Introduction
Diabetic nephropathy (DN) is a major complication of
diabetes which causes thickening of the glomerular basement
membrane, glomerular hypertrophy and mesangial expansion, resulting
in overt renal disease. The filtration barrier of the kidney is
formed by the glomerulus, which is composed of a fenestrated
endothelium, glomerular basement membrane, and podocytes that form
slit diaphragms (1). When
podocytes are damaged, chronic kidney disease (CKD) and end-stage
renal disease (ESRD) occur. The terminally differentiated podocytes
(also known as glomerular visceral epithelial cells) are a group of
highly specialized cells, which are derived from mesenchymal cells
and possess a limited capacity to proliferate (2). Podocytes have been recognized as
critical regulators of glomerular injury and podocyte injury is
clinically characterized by proteinuria and podocyte loss, which
are observed as the apoptosis of podocytes. As podocytes are
terminally differentiated cells, their loss in association with
their inability to be replaced or regenerated, ultimately results
in glomerulosclerosis (2,3). Diabetic glomerular diseases remain
among the major causes of CKD and ESRD (4). With the increasing prevalence of
diabetes, DN and podocyte protection has drawn increasing
attention. Despite extensive research into understanding and
treating DN, investigating the application of herbal medicine in
the treatment of DN as well as the underlying mechanisms, remains a
challenging task.
Notoginsenoside R1 (NR1) is the major component of
Fufang Xiancao Keli (a Chinese herbal medicine compound), which has
been used clinically for the treatment of DN for centuries in China
(6). Previous studies have
demonstrated that NR1, a major component of Panax
notoginseng, attenuated renal ischemia-reperfusion injury in
rats (5). A study on the
application of NR1 in a rat model of DN indicated that NR1
ameliorated the glucose-induced impairment of podocyte adhesive
capacity and subsequent podocyte depopulation, partly through the
upregulation of α3β1 integrin (6). Gui et al showed that in rats
with streptozotocin (STZ)-induced diabetes, treatment with NR1 for
12 weeks partially restored the number of podocytes per glomerular
volume and glomerular α3β1 integrin expression (6). However, further exploration of the
mechanism responsible for NR1-induced podocyte protection is
urgently needed.
To maintain podocyte integrity, nephrin, podocin and
desmin have been demonstrated to play a pivotal role (7–9),
and their expression levels were further analyzed in this study.
Mounting evidence has suggested that there is a decrease in
podocyte number in diabetic glomerular disease (10–13). Accompanying the decreased podocyte
number, the consequences include proteinuria and
glomerulosclerosis. Particularly in DN, studies have shown that
proteinuria increased as the podocyte number decreased (12,14). Thus, in the present study we aimed
to examine the protective effects of NR1 in podocytes, initially
through the measurement of proteinuria. In addition, emerging
experimental and clinical literature suggests that apoptosis is a
major cause of reduced podocyte numbers, which ultimately leads to
proteinuria. Using terminal deoxynucleotidyl transferase-mediated
deoxyuridin triphosphate nick end labeling (TUNEL) staining and
quantifying the number of apoptotic cells per field, the protection
effects of NR1 in podocytes were further assessed. As demonstrated
by Schiffer et al, who were among the first to provide
evidence for the increased apoptosis of podocytes in experiments
performed in transgenic mice with elevated levels of tumor growth
factor-β1 (TGF-β1) (15), the
serum levels of TGF-β1 and tumor necrosis factor-α (TNF-α) were
analyzed. In addition, an increasing body of experimental and
clinical literature shows that the phosphoinositide 3-kinase
(PI3K)/Akt signaling pathway is widely expressed in eukaryotes and
plays essential roles in growth, differentiation, proliferation and
survival (16). Furthermore, a
study of puromycin aminonucleoside (PAN)-induced podocyte injury
revealed that the tyrosine phosphorylation of nephrin and the
nephrin-p85 interaction were reduced, which is indicative of
decreased Akt activity in podocytes (17). Another study investigating the
correlation between the PI3K/Akt signaling pathway and podocyte
numbers showed that dexamethasone inhibited podocyte apoptosis by
stabilizing the PI3K/Akt signaling pathway in vitro
(18). Thus, in the present
study, we aimed to further examine our hypothesis that NR1
ameliorates podocyte injury in rats with STZ-induced DN by
inhibiting the apoptosis of podocytes through the PI3K/Akt
signaling pathway.
