Introduction
Lung cancer remains a leading cause of
cancer-related mortality worldwide. Non-small cell lung cancer
(NSCLC) accounts for 80–85% of all cancer cases, with an overall
5-year survival rate of <20% (1,2).
Targeted molecular therapy and small-molecule competitive epidermal
growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs),
which largely improve the prognosis of patients, has optimized the
therapeutic strategy. Approximately 25% of NSCLC patients harboring
the EGFR active mutation (including exon 19 del and exon 21 L858R)
benefit from TKIs (3). However,
acquired resistance to TKIs inevitably occurs following 8–10 months
of treatment. The acquired resistance positively correlates with a
secondary T790M mutation or amplification of hepatocyte growth
factor receptor (HGFR) (3,4).
In addition, previous studies have demonstrated that the
overexpression of insulin-like growth factor-1 receptor (IGF-1R) is
also involved in this type of drug resistance (5). Recently, a class of small non-coding
RNAs, termed microRNAs (miRNAs or miRs), has also been shown to
play important roles in NSCLC (6). However, whether miRNAs are related
to resistance to EGFR-TKIs remains unknown. The exact mechanisms
involved, thus need to be explored. At the same time, solutions to
improve the sensitivity to TKIs are urgently required.
miRNAs are single-stranded RNAs, 20–22 nucleotides
in length (7). They usually act
as negative regulators of gene expression at the
post-transcriptional level (8),
and are involved in several cellular functions, including
differentiation, proliferation and apoptosis (9–12).
Garofalo et al (13)
demonstrated that numerous miRNAs play important roles in
modulating the cell phenotype of NSCLC. miR-223 is a tumor
suppressor miRNA that has been reported in various types of cancer.
It has been shown to be expressed at low levels in hepatocellular
carcinoma and ovarian cancer (14,15). In our previous study, a low
expression level of miR-223 was observed in Lewis lung carcinoma
(LLC) tissue, and miR-223 was shown to be extensively involved in
the cell cycle regulation. Furthermore, the upregulation of miR-223
inhibited the migration and invasion of LLC cells in vitro
by targeting IGF-1R (16).
miR-223 is involved in regulating a variety of malignant
phenotypes. However, the mechanisms through which miR-223 regulates
the resistance of NSCLC cells to erlotinib remain unknown.
IGF-1R is a member of the insulin receptor (IR)
family, which is well known for its role in the resistance of NSCLC
cells to EGFR-TKIs (17). IGF-1R
is highly expressed in lung adenocarcinoma, breast cancer,
prostatic cancer and pancreatic carcinoma (18). The intracellular signaling of
IGF-1R is mediated by IGF-1, which in turn results in the
activation of the mitogen-activated protein kinase (MAPK) and
phosphatidylinositol 3-kinase (PI3K)/Akt pathways (19). PI3K/Akt is considered to be
predominant in the resistance to EGFR-TKIs. miR-223 has been shown
to suppress cell proliferation by regulating the IGF-1R signaling
pathway in HeLa cells (15).
Moreover, miR-223 plays a central role in degranulation by
targeting the IGF-1R/PI3K/Akt pathway in mast cells (20). Based on the above-mentioned
findings, we hypothesized that miR-223 may modulate apoptosis and
improve the sensitivity of NSCLC cells to erlotinib by targeting
the IGF-1R/Akt signaling pathway. In the present study, miR-223 was
overexpressed in erlotinib-resistant PC-9 (PC-9/ER) cells and the
potential regulatory effects of miR-223 on IGF-1R were
investigated.
Materials and methods
Cell lines, reagents and groups
The lung adenocarcinoma PC-9 cell line was purchased
from the American Type Culture Collection (ATCC, Manassas, VA,
USA), and cultured in RPMI-1640 medium, with 10% fetal bovine serum
(FBS) (both from Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) and 100 U/ml penicillin/streptomycin (Sigma-Aldrich, St.
Louis, MO, USA). All cells were cultured at 37°C in a humidified
atmosphere of 95% air/5% CO2. Erlotinib (Sigma-Aldrich)
was dissolved by dimethyl sulfoxide (DMSO) and stored at −20°C. The
concentration used to maintain the resistance of PC-9/ER cells to
erlotinib was 0.1 µM. The cells were divided into 6 groups
as follows: PC-9, PC-9/ER, PC-9/ER-miR-223, PC-9/ER-miR-223 plus
IGF-1, PC-9/ER-empty vector (EV) and PC-9/ER-EV plus IGF-1.
Establishment of erlotinib-resistant
PC-9/ER cell line
The PC-9 cell line harboring exon 19 del mutation
acquired erlotinib resistance following 6 months of continuous drug
exposure (1 µM erlotinib). The cell counting kit-8 (CCK-8;
Dojindo, Kumamoto, Japan) was used to confirm the resistance of the
PC-9 cells to erlotinib (PC-9/ER), and this was confirmed 3 times.
The cells were allowed to grow under drug-free conditions for at
least 1 week before being used in the experiments. The PC-9/ER
cells did not harbor the T790M mutation, according to our
subsequent EGFR gene detection (data not shown).
Infection with lentivirus
The lentiviral vector GV259, used to induce miR-223
οverexpression, was packaged and purchased from Shanghai GeneChem
Co., Ltd. (Shanghai, China). The PC-9/ER cells (5×104)
were infected with 2×106 lentivirus-transducing units in
the presence of 10 µg/ml polybrene in a 24-well plate
(MOL=40). An empty lentiviral vector was transfected into the
target cells as the control under identical conditions. The EV
group is the control of miR-223 group both in vivo and in
vitro. Therefore, the infected cells were referred to as either
miR-223/EV or as PC-9//ER-miR-223/PC-9/ER-EV, respectively. The
cells were collected and the media were updated following
cultivation for 12 h. The transfection efficiency was observed
under a fluorescent microscope (BX50; Olympus, Tokyo, Japan). The
transfected cells were subsequently sorted using a fluorescence
activated cell sorter (FACS) based on the green fluorescent protein
(GFP) signal.
Cell proliferation assay
Cell proliferation assay was carried out according
to the instructions of the manufacturer of CCK-8 (Dojindo).
