosis (7,8).
It manipulates a number of genes which are tightly involved in the
development and progression of atherosclerosis (7,8).
Previous studies have demonstrated that the activated NF-κB
signaling pathway is directly responsible for promoting leukocyte
adhesion to the endothelium and for the increased expression of
adhesion molecules in TNF-α-stimulated endothelial cells (9,10).
Data have shown that the inhibition of the NF-κB pathway and
inflammatory molecules results in reduced lesion size and reduced
inflammatory cell infiltration in vivo (11,12). Previous studies have also
indicated that the mitogen-activated protein kinase (MAPK)
signaling pathway is involved in monocyte adhesion to human
umbilical vein endothelial cells (HUVECs) (13).
Artemisinin
(C15H22O5), derived from the sweet
wormwood Artemisia annua, has been used in the treatment of
malaria in China for over 2,000 years (14). Due to its superior efficiency and
low toxicity, the World Health Organization has recommended
artemisinin for worldwide malaria control (15). Recently, artemisinin and its
derivatives have been shown to have pharmacological actions beyond
their anti-malarial effects; these other properties include,
immunosuppressive and anti-inflammatory properties and have been
shown to exert anticancer effects by inducing cell cycle arrest,
promoting apoptosis, preventing angiogenesis, and abrogating cancer
invasion and metastasis (16).
Moreover, we have previously demonstrated that artemisinin inhibits
the expression of a number of factors which are important in
inflammation and plaque stability (17,18). As is already known, monocyte
adhesion to endothelial cells is considered the initiation of
atherosclerosis (3). Under
conditions of chronic inflammation, the expression of adhesion
molecules is upregulated in activated endothelial cells, which
mediates the adhesion of moncotytes to endothelial cells (19). However, it would be interesting to
determine whether artemisinin can affect the adhesion of monocytes
to endothelial cells under inflammatory stimuli (e.g. TNF-α), as it
affects the expression of inflammation factors.
In the present study, we examined the effects of
artemisinin on the adhesion of monocytes to HUVECs and the
expression of adhesion molecules in TNF-α-stimulated HUVECs. We
demonstrate that artemisinin decreases monocyte adhesion to HUVECs
at least in part through the inhibition of the activation of the
NF-κB and MAPK signaling pathways.
Materials and methods
Cell culture and treatment
HUVECs (PCS-100-013™) were purchased from ATCC
(Manassas, VA, USA) and maintained in M199 medium (Gibco, Grand
Island, NY, USA) supplemented with 10% fetal buffer saline (FBS),
10 mM HEPES (Sigma-Aldrich, St. Louis, MO, USA) and 1%
penicillin/streptomycin solution. THP-1 cells were purchased from
the Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China) and maintained in RPMI-1640 medium (Gibco) with
10% FBS, 10 mM HEPES (Sigma-Aldrich) and 1% penicillin/streptomycin
solution. Both the HUVECs and THP-1 cells were maintained at 37°C
with 5% CO2 and passaged 2–6 times before use.
Cytotoxicity assay
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) assay was performed as previously described (10). Following culture in 96-well
plates, the HUVECs were pre-incubated with increasing
concentrations of atre-misinin (0-300 µM) for 4 h followed
by stimulation with TNF-α (PeproTech, Rocky Hill, NJ, USA) for a
further 24 h. Subsequently, 20 µl/well of MTT solution (5
mg/ml) (Sigma-Aldrich) were added and the HUVECs were cultured at
37°C with 5% CO2 for 4 h. The medium was then removed
and 100 µl/well dimethyl sulfoxide (DMSO) were added. The
plate was pipetted up and down to dissolve crystals, and the
effects of artemisinin on HUVEC viability were assessed by
measuring the absorbance at 570 nm using a spectrophotometer
(Beckman DU-650, Beckman Coulter, Brea, CA, USA).
Analysis of the adhesion of monocytes to
HUVECs
The HUVECs were seeded and incubated in 12-well
dishes until they reached >85% confluence. Subsequently, the
HUVECs were pre-incubated with various concentrations of
artemisinin (0–200 µM) for 4 h prior to stimulation with
TNF-α (10 ng/ml) for 24 h. For examining the signaling pathways
involved, the HUVECs were pre-treated with 10 µM Bay-11-7082
(Beyotime) for 30 min or 10 µM MAPK inhibitors (SP600125,
SB203580 and U0126) (all purchased from Beyotime) for 1 h and then
cultured with TNF-α for 24 h. The THP-1 cells were labeled with 5
µM 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein,
acetoxymethyl ester (BCECF/AM; Invitrogen, Carlsbad, CA, USA) for
30 min in RPMI-1640. The THP-1 cells labeled with BCECF/AM were
added to the 12-well dishes containing the HUVECs and incubated for
1 h. Subsequently, unbound monocytes were removed by 3 washes with
warm phosphate-buffered saline (PBS). Bound monocytes were
determined using a fluorescence microscope (Olympus BX-51; Olympus,
Tokyo, Japan).