Materials and methods
Chemicals
NR1 (chemical structure
C47H80O18; molecular weight, 933
Da) was purchased from Sigma-Aldrich Chemicals (St. Louis, MO,
USA), and the purity of NR1 was >98%. All other chemicals and
reagents were also purchased from Sigma-Aldrich Chemicals. Podocin
(#SC-21009), nephrin (#SC-19000), desmin (#SC-14026), NF-κB
(#sc-109) and β-actin (#sc-47778) antibodies were purchased from
Santa Cruz Biotechnology (Santa Cruz, CA, USA). Phosphorylated
(p-)PI3K-p85(Y458) (#4228), PI3K-p85 (#4257), p-Akt (S473) (#4060),
Akt (#4685) and p-NF-κB p65 (#3033) were purchased from Cell
Signaling Technologies (Danvers, MA, USA). Goat anti-rabbit
secondary antibody conjugated with HRP (#11-035-003) and goat
anti-mouse antibody conjugated with HRP (#211-035-109) were
purchased from Jackson ImmunoResearch Laboratories (West Grove, PA,
USA).
Experimental design and animal
groups
Six-week-old, healthy male Sprague-Dawley (SD) rats
(200–220 g) were obtained from and kept at the Animal Center of
Sichuan Provincial People's Hospital (Chengdu, China). The rats
were randomly divided into the following five groups (n=10): i)
normal control (NC) group of rats which received 0.1 mol/l citrate
buffer solution (control solution); ii) DN group of rats which
received an intraperitoneal (i.p.) injection of STZ (Sigma-Aldrich
Chemicals), dissolved in 0.1 mol/l citrate buffer (pH 4.5), at a
dosage of 65 mg/kg; iii) rats with DN treated with a low dose of
NR1 [5 mg/kg·day (6); DN + NR1
(low) group]; iv) DN rats treated with a medium dose of NR1 [10
mg/kg·day (6); DN + NR1 (medium)
group]; and v) DN rats treated with a high dose of NR1 [20
mg/kg·day (5); DN + NR1 (high)
dose group]. NR1 was administered orally.
For the DN group of rats, 3 days after the injection
of STZ, their fasting blood glucose levels were measured using a
One Touch Ultra Blood Glucose meter (LifeScan Inc., Milpitas, CA,
USA). Animals with fasting blood glucose levels >16.7 mmol/l
were considered to be diabetic rats, and were used in the present
study. NR1 was administered after the induction of diabetes, and
the control group (DN group) was given an equivalent volume of
vehicle. Every 4 weeks, fasting blood glucose levels and body
weight were measured, and 24 h urine samples were also collected in
metabolic cages, starting at 3 days after STZ injection. All
animals were euthanized by CO2 inhalation at 16 weeks
after the initiation of NR1 or vehicle treatment. At the end of the
experiment and prior to sacrifice, blood samples were obtained from
the inferior vena cava, and the kidneys were harvested immediately.
Animal handling was performed in accordance with the Ethics
Committee of Sichuan Provincial People's Hospital, and all animals
were kept under a 12 h light/dark cycle with free access to water
and food.
Assessment of renal function
To assess renal function, blood urea nitrogen (BUN)
and serum creatinine levels were determined using an automatic
biochemistry analyzer (Hitachi, Tokyo, Japan). Urinary albumin
concentrations were measured using an enzyme-linked immunosorbent
assay (ELISA) kit (Runyu Biotechnology Co., Shanghai, China),
according to the manufacturer's instructions.
Histological analysis
The renal samples were fixed in 10% formalin for 24
h at room temperature, and histological slides were prepared
according to the standardized protocol in our laboratory (19). The sections were then stained with
periodic acid-Schiff (PAS) and Trichrome reagent (Masson kit) (both
from Sigma-Aldrich Chemicals), and examined under a microscope
(Nikon 80i microscope; Nikon, Tokyo, Japan) in a blinded manner.
The extent of renal injury was estimated by the morphometric
assessment of the degree of mesangial matrix expansion and of
glomerular enlargement. A point-counting method was used to
quantify mesangial matrix deposition, and 20 randomly selected
non-overlapping PAS-stained glomeruli from each rat were analyzed
(20).