Briefly, the PC-9 and PC-9/ER cells were seeded in 96-well plates
at a density of 3.5×103 cells/well with 100 µl
cell culture medium. After 24 h of culture, the medium was removed
from the wells and replaced with medium containing erlotinib at
concentrations ranging between 0.01 and 500 µM. Following
further culture for 48 h, 100 µl of medium containing 10%
CCK-8 were added to each well. The cells were then incubated at
37°C for approximately 30 min. The absorbance was detected at 450
nm using a spectrophotometer (Epoch; BioTek, Winooski, VT, USA).
All experiments were set up in triplicate, and confirmed in at
least 3 independent experiments.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the cells using RNAiso
Plus reagent (Takara Bio, Inc., Otsu, Japan), according to the
manufacturer's instructions. The expression of miR-223 was detected
by one-step RT-qPCR with SYBR Premix Ex Taq (Takara Bio., Inc.).
The specific primers of miR-223 and IGF-1R were designed by
Shanghai GeneChem Co., Ltd. The relative expression levels were
calculated by means of the 2−ΔΔCt method, relative to
the internal controls, U6 RNA for miRNA and β-actin for genes,
respectively. The following primers were used: IGF-1R forward,
5′-GGCATACCTCAACGCCAATA-3′ and reverse, 5′-CAGCCCTTTCCCTCCTTT-3′;
β-actin forward, 5′-GTGAAGGTGACAGCAGTCGGTT-3′ and reverse,
5′-GAAGTGGGGTGGCTTTTAGGA-3′. All the RT-qPCR reactions were run in
triplicate and repeated in 3 independent experiments.
Western blot analysis
Total protein was extracted from the cells using
radio immunoprecipitation assay (RIPA) buffer and was quantified by
a bicinchoninic acid assay kit (Beyotime Institute of
Biotechnology, Haimen, China). The proteins were separated by
electrophoresis on 12% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) gels and were transferred onto
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA,
USA). The membranes were then blocked for 1 h at room temperature
with 5% BSA and were subsequently incubated overnight at 4°C with
the following antibodies: Polyclonal rabbit anti-IGF-1R (cat. no.
3027), monoclonal rabbit anti-phosphorylated (p-)IGF-1R (cat. no.
3918), polyclonal rabbit anti-Akt (cat. no. 4691), polyclonal
rabbit anti-p-Akt (cat. no. 4060), monoclonal rabbit anti-S6 (cat.
no. 2217), monoclonal rabbit anti-p-S6 (cat. no. 4858) and
polyclonal mouse anti-β-actin (cat. no. 3700; 1:1,000) (Cell
Signaling Technology, Inc., Beverly, MA, USA). Following washing 3
times with Tris-buffered saline containing Tween-20 (1:500), the
membranes were incubated with secondary antibodies-anti-rabbit IgG
(cat. no. SA00001-2; 1:5,000) and anti-actin antibody (cat. no.
20536-1-AP; 1:5,000) (both from Proteintech Group, Chicago, IL,
USA) for 1 h at room temperature as previously described (21). The blots were visualized using
enhanced chemiluminescence, as previously described (22). The density of the bands was
quantified using Quantity One software (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Analysis of apoptosis
A total of 5×105 cells was resuspended in
phosphate-buffered saline (PBS) following exposure to 1 µM
erlotinib for 48 h. The cells were stained using the Annexin
V-APC/7-AAD Apoptosis Detection kit (KeyGen Biotech, Nanjing,
China) and subsequently analyzed using a flow cytometer (Beckman
Coulter, Brea, CA, USA) equipped with Summit software according to
the manufacturer's instructions.
IGF-1 stimulation experiments in
vitro
To induce exogenous IGF-1 expression, the
PC-9/ER-miR-223 and PC-9/ER-EV cells were seeded in 6-well plates
with RPMI-1640 medium containing 10% FBS. Once the cells reached
70% confluence, they were washed twice with serum-free medium to
remove the FBS. Following overnight serum starvation, the cells
were stimulated for 30 min at 37°C, 5% CO2 in serum-free
medium containing 100 ng/ml of IGF-1 (cat. no. CYT-216; ProSpec,
Rehovot, Israel), as previously described (17). The cells were washed with 1X
ice-cold PBS and incubated on ice in RIPA lysis buffer or RNAiso
Plus reagent to continue the experiment.
In vivo experiments
Nude mice (male, 4–6 weeks old) were purchased from
the Chinese Academy of Medical Sciences (Beijing, China). They were
housed in a laminar flow room under specific pathogen-free
conditions at room temperature (22±2°C) and humidity (<40%) with
free access to food and water. In tumor growth assay, a total of 18
mice was randomly divided into 6 groups. A total of
2×106 PC-9, PC-9/ER, PC-9/ER-miR-223 and PC-9/ER-EV
cells was subcutaneously injected into the lower quadrant of the
rats. When the tumor volumes reached approximately 100–150
mm3, erlotinib (60 mg/kg) was intragastrically
administered from the 14th day to each mouse daily for 14 days.
Moreover, the mice in the PC-9/ER-miR-223 and PC-9/ER-EV groups
received a peritoneal perfusion of IGF-1 (50 µg/kg) daily
for 14 days and were classified as groups PC-9/ER-miR-223 plus
IGF-1 and PC-9/ER-EV plus IGF-1. We evaluated tumor sizes every 3
days for 4 weeks using calipers. Tumors were calculated according
to the formula V = (length × width2)/2. Additionally, to
observe survival, another 30 nude mice were randomly divided into 6
groups in this study. The grouping, erlotinib administration and
IGF-1 injection were the same as mentioned above. These mice were
not sacrificed by cervical dislocation until they suffered from
limitations in activities and significant weight loss. Efforts were
made to reduce the animal suffering and the number of animals used.
All the animal care and experimentation were approved by the
Institutional Animal Care and Use Committee of the Third Military
Medical University (Chongqing, China).