Western blot analysis
Protein expression was determined by western blot
analysis as previously described (17). To detect the time course of NF-κB
nuclear translocation, the HUVECs were incubated with TNF-α (10
ng/ml) for different periods of time (15 min to 3 h). In another
experiment, the HUVECs were pre-incubated with increasing
concentrations of artemisinin (0–200 µM) for 4 h followed by
stimulation with TNF-α (10 ng/ml) for 3 h. For examining the
signaling pathways involved, the HUVECs were pre-treated with 10
µM Bay-11-7082 (Beyotime) for 30 min or with 10 µM
MAPK inhibitors (SP600125, SB203580 and U0126) (all purchased from
Beyotime) for 1 h and then cultured with TNF-α for 3 h. Protein
from the cytoplasm or nucleus was collected using a Nuclear and
Cytoplasmic Protein Extraction kit (Beyotime Institute of
Biotechnology, Shanghai, China) as previously described (17). Subsequently, 20 µg of
protein were loaded onto a 10% polyacrylamide gel, separated by
electrophoresis, and transferred onto polyvinylidene difluoride
membranes. After blocking with albumin from bovine serum for 1 h,
the membranes were reacted with primary antibodies, including
anti-ICAM-1 (ab20), anti-VCAM-1 (Ab174279) (both from Abcam
Cambridge, UK), anti-p65 (#6956), anti-IκB (#4814), anti-ERK
(#4695), anti-p-ERK (#4370), anti-JNK (#9252), anti-p-JNK (#4668),
anti-p38 (#8690), anti-p-p38 (#4511) and anti-GAPDH (#5174) (all
from Cell Signaling Technology, Danvers, MA, USA). The membranes
were then washed and incubated with secondary antibody conjugated
with HRP (Cell Signaling Technology). Protein signals were detected
using chemiluminescence and band intensities were analyzed using
Quantity One software (Bio-Rad, Hercules, CA, USA).
Reverse transcription-quantitative PCR
(RT-qPCR)
mRNA expression levels were measured by RT-qPCR as
previously described (17).
Briefly, the HUVECs were treated with various concentrations of
artemisinin for 4 h followed by stimulation with TNF-α for 24 h.
The HUVECs were collected and total RNA was extracted using TRIzol
reagent (Invitrogen) according to the manufacturer's instructions.
These RNA samples were converted into cDNA by reverse transcription
and quantitative (real-time PCR; qPCR) was carried out using the
GoTaq® 2-Step RT-qPCR System (Promega) with a Roche
LightCycler 480 system. The primers used are listed in Table I.
 | Table IPrimers used for RT-PCR. |
Table I
Primers used for RT-PCR.
| Gene | | Primer
sequences |
|---|
| ICAM-1 | Forward |
CCCTTGACCGGCTGGAGATT |
| Reverse |
CTGGGGGCAACATTGACATAAAGTG |
| VCAM-1 | Forward |
CTGTCACTCGAGATCTTGAGG |
| Reverse |
CCTGCAGTGCCCATTATGA |
| GAPDH | Forward |
ACCCAGAAGACTGTGGATGG |
| Reverse |
TTCTAGACGGCAGGTCAGGT |
Confocal immunofluorescence analysis of
NF-κB nuclear translocation
The HUVECs were pre-incubated with different doses
of artemisinin (0-200 µM) for 4 h and followed by TNF-α (10
ng/ml) for 3 h. Immunofluorescence assay was carried out as
described previously using a cellular NF-κB translocation kit
(Beyotime Institute of Biotechnology) according to the
manufacturer's instructions. Briefly, the HUVECs were fixed and
washed with PBS 3 times. The HUVECs were then blocked with 10% goat
serum at room temperature for 1 h before rabbit anti-p65 antibody
(SN368, Beyotime Institute of Biotechnology) was added followed by
incubation for a further 1 h at room temperature. The HUVECs were
then washed and reacted with anti-rabbit IgG Cy3 conjugated
secondary antibody (SN368, Beyotime Institute of Biotechnology) for
1 h. Subsequently, the HUVECs were washed 3 times before
4′,6-diamidino-2-phenylindole (DAPI) was added and washed again.
The location of NF-κB p65 and nuclei were determined using an
Olympus FluoView™ FV1000 confocal microscope (Olympus America Inc.,
Center Valley, PA, USA).
NF-κB transcription factor activity
assay
The HUVECs were pre-incubated with various
concentrations of artemisinin for 4 h followed by stimulation with
TNF-α for 3 h. The HUVECs were collected and the nuclear extract
was prepared using a Nuclear Extract kit (Active Motif, Carlsbad,
CA, USA) according to the manufacturer's instructions. The DNA
binding activity of NF-κB (p50/p65) was then analyzed using the
Trans-AM NF-κB enzyme-linked immunosorbent assay (ELISA) kit
(Active Motif). Briefly, nuclear proteins (10 µg/well) were
added to a 96-well plate coated with an oligonucleotide containing
the NF-κB consensus binding site (5′-GGGACTTTCC-3′) and incubated
for 1 h. After washing, 100 µl of NF-κB antibody (1:1,000;
Cat. no. 40096, Active Motif) was added and the incubation lasted
for 1 h. After that, the plate was washed 3 times before a
horseradish peroxidase-conjugated secondary antibody (Cat. no.
40096, Active Motif) was added to the plate and incubated for 1 h.
The absorbance was determined using a spectrophotometer (Beckman
DU-650, Beckman Coulter) at OD450 nm.
Statistical analysis
All values are presented as the means ± SD.
Statistical analysis was performed by one-way ANOVA (LSD) using
SPSS 9.0 software. A value of P<0.05 was considered to indicate
a statistically significant difference. All experiments were
performed at least 3 times.
Results
Cell viability
The HUVECs were cultured with increasing
concentrations of artemisinin (0-300 µM) for 24 h. The
toxicity of artemisinin was determined by MTT assay. The results of
MTT assay revealed that the viability of the HUVECs was >90%
when the concentration of artemisinin was 200 µM. Cell
viability only decreased significantly when the concentration of
artemisinin increased to 250 µM (Fig. 1A). Therefore, the highest
concentration of artemisinin selected for use was 200 µM in
the subsequent experiments.