Evaluation of apoptosis by the TUNEL
assay
Paraffin-embedded kidney tissues were cut into 4
µm thick sections. A TUNEL assay was performed to assess DNA
fragmentation. The assay was performed according to the
manufacturer's instructions (Promega, Madison, Wl, USA) as
previously described (21). The
total cell population and the number of TUNEL-positive cells were
manually counted, and double checked using Image Pro Plus 6.3
software (Media Cybernetics, Silver Spring, MD, USA).
Measurement of cytokines
For measuring pro-inflammatory mediators [TNF-α,
TGF-β1, interleukin (IL)-1 and IL-6] and an anti-inflammatory
mediator (IL-10) in the serum, blood samples were collected at the
beginning and end of NR1 treatment. The serum cytokines were
assayed using specific ELISA kits for rats according to the
manufacturer's instructions (R&D Systems, Minneapolis, MN,
USA).
Western blot analysis
The proteins were lysed in 50 mM Tris-HCl, pH 8.0,
150 mM NaCl, 5 mM ethylenediaminetetraacetic acid (EDTA), 1% NP-40,
and protease inhibitor cocktail (22). The protein lysates were
centrifuged at 13,400 × g for 10 min. Supernatants were collected
and loaded on a NuPage Novex 10% Bis-Tris gel (Life Technologies,
Carlsbad, CA, USA) for electrophoresis. Following electrophoresis,
the proteins were transferred onto polyvinylidene fluoride
membranes (Pall Corporation, Port Washington, NY, USA) in NuPage
transfer buffer (Life Technologies). The membranes were blocked
with 5% bovine serum albumin in TBST for 1 h. The blots were
incubated overnight at 4°C with primary antibodies at the dilutions
recommended by the manufacturer, followed by incubation with a
horseradish peroxidase (HRP)-conjugated secondary antibody.
Chemiluminescence detection was performed using Immobilon Western
Chemiluminescent HRP substrate (Millipore, Billerica, MA, USA), and
measured directly by a BioSpectrum Imaging System (UVP, Upland, CA,
USA). Equal protein loading was confirmed by immunostaining against
β-actin. The signal intensity was analyzed by ImageQuant software
(Molecular Dynamics, Sunnyvale, CA, USA) and normalized to
β-actin.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from samples of isolated rat
glomeruli using TRIzol reagent (Life Technologies). Reverse
transcription was performed using a high capacity cDNA reverse
transcription kit according to the manufacturer's instructions
(Life Technologies). Two-step RT-qPCR analysis of the expression of
nephrin, podocin and desmin was performed using Applied Biosystems
Power SYBR®-Green PCR master mix (Life Technologies)
with 40 cycles of 45 sec at 95°C and 1.5 min at 55°C on an Applied
Biosystems 7900 Real-Time PCR system using primers as described
previously (23–25). Fluorescence data were collected at
55°C after each cycle. After the final cycle, melting curve
analysis of all samples was conducted within the range of 55–95°C.
Relative quantification of gene expression was performed using
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal
control. The threshold cycle and 2−ΔΔCt method were used
for calculating the relative amount of the target RNA (26). RT-qPCR always included a
no-template sample as a negative control. The experiments were
repeated twice, and each sample was tested in triplicate.
Statistical analysis
The statistical analyses were performed using
GraphPad Prism (version 5.0 for windows) statistical software
package (GraphPad Software, San Diego, CA, USA). All results are
expressed as the means ± standard error of mean (SEM) for each
group. One-way analysis of variance (ANOVA) and the Mann-Whitney U
test were used to determine the level of significance among the
groups, and P<0.05 was considered to indicate a statistically
significant difference.