Statistical analysis
Quantitative data are presented as the means ±
standard deviation (SD). The Student's t-test was used to analyze
data following testing for normal distribution and
homoscedasticity. Statistical analysis was performed and the half
inhibitory concentration (IC50) of erlotinib was
calculated using GraphPad Prism 5 (GraphPad Software, Inc., San
Diego, CA, USA). A value of P<0.05 was considered to indicate a
statistically significant difference. All the experiments were
independently repeated at least 3 times.
Results
Lentivirus-mediated miR-223
οverexpression and IGF-1R expression in the PC-9 cell line
Lentiviral gene transfer is capable of inducing a
stable gain- and loss-of-function phenotype of cells. It is a
critical tool to explore miRNA function in cell culture and animal
models (23). In the present
study, lentiviral vectors overexpressing miR-223
(lentivirus-miR-223) and EV were transfected into the PC-9/ER
cells. The transfected cells were subsequently sorted using a
fluorescence activated cell sorter (FACS) based on the green
fluorescent protein (GFP) signal. Prior to sorting, the
transfection efficiencies were found to be approximately 35 and
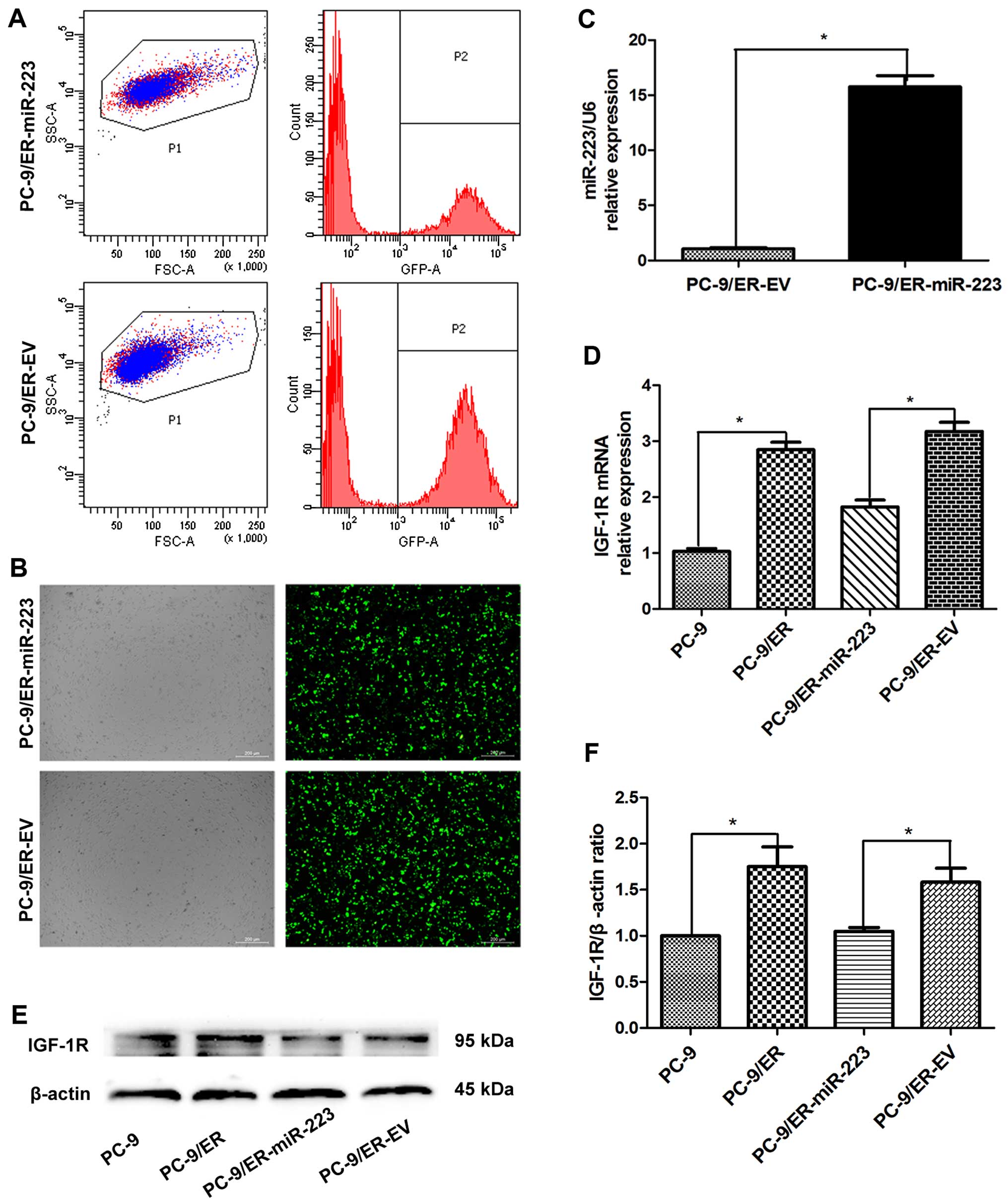
55%, respectively (Fig. 1A).
However, through FACS sorting, the GFP-positive subpopulation was
highly purified (purity >95%) (Fig. 1B), similar to our previous study
(15). Using RT-qPCR, the levels
of miR-223 were detected in the PC-9/ER-miR-223 and PC-9/ER-EV
cells. The expression of miR-223 in the miR-223 overexpression
group was upregulated approximately 16-fold compared with that in
the EV group (Fig. 1C). Using
RT-qPCR, the mRNA expression of IGF-1R was also detected in the 4
groups of PC-9 cells (Fig. 1D).
The expression of IGF-1R was revealed to be significantly increased
in the PC-9/ER cells versus the PC-9 cells (3.14-fold).
Furthermore, this upregulation was suppressed by the overexpression
of miR-223. The expression of total IGF-1R was also determined in
the PC-9 cells (Fig. 1E and F).
The increased expression of miR-223 in the PC-9/ER cells led to the
downregulation of IGF-1R at both the mRNA and protein level.