Artemisinin inhibits monocyte adhesion to
HUVECs
We first quantified the number of THP-1 cells that
adhered to the HUVECs. The HUVECs were pre-incubated with
increasing concentrations of artemisinin for 4 h and TNF-α was
added then for 24 h. The adherent THP-1 cells labeled with BCECF-AM
dye were examined under a fluorescence microscope, and digital
images were captured at ×200 magnification. TNF-α (10 ng/ml)
markedly increased adhesion between monocytes and HUVECs following
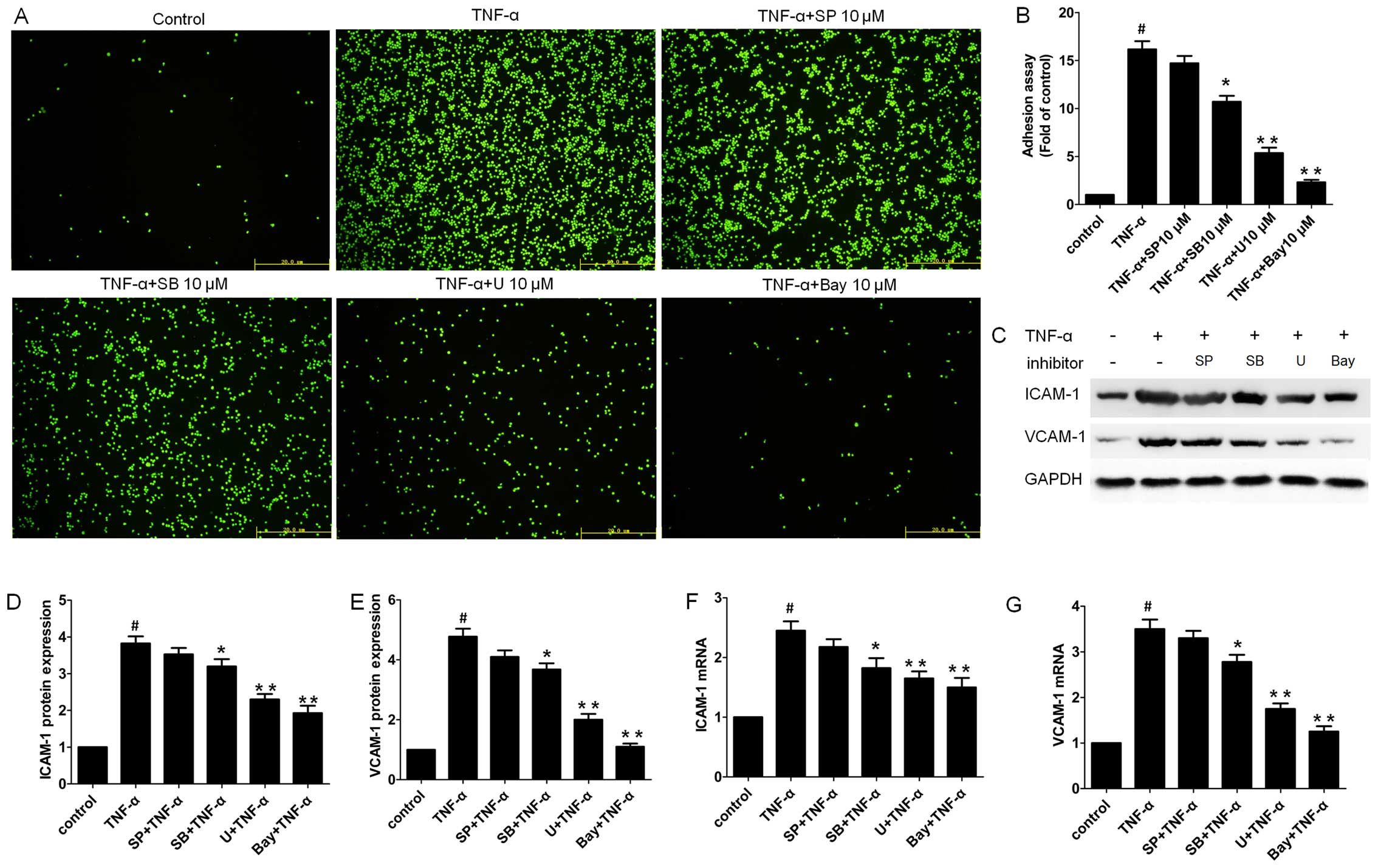
24 h of incubation with the HUVECS (Fig. 1B and C). Of note, pre-treatment of
the HUVECs with 50, 100 and 200 µM artemisinin markedly
suppressed the TNF-α-induced adhesion between the monocytes and
HUVECs in a dose-dependent manner.
Artemisinin decreases the expression of
ICAM-1 and VCAM-1
ICAM-1 and VCAM-1 are considered the main adhesion
molecules which mediate the adhesion of monocytes to HUVECs
(20,33). Thus, we wished to determine
whether artemisinin affects the expression of ICAM-1 and VCAM-1.
The HUVECs were first pre-incubated with or without arte-misinin
(0-200 µM) for 4 h prior to incubation with or without TNF-α
for 24 h. Cell pellets were lysed, and western blot analysis was
carried out. TNF-α alone significantly increased the protein
expression of ICAM-1 and VCAM-1 (Fig.
2A). However, ICAM-1 and VCAM-1 protein levels were markedly
downregulated following pre-treatment with artemisinin (Fig. 2A–C).