Results
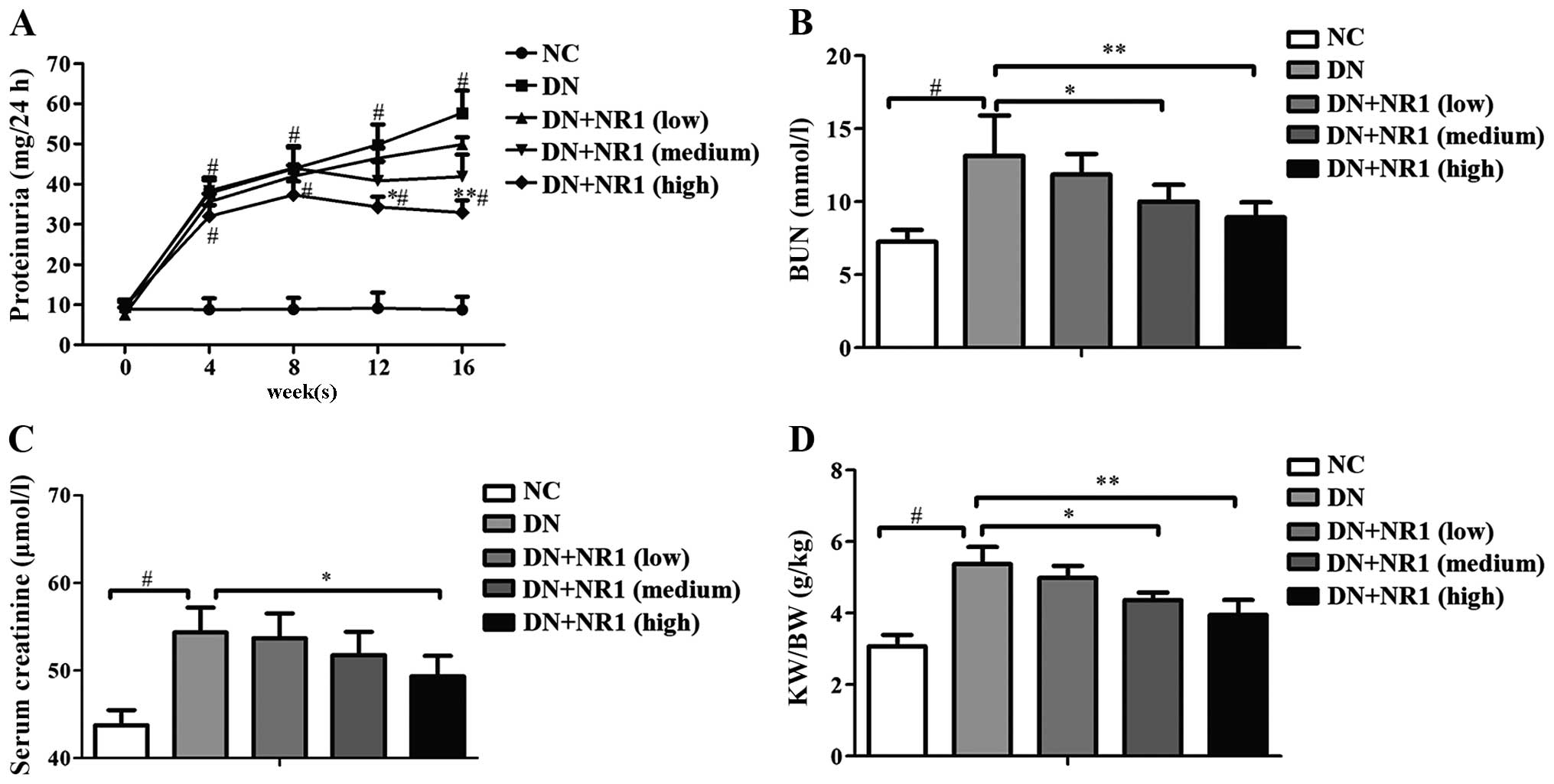
NR1 treatment reduces proteinuria in rats
with DN
To assess the renal protective effect of NR1,
fasting blood glucose levels and body weights were measured every 4
weeks starting from 3 days after STZ injection. As shown in
Table I, the rats in the DN group
demonstrated elevated blood glucose levels, which were
significantly higher than those of the non-diabetic rats
(P<0.05). During the study period, a low dose of NR1 did not
induce a decrease in blood glucose levels (P>0.05), whereas with
the elevated dosage of NR1, the blood glucose levels were
significantly lower than those of the DN group (P<0.05),
particularly from 12 weeks onwards. Moreover, the body weight of
the NR1-treated (medium dose and high dose) rats increased
steadily, compared with those of the rats in the DN group
(P<0.05) (Table II).