Overexpression of miR-223 enhances the
sensitivity of PC-9/ER cells to erlotinib by inducing
apoptosis
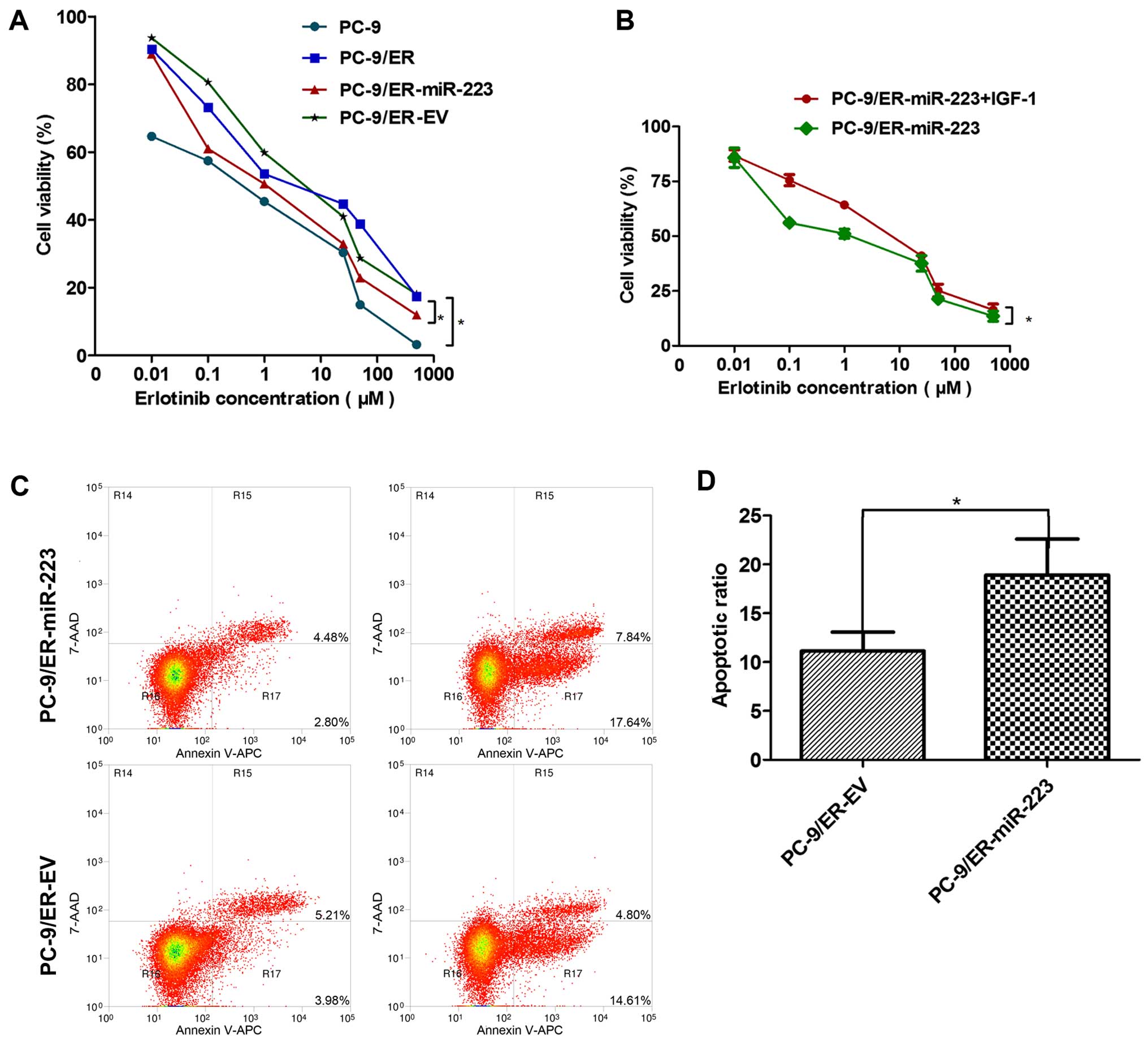
The CCK-8 assay revealed the sensitivity of the PC-9
and PC-9/ER cells (or miR-223 and EV) groups to erlotinib. The
PC-9/ER cells were clearly more resistant to erlotinib than the
PC-9 cells. The over-expression of miR-223 in the PC-9/ER cells
markedly enhanced the sensitivity to erlotinib. The IC50
values of the PC-9, PC-9/ER, PC-9/ER-miR-223 and PC-9/ER-EV cells
were: 0.25, 5.16, 1.19 and 5.29, respectively (Fig. 2A). However, the addition of 100
ng/ml of IGF-1 significantly re-enhanced the resistance of the
PC-9/ER-miR-223 cells to erlotinib. The IC50 values of
the PC-9/ER-miR-223 and PC-9/ER-miR-223 + IGF-1 cells were 1.04 and
4.29, respectively (Fig. 2B). The
differences were statistically significant (P<0.05). In order to
explore the mechanisms underlying this phenomenon, the apoptosis of
the cells was assessed. The οverexpression of miR-223 was revealed
to induce the apoptosis of the PC-9/ER cells treated with 1
µM erlotinib for 48 h. Following staining with Annexin V-APC
and 7-AAD, the number of apoptotic cells was detected and we
observed a significant difference in their percentage between the
groups (Fig. 2C). The differences
were statistically significant (P<0.05) (Fig. 2D).