We also examined the mRNA levels of ICAM-1 and
VCAM-1 by RT-qPCR. ICAM-1 and VCAM-1 mRNA expression levels were
markedly elevated following stimulation with TNF-α (Fig. 2D and E). Consistent with the
results of western blot analysis for protein expression,
pre-treatment with artemisinin significantly decreased the mRNA
levels of ICAM-1 and VCAM-1 which were increased by TNF-α in a
dose-dependent manner.
NF-κB and MAPK inhibitors block
TNF-α-induced monocyte adhesion to HUVECs and decrease the
expression of ICAM-1 and VCAM-1 in HUVECs
It is well known that the NF-κB signaling pathway
plays a pivotal role in the pathogenesis of atherosclerosis by
regulating a series of inflammation-associated genes and adhesion
molecules (7,8). Thus, to further elucidate the
potential mechanisms of action of artemisinin, we first examined
monocyte adhesion to HUVECs using the NF-κB-specific inhibitor, Bay
11-7082. Pre-treatment with Bay 11-7082 (10 µM)
significantly decreased monocyte adhesion, and the effects were
similar to those observed with the high concentration of
artemisinin (Fig. 3A and B).
We further examined the mRNA and protein level of
ICAM-1 and VCAM-1 following pre-treatment with Bay 11-7082 by
western blot analysis and RT-qPCR, respectively. As shown in
Fig. 3C–E, pre-treatment of the
HUVECs with Bay 11-7082 significantly decreased the protein
expression of ICAM-1 and VCAM-1 which was induced by TNF-α, which
correlated with reduced monocyte adhesion to the HUVECs (Fig. 3A and B). A similar pattern was
observed with the mRNA expression of ICAM-1 and VCAM-1 in the Bay
11-7082-pre-treated group (Fig. 3F
and G).
Previous studies have indicated that the MAPK
signaling pathway is also activated in TNF-α-stimulated HUVECs
(21,22). In this study, to determine which
MAPK signaling pathway (ERK1/2, p38 or JNK) is involved in the
increased monocyte adhesion to HUVECs, the HUVECs were pre-treated
with an ERK-specific inhibitor (U0126, 10 µM), a
p38-specific inhibitor (SB203580, 10 µM) and a JNK-specific
inhibitor (SP600125, 10 µM) for 1 h prior to incubation with
TNF-α for 24 h. Although SP600125 did not exert a significant
effect, SB203580 (P<0.05) and U0126 (P<0.01) significantly
decreased the number of adherent monocytes to HUVECs stimulated
with TNF-α (Fig. 3A and B).
Consistently, although SP600125 exerted no significant effect on
the protein levels of ICAM-1 and VCAM-1, SB203580 (P<0.05) and
U0126 (P<0.01) markedly downregulated TNF-α-induced ICAM-1 and
VCAM-1 mRNA expression in the HUVECs (Fig. 3C–E). A similar pattern was
observed with the mRNA levels of ICAM-1 and VCAM-1 in the MAPK
inhibitor-pre-treated group (Fig. 3F
and G), suggesting that the p38 and ERK pathways are the major
MAPK pathways responsible for this process.
Artemisinin blocks the activation of the
NF-κB and MAPK signaling pathways in TNF-α-stimulated HUVECs
Our previous study demonstrated that artemisinin
inhibited pro-inflammatory factors through the NF-κB signaling
pathway in phorbol 12-myristate 13-acetate (PMA)-stimulated
monocytes (17). As artemisinin
and Bay 11-7082 had a similar effect on monocyte adhesion to
HUVECs, we hypothesized that artemisinin decreased the adhesion of
monocytes to HUVECs through the NF-κB signaling pathway. Therefore,