Furthermore, general parameters, namely BUN, serum creatinine, the
kidney-to-body weight ratio and proteinuria were measured. With the
progression of DN, proteinuria was increased in the diabetic rats
(P<0.05) (Fig. 1A). As
expected, the low dose NR1-treated rats did not show significantly
decreased proteinuria levels (P>0.05), and neither did the
medium dose NR1-treated rats. However, the proteinuria levels of
rats treated with a high dose of NR1 were significantly lower than
those of the rats in the DN group. Further evidence of NR1-induced
podocyte protection was obtained from the BUN concentrations
(Fig. 1B) and serum creatinine
levels (Fig. 1C). As expected,
the kidney-to-body weight ratios increased approximately 2-fold in
the rats of the DN group compared with those of the non-diabetic
controls, whereas in those treated with NR1, those ratios were
decreased significantly (Fig. 1D)
(P<0.05 for the medium and the high dose NR1-treated groups vs.
the DN group). The above data suggest that NR1 treatment
significantly reduced proteinuria and protected podocytes in the
rats with experimentally-induced diabetes.
 | Table IEffect of treatment on blood glucose
levels in all experimental groups (mmol/l, means ± SEM). |
Table I
Effect of treatment on blood glucose
levels in all experimental groups (mmol/l, means ± SEM).
| Group | 0 week | 4 weeks | 8 weeks | 12 weeks | 16 weeks |
|---|
| NC |
5.82±0.12 |
5.56±0.26 |
5.23±0.16 |
5.44±0.09 |
5.47±0.14 |
| DN | 25.13±2.22a | 26.48±1.57a | 27.11±3.28a | 28.54±2.14a | 26.42±2.64a |
| DN + NR1 (low) | 24.98±1.35 | 25.73±2.33 | 26.75±2.50 | 25.38±1.79 | 25.10±2.34 |
| DN + NR1
(medium) | 25.06±2.16 | 24.96±1.89 | 23.67±1.84 | 21.43±2.17b | 18.62±1.59c |
| DN + NR1