miR-223 inhibits IGF-1R/Akt/S6 signaling,
and this effect is reversed by the exogenous expression of
IGF-1
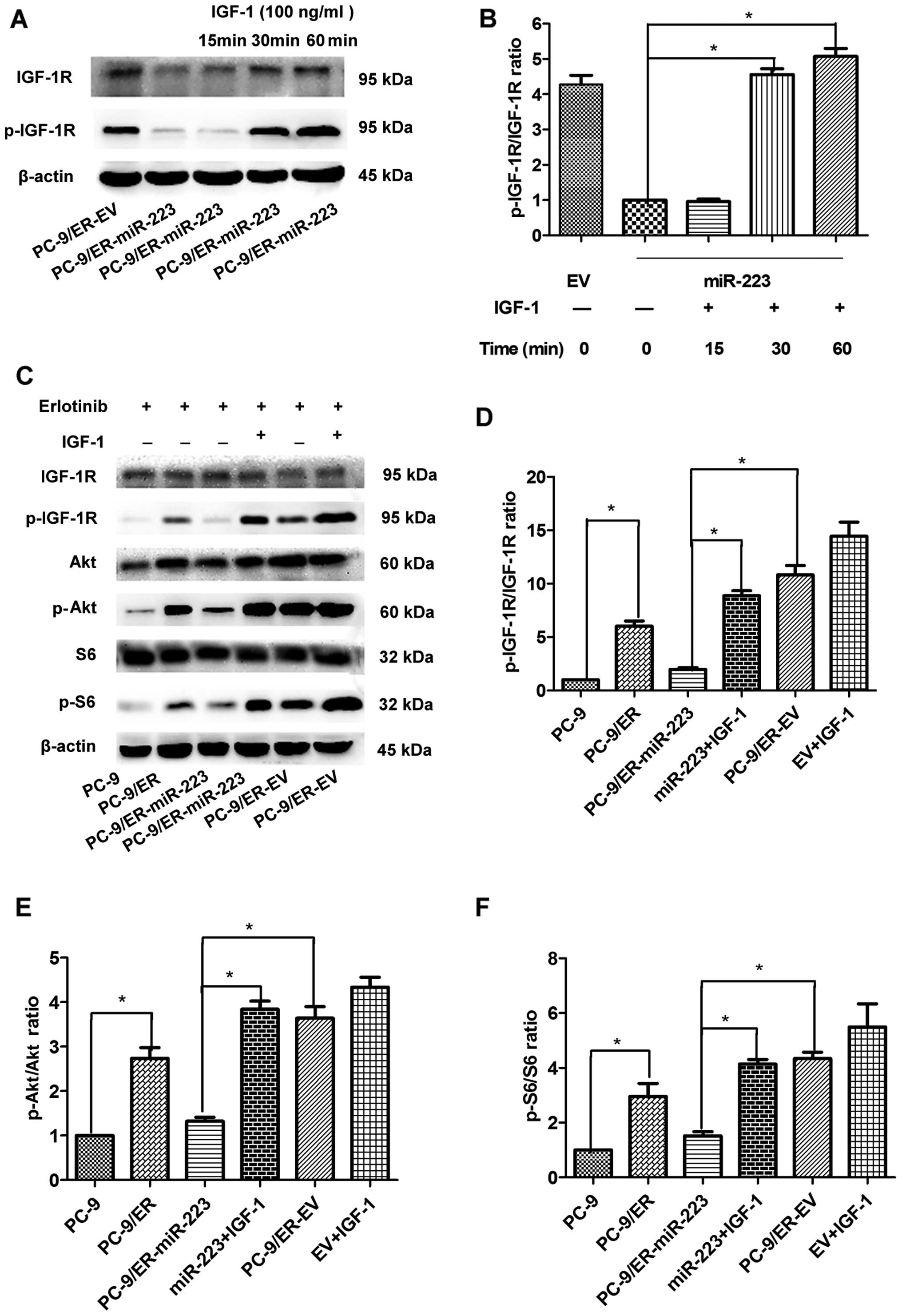
IGF-1R can be activated by the ligand IGF-1. The
concentration and exposure time to IGF-1 were determined as
follows: 60 min was the most appropriate duration to induce the
re-expression of p-IGF-1R in the PC-9 cell line. The most
appropriate concentration was 100 ng/ml (data not shown). We thus
performed the following experiments under this condition. The
levels of p-IGF-1R were significantly increased following
stimulation with IGF-1 (Fig. 3A and
B). As the mRNA and protein expression levels of IGF-1R were
suppressed by miR-223, we wished to determine whether the
IGF-1R-mediated downstream signaling pathway is also regulated by
miR-223. The levels of total Akt (t-Akt) were unaffected; however,
the levels of its active form (p-Akt) in the miR-223 overexpression
group were reduced by approximately 65% compared with those of the
EV group (Fig. 3E). Furthermore,
the levels of S6 and its active form, p-S6, were also detected. The
levels of p-S6 in the miR-223 group were markedly reduced by 45%
compared with the EV group; however, the levels of total S6 were
unaffected (Fig. 3F). The
suppression of the Akt/S6 signaling pathway was consistently
abolished by the exogenous expression of IGF-1. Therefore, miR-223
inhibited the IGF-1R/AKT/S6 signaling pathway by targeting IGF1R,
and this inhibitory effect was reversed by IGF-1 in vitro
(Fig. 3C).