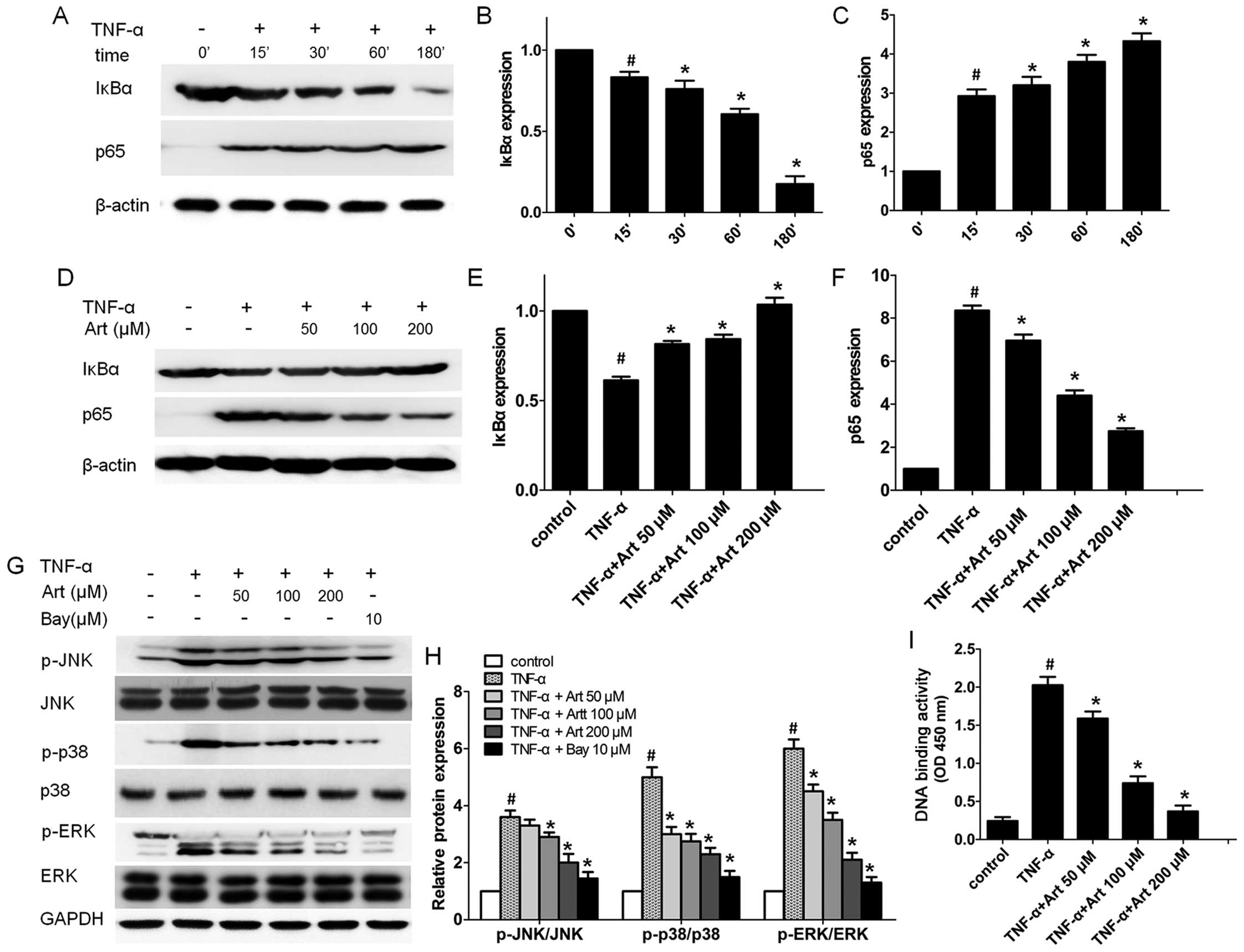
we first examined the time-course (15 min to 3 h) of the activation
of the NF-κB signaling pathway in HUVECs. After the HUVECs were
stimulated with TNF-α, the expression levels of IκBα in the
cytoplasm and p65 in the nucleus were examined at different time
points by western blot analysis. The decreased IκBα protein level
in the cytoplasm was observed as early as 15 min following the
addition of TNF-α and the IκBα protein level reached its lowest
level at 180 min (Fig. 4A–C).
TNF-α increased the p65 protein level in the nuclear fractions
after 15 min and its level peaked at 180 min. These data suggest
that TNF-α activates the NF-κB signal transduction pathway.
Subsequently, we wished to determine whether
artemisinin blocks the NF-κB signal transduction pathway activated
by TNF-α. Consistent with our hypothesis, pre-treatment with
artemisinin significantly increased the IκBα protein level in the
cytoplasm, while it abrogated the NF-κB p65 subunit level in the
nucleus of the HUVECs in a dose-dependent manner (Fig. 4D–F).
We further performed p65 DNA binding activity assay
as activated NF-κB binds to a specific sequence of DNA to regulate
downstream gene transcription, such as ICAM-1 and VCAM-1.
p65-mediated NF-κB DNA-binding activity was markedly increased by
TNF-α (Fig. 4I). Of note, the
enhanced DNA binding activity of NF-κB p65 was attenuated by
artemisinin (50–100 µM) in a dose dependent manner (Fig. 4I), indicating that artemisinin
indeed suppresses the activation of the NF-κB signaling pathway
induced by TNF-α.
To determine whether artemisinin blocks MAPK
activation in TNF-α-stimulated HUVECs, we pre-treated the HUVECs
with artemisinin (0–200 µM) for 4 h before stimulating the
cells with TNF-α for a further 30 min, and examined the expression
of phosphorylated and total proteins during MAPK pathway
activation. Artemisinin significantly inhibited the protein level
of phosphorylated ERK1/2 and p38 MAPK induced by TNF-α in the
HUVECs at all concentrations tested, while the inhibition of the
phosphorylation of JNK was only observed at the high concentrations
(100 and 200 µM) (Fig. 4G and
H). Of note, the inhibition of the NF-κB signaling pathway by
treatment of the HUVECs with 10 µM Bay 11-7082 led to
significant decrease in the levels of phosphorylated ERK1/2, p38
and JNK, while the total protein level of ERK1/2, p38 and JNK
remained unaltered (Fig. 4G and
H), suggesting that MAPK is downstream of the NF-κB signal
transduction pathway in TNF-α-stimulated HUVECs.
Artemisinin blocks NF-κB
translocation
Once activated, the NF-κB p65 subunit translocates
from the cytoplasm to the nucleus and regulates target gene
expression (8,9). Thus, we traced the translocation
process in the HUVECs using NF-κB-specific antibody and a confocal
laser scanning microscope. The NF-κB p65 subunit in the nuclei was
significantly increased following stimulation with TNF-α, while in
the untreated control group it was predominantly located in the
cytoplasm (Fig. 5). Compared with
the cells stimulated with TNF-α alone, a weaker p65 fluorescence
signal in the nuclei was detected in the cells pre-treated with
artemisinin, suggesting that artemisinin impeded the translocation
of NF-κB p65 to the nucleus (Fig.
5).