(high) | 24.92±1.05 | 25.73±2.15 | 21.58±1.49b | 16.35±3.26c | 13.75±3.64c |
| Table IIEffect of treatment on body weight in
all experimental groups (g, means ± SEM). |
Table II
Effect of treatment on body weight in
all experimental groups (g, means ± SEM).
| Group | 0 week | 4 weeks | 8 weeks | 12 weeks | 16 weeks |
|---|
| NC | 214±26 | 389±31 | 459±26 | 516±34 | 547±48 |
| DN | 210±23 | 218±17a | 215±18b | 207±11b | 198±13b |
| DN + NR1 (low) | 218±13 | 220±21 | 218±15 | 216±19 | 219±25 |
| DN + NR1
(medium) | 216±21 | 218±23 | 225±19 | 237±28c | 239±36d |
| DN + NR1
(high) | 218±18 | 229±17 | 239±15c | 245±15d | 258±19d |
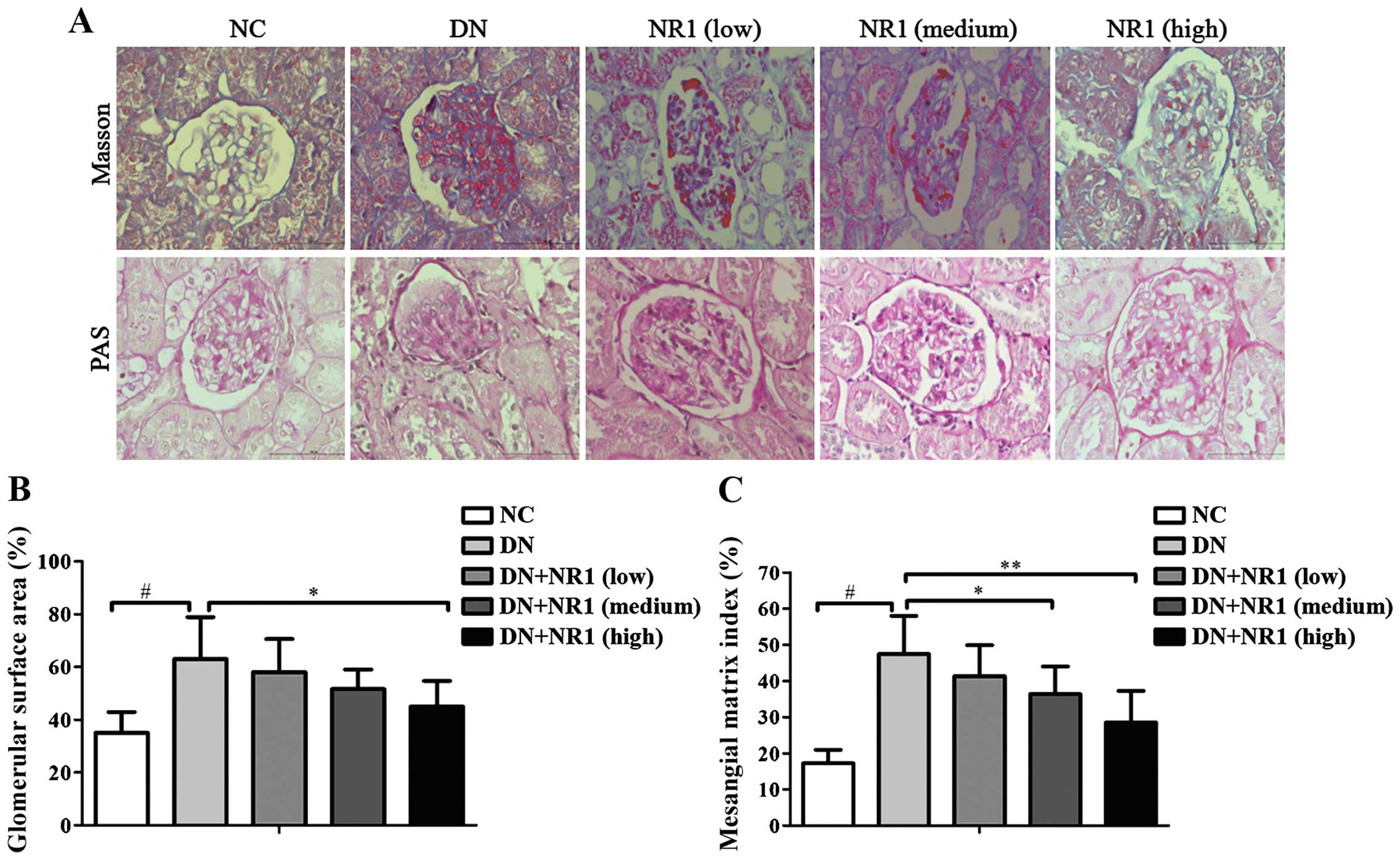
NR1 treatment ameliorates the
diabetes-induced histological alterations in rat kidneys
To further analyze the efficacy of NR1 in
controlling DN in STZ-exposed rats, the histological changes in
each group of rats were observed under a light microscope. As shown
in Fig. 2A [upper panels, Masson
staining; lower panels, PAS staining] as well as Fig. 2B and C, compared with the
non-diabetic control group, the glomerular surface area increased
2-fold and the glomerular mesangial matrix expanded significantly
in the DN group (P<0.05, DN group vs. NC group). The
quantitative histological analysis revealed that with the increased
dosage of NR1, the percentage of the glomerular surface area
decreased significantly (Fig. 2B)
(P<0.05, high dose NR1-treated group vs. DN group). Moreover,
the expansion of the glomerular mesangial matrix in the NR1-treated
groups (medium dose and high dose) decreased as well (Fig. 2C) (P<0.05, medium and high dose
NR1-treated groups vs. DN group). The above-mentioned histological
evaluation confirmed that NR1 treatment ameliorates the structural
changes in the diabetic kidney.