miR-223 enhances the sensitivity to
erlotinib and inhibits tumor growth in nude mice
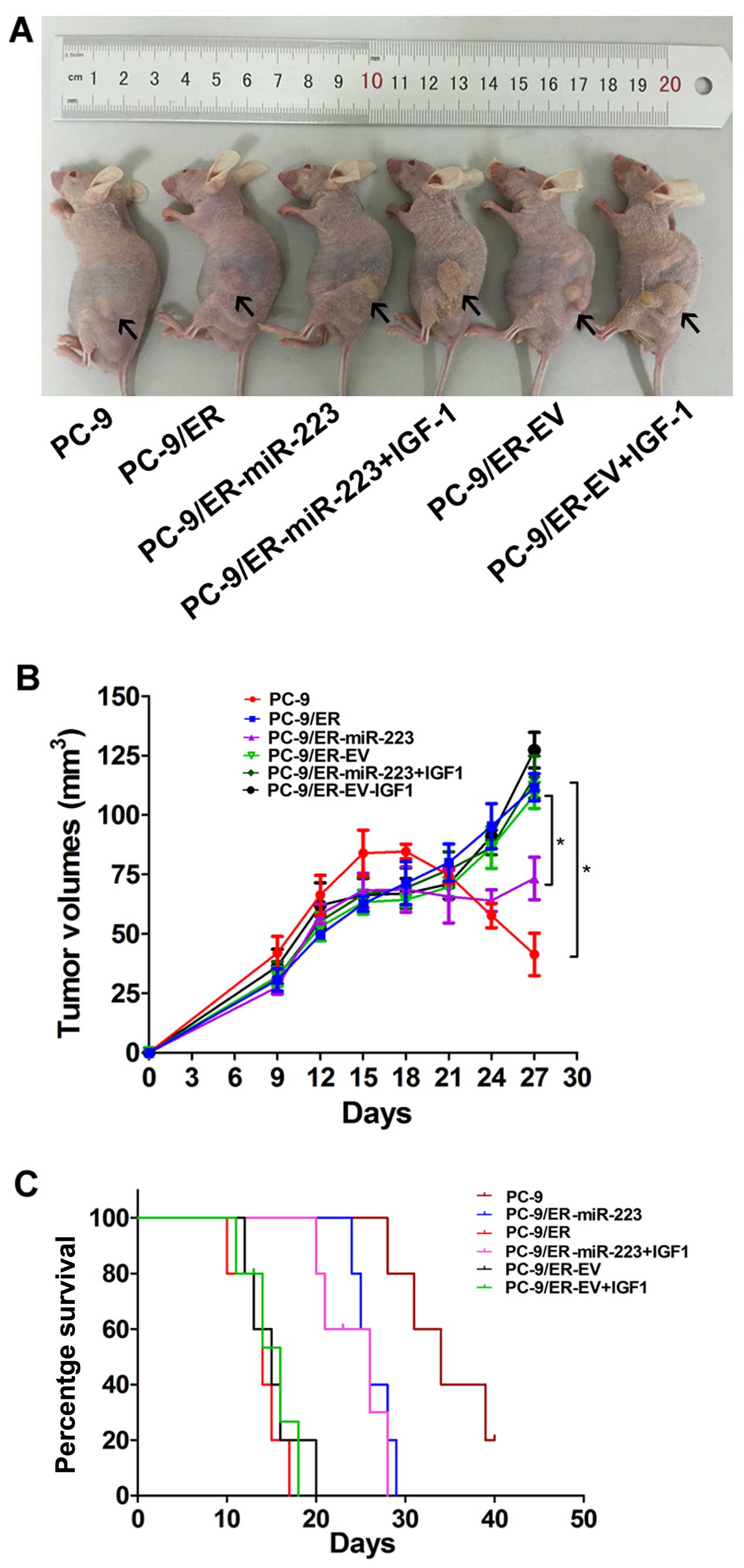
To further confirm the above findings, an in
vivo model was established by the subcutaneous injection of
2×106 PC-9/ER-miR-223- or PC-9/ER-EV-infected cells into
the mouse skins under the left lower quadrants. Tumor sizes in the
6 groups were measured every 3 days. The tumor mass became palpable
6–9 days following inoculation in all groups. All the nude mice
began to receive erlotinib treatment from the 14th day. Following
the intragastric administration of erlotinib for 2 weeks, all mice
were sacrificed and the tumor mass was measured (Fig. 4A). Tumor growth curves were
plotted using GraphPad Prism 5 (Fig.
4B). The tumor volume of the PC-9/ER-injected mice was
approximately 5-fold larger than that of the PC-9-injected mice.
The inhibition of tumor growth was observed in the PC-9/ER-miR-223
tumor-bearing nude mice. The tumor mass in the PC-9/ER-miR-223
group was 60% of the control (P<0.05). The PC-9/ER-miR-223 group
with IGF-1 intervention exhibited a significant difference in tumor
volume when compared with the control. The upregulation of miR-223
enhanced sensitivity to erlotinib and this was reversed by the
exogenous expression of IGF-1. In survival analysis, 30 mice were
used. Survival curves were produced using GraphPad Prism 5. A high
expression of IGF-1R correlated with a poor survival rate
(P<0.05). However, there was no difference between the exogenous
IGF-1 group and the PC-9/ER-EV group in survival (Fig. 4C).
Discussion
miRNAs have been reported to play important roles in
tumor initiation and progression (24). The up- or downregulation of miRNAs
are predominantly investigated in cancer biology. miR-223 was
initially identified and subsequently characterized in the
hematopoietic system (25).
Previous studies have revealed that miR-223 is expressed at a low
level in nasopharyngeal carcinoma cells (26), undifferentiated human embryonic
stem cells (hESCs) (27) and HeLa
cells (15). Furthermore, miR-223
often suppresses cell growth, colony formation and proliferation
(15). Numerous studies have
confirmed that miR-223 plays an important role in lung cancer
(28,29). However, its role in resistance to
TKIs in targeted molecular therapy in NSCLC remains unknown. The
mechanisms by which miR-223 regulates resistance to erlotinib also
remain to be elucidated. In the present study, we established a
lentivirus-based miR-223 overexpression system. Several important
methods were used to examine the association between miR-223 and
IGF-1R, and the downstream signaling pathway of miR-223.
In our previous study, we found that IGF-1R is a
target of miR-223 (16). A
significant difference was observed in the response to erlotinib
between the PC-9 and PC-9/ER cells, or the miR-223- and EV-infected
groups. We found that miR-223 was a positive regulator or promoter
of tumor cell apoptosis, which enhanced the sensitivity of the
cells to erloninib by targeting the IGF-1R/Akt/S6 signaling pathway
in vitro. In vivo, miR-223 overexpression enhanced
the sensitivity to erlotinib, as the tumor volume of mice injected
with miR-223-infected cells was lower than that of the mice
injected with PC-9/ER cells. In addition, the nude mice injected
with miR-223-infected cells survived longer than the control
mice.
Despite the promising effects of TKIs, the acquired
resistance must be overcome. The persistent activation of
downstream signaling pathways, particularly PI3K/Akt, is sufficient
to confer resistance to cells against EGFR-TKIs by bypassing EGFR
blocking (30). To elucidate the
underlying mechanisms involved in the miR-223-induced apoptosis of
PC-9/ER cells, we used different prediction algorithms to predict
the target genes of miR-223. IGF-1R was predicted to be one such
target. IGF-1R has been well characterized in resistance to TKIs in
NSCLC. The regulation of IGF-1R signaling is receiving increasing
attention in the development of targeted therapeutics to improve
human health. IGF-1R activates the Ras/Erk- and PI3K/Akt-associated
signal transduction pathways (31). The present study revealed that
IGF-1R expression was upregulated in the cells with acquired
resistance to erlotinib at the mRNA and protein level. By contrast,
the mRNA expression level of IGF-1R in H1975 cells with T790M
mutation was lower than that in PC-9/ER cells (data not shown).