Discussion
Inflammatory stimuli, such as TNF-α, lead to
endothelial cell activation and the upregulation of adhesion
molecules (4,23). ICAM-1 and VCAM-1 are the main
adhesion molecules which are crucial for the firm adhesion of
leukocytes to the endothelium (20,33). Continuous adhesion and migration
result in the infiltration of inflammatory cells, the release of
inflammatory factors and lipid overloaded, which ultimately
aggravates plaque instability. Accordingly, impeding monocyte
adhesion to the endothelium is of great importance in early
atherosclerosis (24,25). In addition, a previous study
linked the expression of ICAM-1 and VCAM-1 to an increased risk of
the incidence of clinical coronary artery disease (26). In the present study, we
demonstrated that artemisinin signifi-cantly decreased monocyte
adhesion to TNF-α-stimulated HUVECs, and suppressed ICAM-1 and
VCAM-1 expression in TNF-α-stimulated HUVECs. Thus, artemisinin may
prove to be efficient in the protection against the development of
early atherosclerotic lesions.
The NF-κB pathway is the major signaling pathway
involved in the activation of HUVECs induced by TNF-α (27,28). NF-κB is known to play a critical
role in the regulation of genes which is tightly involved in
atherosclerosis (29,30). Under physiological conditions,
NF-κB is sequestered into the cytoplasm by IκB protein. Upon
inflammatory stimuli, including TNF-α, IκBα is phosphorylated and
degraded, which enables NF-κB to translocate to the nucleus. NF-κB
then binds to its specific promoter region, and initiates the
transcription of numerous genes, including inflammatory factors
[such as IL-1β, IL-6, TNF-α and matrix metallopeptidase (MMP)-9]
and adhesion molecules (such as ICAM-1 and VCAM-1) (31,32). In the present study, we observed
that pre-treatment with artemisinin significantly increased the
cytosolic level of IκBα, while it reduced NF-κB p65 expression in
the nucleus of TNF-α-stimulated HUVECs. Using a confocal laser
scanning fluorescence microscope, we found that artemisinin
inhibited NF-κB p65 subunit translocation from the cytoplasm to the
nucleus. Moreover, the DNA binding activity of NF-κB was also
inhibited by artemisinin in the TNF-α-stimulated cells. This study
suggests that artemisinin impedes NF-κB activation in
TNF-α-stimulated HUVECs.
Previous studies have indicated that the MAPK
signaling pathway is also activated in TNF-α-stimulated HUVECs. Lu
et al reported that the NF-κB and JNK pathways are related
to VCAM-1 expression in lipopolysaccharide (LPS)-stimulated HUVECs
(33), and Ju et al
reported that p38 MAPK is involved in TNF-α-induced ICAM-1 and
VCAM-1 expression in HUVECs (34). Moreover, in another study, p38
inhibitor decreased the protein level of ICAM-1 and VCAM-1 in
TNF-α-stimulated HUVECs, while the ERK inhibitor had no effect on
ICAM-1 and VCAM-1 expression (35). In this study, we investigated
whether the MAPK signaling pathway is related to the adhesion of
monocytes to HUVECs. We also examined the association between the
MAPK signaling pathway and the expression of ICAM-1 and VCAM-1 in
TNF-α-stimulated HUVECs. Both artemisinin and the NF-κB inhibitor,
Bay 11-7028, inhibited MAPK signaling pathway activation in
TNF-α-stimulated HUVECs. Using specific inhibitors of MAPK (ERK,
JNK and p38), we found that U0126 (ERK1/2 inhibitor) significantly
decreased the adhesion of monocytes to HUVECs and the expression of
ICAM-1 and VCAM-1, while SB203580 had a weaker effect and SP600125
had no effect, which indicated that ERK1/2 is the major MAPK
responsible for the decreased adhesion of monocytes to HUVECs and
the expression of ICAM-1 and VCAM-1 by artemisinin.
Recently, artemisinin and its derivatives have
attracted increasing attention due to their effects beyond their
antimalarial properties. Our previous studies have demonstrated
that artemisinin exerts anti-inflammatory effects in
monocytes/macrophages through the MAPK and NF-κB pathways (17,18). Cao et al reported that
artemisinin blocked the proliferation, migration and inflammatory
reaction induced by TNF-α in vascular smooth muscle cells through
the NF-κB pathway (36). Tripathi
et al reported that artemisinin reduced ICAM-1 expression in
human brain microvascular endothelial cells (37). Artesunate, an artemisinin
derivative, has been reported to abrogate the expression of ICAM-1
in parasitized red blood cell (pRBC)-stimulated endothelial cells
and prevent pRBCs adhesion to vascular endothelial cells by
impairing NF-κB translocation to the nucleus (38). Another study demonstrated that
dihydroarteannuin inhibited NF-κB translocation and ameliorated
lupus symptoms in BXSB mice (39). Our data further indicated that
artemisinin significantly decreased monocyte adhesion to
TNF-α-stimulated HUVECs, and suppressed the mRNA and protein level
of ICAM-1 and VCAM-1 in TNF-α-stimulated HUVECs through the NF-κB
and MAPK pathways. All these data indicate that artemisinin plays a
significant role in atherosclerosis-related inflammation and lipid
uptake, which exert protective effects against the development and
progression of atherosclerosis.
In conclusion, in this study, we demonstrated that
artemisinin inhibited the adhesion of monocytes to HUVECs and
suppressed the expression of ICAM-1 and VCAM-1 in TNF-α-stimulated
HUVECs (Fig. 6). The protective
effects of artemisnin against adhesion are likely mediated through
the suppression of the NF-κB and MAPK pathways. These findings not
only shed new light on the mechanisms of action of artemisinin, but
also suggest that artemisinin may prove to be useful in the
protection against the development of early atherosclerotic
lesions.