These results suggested that IGF-1R may play an important role in
the acquired resistance to TKIs than in intrinsic resistance. In
addition, it was demonstrated that the binding of IGF-1 to IGF-1R
stimulated tumor growth by activating anti-apoptotic signaling
pathways.
Techniques to inhibit IGF-1R have been investigated
as promising therapeutic strategies against resistance to TKIs in
NSCLC. To date, IGF-1R antibodies, including dalotuzumab (MK-0646),
have been reported to be safe and able to reduce IGF-1R signaling
in phase I and II clinical trials. The clinical activity of IGF-1R
inhibitors has been demonstrated with sustained responses in a
number of patients. However, in adult patients with tumors,
including NSCLC, breast cancer and pancreatic cancer, have failed
to reveal the clinical benefit of IGF-1R inhibitors in the overall
patient population (32,33). In those studies, it was assumed
that the level of IGF-1R activation in patients correlated with the
efficacy of dalotuzumab. Therefore, we hypothesized that a highly
activate IGF-1R is essential for developing its functions. However,
in the present study, IGF-1R expression was much higher in the
PC-9/ER cells than in the PC-9 cells, indicating that TKI-resistant
patients are possibly suitable candidates for MK-0646. Patients
with TKI-resistant NSCLC may benefit from dual drug therapy with
EGFR-TKIs and dalotuzumab. High levels of circulating IGF-1 are
known to correlate with increased risks of breast, prostate and
colon cancers (34). IGF-1 is an
ubiquitously produced protein hormone, which interacts with IGF-1R
to regulate cell growth, differentiation and survival (35). Ligand binding fails to stimulate
the kinase activity of phosphorylated IGF-1R; however, it does
stimulate IGF-1R autophosphorylation (36). Removing or inhibiting the
secretion of IGF-1 in plasma may reverse cell tolerance to
TKIs.
In conclusion, the present study hypothesized that
combined EGFR/IGF-1R inhibition may improve the efficacy of
targeted molecular therapy in erlotinib-resistant NSCLC. IGF-1R is
a valid target for selected tumor types, including
erlotinib-resistant lung cancer with a low expression of miR-223.
By contrast, miR-223, an evolutionarily conserved miRNA, represents
a potential biomarker for erlotinib-resistant NSCLC. Thus, the
overexpression of miR-223 in TKI-resistant NSCLC may prove to be
beneficial. The present study described the role of the IGF/IGF-1R
system, and proposed additional novel strategies for targeting this
system. Strategies to target this system have also been proposed
previously (37). These results
indicate the great potential of miRNAs in the development of
therapeutics. Despite the limitations in delivery and toxicity,
this study may provide a vital miRNA target for combating
resistance to TKIs in the treatment of NSCLC.
Acknowledgments
The authors would like to express their gratitude to
Mr. Dian-Gang Chen (Cancer Institute of PLA, Xinqiao Hospital,
Third Military Medical University, China) for kindly providing the
H1975 cell line and instructing the experimental skills. We should
express our gratitude to the premium language editing service of
Spandidos Publications. The present study was supported by the
National Natural Science Foundation of China (grant no. 81172070),
the Chongqing Science and Technology Key Project Fund (grant no.
2011AB5032), the Military Clinical Key Projects of New and High
Technology (grant no. 2010gxjs070) and the Natural Science
Foundation of Chongqing (grant no. cstc2012jjA10096).
References
|
1
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Allemani C, Weir HK, Carreira H, Harewood
R, Spika D, Wang XS, Bannon F, Ahn JV, Johnson CJ, Bonaventure A,
et al CONCORD Working Group: Global surveillance of cancer survival
1995–2009: Analysis of individual data for 25,676,887 patients from
279 population-based registries in 67 countries (CONCORD-2).
Lancet. 385:977–1010. 2015. View Article : Google Scholar
|
|
3
|
Larsen JE and Minna JD: Molecular biology
of lung cancer: Clinical implications. Clin Chest Med. 32:703–740.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Morgillo F, Kim WY, Kim ES, Ciardiello F,
Hong WK and Lee HY: Implication of the insulin-like growth
factor-IR pathway in the resistance of non-small cell lung cancer
cells to treatment with gefitinib. Clin Cancer Res. 13:2795–2803.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Del Vescovo V and Denti MA: microRNA and
lung cancer. Adv Exp Med Biol. 889:153–177. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lagos-Quintana M, Rauhut R, Lendeckel W
and Tuschl T: Identification of novel genes coding for small
expressed RNAs. Science. 294:853–858. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Del Vescovo V, Grasso M, Barbareschi M and
Denti MA: MicroRNAs as lung cancer biomarkers. World J Clin Oncol.
5:604–620. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee CT, Risom T and Strauss WM: MicroRNAs
in mammalian development. Birth Defects Res C Embryo Today.
78:129–139. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bueno MJ, Pérez de Castro I and Malumbres
M: Control of cell proliferation pathways by microRNAs. Cell Cycle.
7:3143–3148. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jovanovic M and Hengartner MO: miRNAs and
apoptosis: RNAs to die for. Oncogene. 25:6176–6187. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mestdagh P, Hartmann N, Baeriswyl L,
Andreasen D, Bernard N, Chen C, Cheo D, D'Andrade P, DeMayo M,
Dennis L, et al: Evaluation of quantitative miRNA expression
platforms in the microRNA quality control (miRQC) study. Nat
Methods. 11:809–815. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Garofalo M, Quintavalle C, Di Leva G,
Zanca C, Romano G, Taccioli C, Liu CG, Croce CM and Condorelli G:
MicroRNA signatures of TRAIL resistance in human non-small cell
lung cancer. Oncogene. 27:3845–3855. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wong QW, Lung RW, Law PT, Lai PB, Chan KY,
To KF and Wong N: MicroRNA-223 is commonly repressed in
hepatocellular carcinoma and potentiates expression of Stathmin1.