Acknowledgments
This study was supported by the Fund of Science and
Technology Commission of Shanghai Municipality (grants no.
12401905200) and the Fund of National Natural Science Foundation of
China (grant nos. 81270376, 81470546 and 81500392).
Abbreviations:
|
HUVECs
|
human umbilical vein endothelial
cells
|
|
ICAM-1
|
intercellular adhesion molecule-1
|
|
VCAM-1
|
vascular cell adhesion molecule-1
|
|
NF-κB
|
nuclear factor-κB
|
|
TNF-α
|
tumor necrosis factor-α
|
|
pRBCs
|
parasitized red blood cells
|
References
|
1
|
Libby P, Ridker PM and Hansson GK; Leducq
Transatlantic Network on Atherothrombosis: Inflammation in
atherosclerosis: From pathophysiology to practice. J Am Coll
Cardiol. 54:2129–2138. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Galkina E and Ley K: Vascular adhesion
molecules in atherosclerosis. Arterioscler Thromb Vasc Biol.
27:2292–2301. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Davignon J and Ganz P: Role of endothelial
dysfunction in atherosclerosis. Circulation. 109(Suppl 1):
III27–III32. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hou HF, Yuan N, Guo Q, Sun T, Li C, Liu
JB, Li QW and Jiang BF: Citreoviridin enhances atherogenesis in
hypercholesterolemic ApoE-deficient mice via upregulating
inflammation and endothelial dysfunction. PLoS One.
10:e01259562015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xu S, Song H, Huang M, Wang K, Xu C and
Xie L: Telmisartan inhibits the proinflammatory effects of
homocysteine on human endothelial cells through activation of the
peroxisome proliferator-activated receptor-δ pathway. Int J Mol
Med. 34:828–834. 2014.PubMed/NCBI
|
|
6
|
Carluccio MA, Siculella L, Ancora MA,
Massaro M, Scoditti E, Storelli C, Visioli F, Distante A and De
Caterina R: Olive oil and red wine antioxidant polyphenols inhibit
endothelial activation: Antiatherogenic properties of Mediterranean
diet phytochemicals. Arterioscler Thromb Vasc Biol. 23:622–629.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baker RG, Hayden MS and Ghosh S: NF-κB
inflammation, and metabolic disease. Cell Metab. 13:11–22. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brown JD, Lin CY, Duan Q, Griffin G,
Federation AJ, Paranal RM, Bair S, Newton G, Lichtman AH, Kung AL,
et al: NF-κB directs dynamic super enhancer formation in
inflammation and atherogenesis. Mol Cell. 56:219–231. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Barnes PJ and Karin M: Nuclear
factor-kappaB: a pivotal transcription factor in chronic
inflammatory diseases. N Engl J Med. 336:1066–1071. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Collins T: Endothelial nuclear
factor-kappa B and the initiation of the atherosclerotic lesion.
Lab Invest. 68:499–508. 1993.PubMed/NCBI
|
|
11
|
Mallavia B, Recio C, Oguiza A, Ortiz-Muñoz
G, Lazaro I, Lopez-Parra V, Lopez-Franco O, Schindler S, Depping R,
Egido J and Gomez-Guerrero C: Peptide inhibitor of NF-κB
translocation ameliorates experimental atherosclerosis. Am J
Pathol. 182:1910–1921. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pamukcu B, Lip GY and Shantsila E: The
nuclear factor-kappa B pathway in atherosclerosis: a potential
therapeutic target for atherothrombotic vascular disease. Thromb
Res. 128:117–123. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Usatyuk PV and Natarajan V: Role of
mitogen-activated protein kinases in 4-hydroxy-2-nonenal-induced
actin remodeling and barrier function in endothelial cells. J Biol
Chem. 279:11789–11797. 2004. View Article : Google Scholar
|
|
14
|
Meshnick SR, Taylor TE and
Kamchonwongpaisan S: Artemisinin and the antimalarial
endoperoxides: From herbal remedy to targeted chemotherapy.
Microbiol Rev. 60:301–315. 1996.PubMed/NCBI
|
|
15
|
World Health Organization: Guidelines for
the Treatment of Malaria. ISBN: 92-4-154694-8Geneva; 2006
|
|
16
|
Ho WE, Peh HY, Chan TK and Wong WS:
Artemisinins: Pharmacological actions beyond anti-malarial.
Pharmacol Ther. 142:126–139. 2014. View Article : Google Scholar
|
|
17
|
Wang Y, Huang Z, Wang L, Meng S, Fan Y,
Chen T, Cao J, Jiang R and Wang C: The anti-malarial artemisinin
inhibits pro-inflammatory cytokines via the NF-κB canonical
signaling pathway in PMA-induced THP-1 monocytes. Int J Mol Med.
27:233–241. 2011. View Article : Google Scholar
|
|
18
|
Wang Y, Huang ZQ, Wang CQ, Wang LS, Meng
S, Zhang YC, Chen T and Fan YQ: Artemisinin inhibits extracellular
matrix metalloproteinase inducer (EMMPRIN) and matrix
metallopro-teinase-9 expression via a protein kinase
Cδ/p38/extracellular signal-regulated kinase pathway in phorbol
myristate acetate-induced THP-1 macrophages. Clin Exp Pharmacol
Physiol. 38:11–18. 2011. View Article : Google Scholar
|
|
19
|
Lamon BD and Hajjar DP: Inflammation at
the molecular interface of atherogenesis: an anthropological
journey. Am J Pathol. 173:1253–1264. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Blankenberg S, Barbaux S and Tiret L:
Adhesion molecules and atherosclerosis. Atherosclerosis.
170:191–203. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Davies MJ, Gordon JL, Gearing AJH, Pigott
R, Woolf N, Katz D and Kyriakopoulos A: The expression of the
adhesion molecules ICAM-1, VCAM-1, PECAM, and E-selectin in human
atherosclerosis. J Pathol. 171:223–229. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xia F, Wang C, Jin Y, Liu Q, Meng Q, Liu K
and Sun H: Luteolin protects HUVECs from TNF-α-induced oxidative
stress and inflammation via its effects on the Nox4/ROS-NF-κB and
MAPK pathways. J Atheroscler Thromb. 21:768–783. 2014. View Article : Google Scholar
|
|
23
|
Koo HJ, Sohn EH, Pyo S, Woo HG, Park DW,
Ham YM, Jang SA, Park SY and Kang SC: An ethanol root extract of
Cynanchum wilfordii containing acetophenones suppresses the
expression of VCAM-1 and ICAM-1 in TNF-α-stimulated human aortic
smooth muscle cells through the NF-κB pathway. Int J Mol Med.
35:915–924. 2015.PubMed/NCBI
|
|
24
|
Libby P: Inflammation in atherosclerosis.
Arterioscler Thromb Vasc Biol. 32:2045–2051. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hansson GK: Inflammation, atherosclerosis,
and coronary artery disease. N Engl J Med. 352:1685–1695. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Blankenberg S, Rupprecht HJ, Bickel C,
Peetz D, Hafner G, Tiret L and Meyer J: Circulating cell adhesion
molecules and death in patients with coronary artery disease.
Circulation. 104:1336–1342. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gustin JA, Pincheira R, Mayo LD, Ozes ON,
Kessler KM, Baerwald MR, Korgaonkar CK and Donner DB: Tumor
necrosis factor activates CRE-binding protein through a p38
MAPK/MSK1 signaling pathway in endothelial cells. Am J Physiol Cell
Physiol. 286:C547–C555. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Monaco C and Paleolog E: Nuclear factor
kappaB: A potential therapeutic target in atherosclerosis and
thrombosis. Cardiovasc Res. 61:671–682. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
de Winther MP, Kanters E, Kraal G and
Hofker MH: Nuclear factor kappaB signaling in atherogenesis.
Arterioscler Thromb Vasc Biol. 25:904–914. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang Y, Yang X, Bian F, Wu P, Xing S, Xu
G, Li W, Chi J, Ouyang C, Zheng T, et al: TNF-α promotes early
atherosclerosis by increasing transcytosis of LDL across
endothelial cells: Crosstalk between NF-κB and PPAR-γ. J Mol Cell
Cardiol. 72:85–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Anderson MT, Staal FJ, Gitler C and
Herzenberg LA and Herzenberg LA: Separation of oxidant-initiated
and redox-regulated steps in the NF-kappa B signal transduction
pathway. Proc Natl Acad Sci USA. 91:11527–11531. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Collins T, Read MA, Neish AS, Whitley MZ,
Thanos D and Maniatis T: Transcriptional regulation of endothelial
cell adhesion molecules: NF-kappa B and cytokine-inducible
enhancers. FASEB J. 9:899–909. 1995.PubMed/NCBI
|
|
33
|
Lu Y, Zhu X, Liang GX, Cui RR, Liu Y, Wu
SS, Liang QH, Liu GY, Jiang Y, Liao XB, et al: Apelin-APJ induces
ICAM-1, VCAM-1 and MCP-1 expression via NF-κB/JNK signal pathway in
human umbilical vein endothelial cells. Amino Acids. 43:2125–2136.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ju H, Behm DJ, Nerurkar S, Eybye ME,
Haimbach RE, Olzinski AR, Douglas SA and Willette RN: p38 MAPK
inhibitors ameliorate target organ damage in hypertension: Part 1.
p38 MAPK-dependent endothelial dysfunction and hypertension. J
Pharmacol Exp Ther. 307:932–938. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Surapisitchat J, Hoefen RJ, Pi X,
Yoshizumi M, Yan C and Berk BC: Fluid shear stress inhibits
TNF-alpha activation of JNK but not ERK1/2 or p38 in human
umbilical vein endothelial cells: Inhibitory crosstalk among MAPK
family members. Proc Natl Acad Sci USA. 98:6476–6481. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cao Q, Jiang Y, Shi J, Xu C, Liu X, Yang
T, Fu P and Niu T: Artemisinin inhibits the proliferation,
migration, and inflammatory reaction induced by tumor necrosis
factor-α in vascular smooth muscle cells through nuclear factor
kappa B pathway. J Surg Res. 194:667–678. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tripathi AK, Sullivan DJ and Stins MF:
Plasmodium falciparum-infected erythrocytes increase intercellular
adhesion molecule 1 expression on brain endothelium through
NF-kappaB. Infect Immun. 74:3262–3270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Souza MC, Paixão FH, Ferraris FK, Ribeiro
I and Henriques Md: Artesunate exerts a direct effect on
endothelial cell activation and NF-κB translocation in a mechanism
independent of plasmodium killing. Malar Res Treat.
679090(2012)2012.
|
|
39
|
Li WD, Dong YJ, Tu YY and Lin ZB:
Dihydroarteannuin ameliorates lupus symptom of BXSB mice by
inhibiting production of TNF-alpha and blocking the signaling
pathway NF-kappa B translocation. Int Immunopharmacol. 6:1243–1250.
2006. View Article : Google Scholar : PubMed/NCBI
|