Gastroenterology. 135:257–269. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jia CY, Li HH, Zhu XC, Dong YW, Fu D, Zhao
QL, Wu W and Wu XZ: MiR-223 suppresses cell proliferation by
targeting IGF-1R. PLoS One. 6:e270082011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nian W, Ao X, Wu Y, Huang Y, Shao J, Wang
Y, Chen Z, Chen F and Wang D: miR-223 functions as a potent tumor
suppressor of the Lewis lung carcinoma cell line by targeting
insulin-like growth factor-1 receptor and cyclin-dependent kinase
2. Oncol Lett. 6:359–366. 2013.PubMed/NCBI
|
|
17
|
Peled N, Wynes MW, Ikeda N, Ohira T,
Yoshida K, Qian J, Ilouze M, Brenner R, Kato Y, Mascaux C and
Hirsch FR: Insulin-like growth factor-1 receptor (IGF-1R) as a
biomarker for resistance to the tyrosine kinase inhibitor gefitinib
in non-small cell lung cancer. Cell Oncol (Dordr). 36:277–288.
2013. View Article : Google Scholar
|
|
18
|
Baserga R, Peruzzi F and Reiss K: The
IGF-1 receptor in cancer biology. Int J Cancer. 107:873–877. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pollak MN, Schernhammer ES and Hankinson
SE: Insulin-like growth factors and neoplasia. Nat Rev Cancer.
4:505–518. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Q, Zhao DY, Xu H, Zhou H, Yang QY,
Liu F and Zhou GP: Downregulation of microRNA-223 promotes
degranulation via the PI3K/Akt pathway by targeting IGF-1R in mast
cells. PLoS One. 10:e01235752015. View Article : Google Scholar
|
|
21
|
Yuan Y, Shen Y, Xue L and Fan H: miR-140
suppresses tumor growth and metastasis of non-small cell lung
cancer by targeting insulin-like growth factor 1 receptor. PLoS
One. 8:e736042013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Agulló-Ortuño MT, Díaz-García CV,
Agudo-López A, Pérez C, Cortijo A, Paz-Ares L, López-Ríos F, Pozo
F, de Castro J and López Martín JA: Relevance of insulin-like
growth factor 1 receptor gene expression as a prognostic factor in
non-small-cell lung cancer. J Cancer Res Clin Oncol. 141:43–53.
2015. View Article : Google Scholar
|
|
23
|
Scherr M, Venturini L and Eder M:
Lentiviral vector-mediated expression of pre-miRNAs and antagomiRs.
Methods Mol Biol. 614:175–185. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ryan BM, Robles AI and Harris CC: Genetic
variation in microRNA networks: The implications for cancer
research. Nat Rev Cancer. 10:389–402. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen CZ, Li L, Lodish HF and Bartel DP:
MicroRNAs modulate hematopoietic lineage differentiation. Science.
303:83–86. 2004. View Article : Google Scholar
|
|
26
|
Yang W, Lan X, Li D, Li T and Lu S:
MiR-223 targeting MAFB suppresses proliferation and migration of
nasopharyngeal carcinoma cells. BMC Cancer. 15:4612015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu YH, Zhang L, Wu DS, Zhang Z, Huang FF,
Zhang J, Chen XP, Liang DS, Zeng H and Chen FP: MiR-223 regulates
human embryonic stem cell differentiation by targeting the
IGF-1R/Akt signaling pathway. PLoS One. 8:e787692013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sanfiorenzo C, Ilie MI, Belaid A, Barlési
F, Mouroux J, Marquette CH, Brest P and Hofman P: Two panels of
plasma microRNAs as non-invasive biomarkers for prediction of
recurrence in resectable NSCLC. PLoS One. 8:e545962013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Heegaard NH, Schetter AJ, Welsh JA, Yoneda
M, Bowman ED and Harris CC: Circulating micro-RNA expression
profiles in early stage nonsmall cell lung cancer. Int J Cancer.
130:1378–1386. 2012. View Article : Google Scholar
|
|
30
|
Yamasaki F, Johansen MJ, Zhang D,
Krishnamurthy S, Felix E, Bartholomeusz C, Aguilar RJ, Kurisu K,
Mills GB, Hortobagyi GN and Ueno NT: Acquired resistance to
erlotinib in A-431 epidermoid cancer cells requires downregulation
of MMAC1/PTEN and upregulation of phosphorylated Akt. Cancer Res.
67:5779–5788. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bohula EA, Playford MP and Macaulay VM:
Targeting the type 1 insulin-like growth factor receptor as
anti-cancer treatment. Anticancer Drugs. 14:669–682. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Scartozzi M, Bianconi M, Maccaroni E,
Giampieri R, Berardi R and Cascinu S: Dalotuzumab, a recombinant
humanized mAb targeted against IGFR1 for the treatment of cancer.
Curr Opin Mol Ther. 12:361–371. 2010.PubMed/NCBI
|
|
33
|
Reidy-Lagunes DL, Vakiani E, Segal MF,
Hollywood EM, Tang LH, Solit DB, Pietanza MC, Capanu M and Saltz
LB: A phase 2 study of the insulin-like growth factor-1 receptor
inhibitor MK-0646 in patients with metastatic, well-differentiated
neuro-endocrine tumors. Cancer. 118:4795–4800. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pollak M: Insulin and insulin-like growth
factor signalling in neoplasia. Nat Rev Cancer. 8:915–928. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jameson MJ, Beckler AD, Taniguchi LE,
Allak A, Vanwagner LB, Lee NG, Thomsen WC, Hubbard MA and Thomas
CY: Activation of the insulin-like growth factor-1 receptor induces
resistance to epidermal growth factor receptor antagonism in head
and neck squamous carcinoma cells. Mol Cancer Ther. 10:2124–2134.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kavran JM, McCabe JM, Byrne PO, Connacher
MK, Wang Z, Ramek A, Sarabipour S, Shan Y, Shaw DE, Hristova K, et
al: How IGF-1 activates its receptor. Elife. 3:32014. View Article : Google Scholar
|
|
37
|
Brahmkhatri VP, Prasanna C and Atreya HS:
Insulin-like growth factor system in cancer: Novel targeted
therapies. BioMed Res Int. 2015:5380192015. View Article : Google Scholar : PubMed/NCBI
|