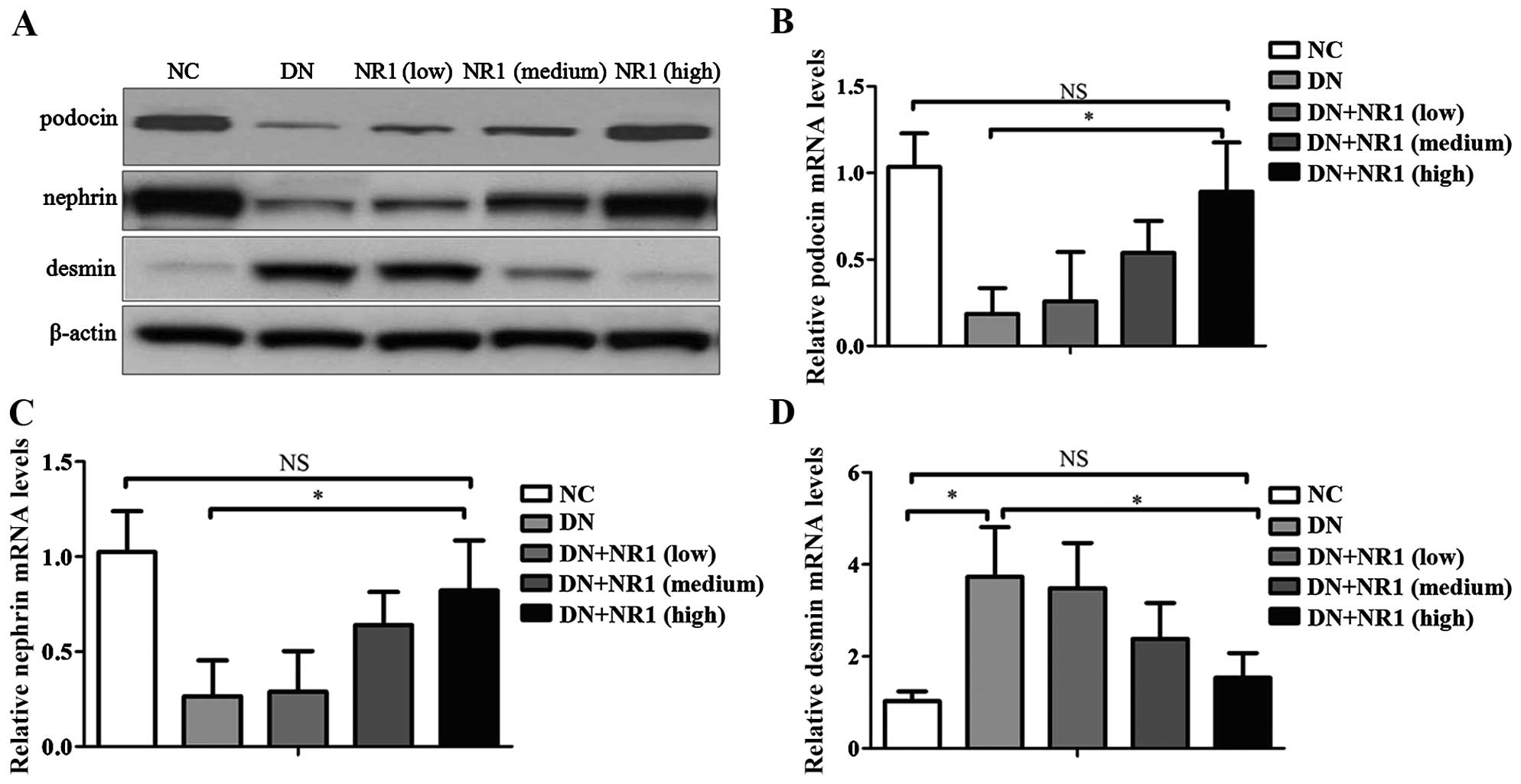
NR1 induces changes in the expression
levels of nephrin, podocin and desmin in the kidneys of rats with
DN
Previous studies have demonstrated that nephrin and
podocin are important slit diaphragm proteins in podocytes,
therefore they are of vital importance in the maintenance of
podocyte integrity (27). In the
present study, in order to elucidate the mechanism responsible for
the NR1-mediated improvement in the glomerular barrier function of
rats with DN, we quantified the expression levels of nephrin and
podocin using western blot analysis and RT-qPCR. Western blot
analysis revealed a reduction in the expression of nephrin and
podocin proteins in the rats with DN, whereas with the
administration of increasing doses of NR1, the protein levels of
nephrin and podocin were markedly restored (Fig. 3A). In addition, the mRNA levels of
nephrin and podocin were in agreement with the protein expression
levels, indicating that both nephrin and podocin were involved in
NR1-medicated podocyte protection (Fig. 3B and C). Moreover, in order to
elucidate the possibility that the protective effect of NR1
occurred through the maintenance of podocyte integrity, we examined
the expression levels of desmin, a marker of podocyte injury.
Judging from Fig. 3A and D, the
expression level of desmin was hardly observed in the podocytes of
non-diabetic rats; however, it increased by almost four times
compared with that in the normal control (P<0.05) (Fig. 3D). As expected, NR1 treatment
markedly decreased desmin expression levels, as shown by the
results of western blot analysis (Fig. 3A) and RT-qPCR (Fig. 3D). These results indicated that
NR1 protected against podocyte injury by increasing the expression
of nephrin and podocin, and by decreasing the expression of desmin
in rats with DN.
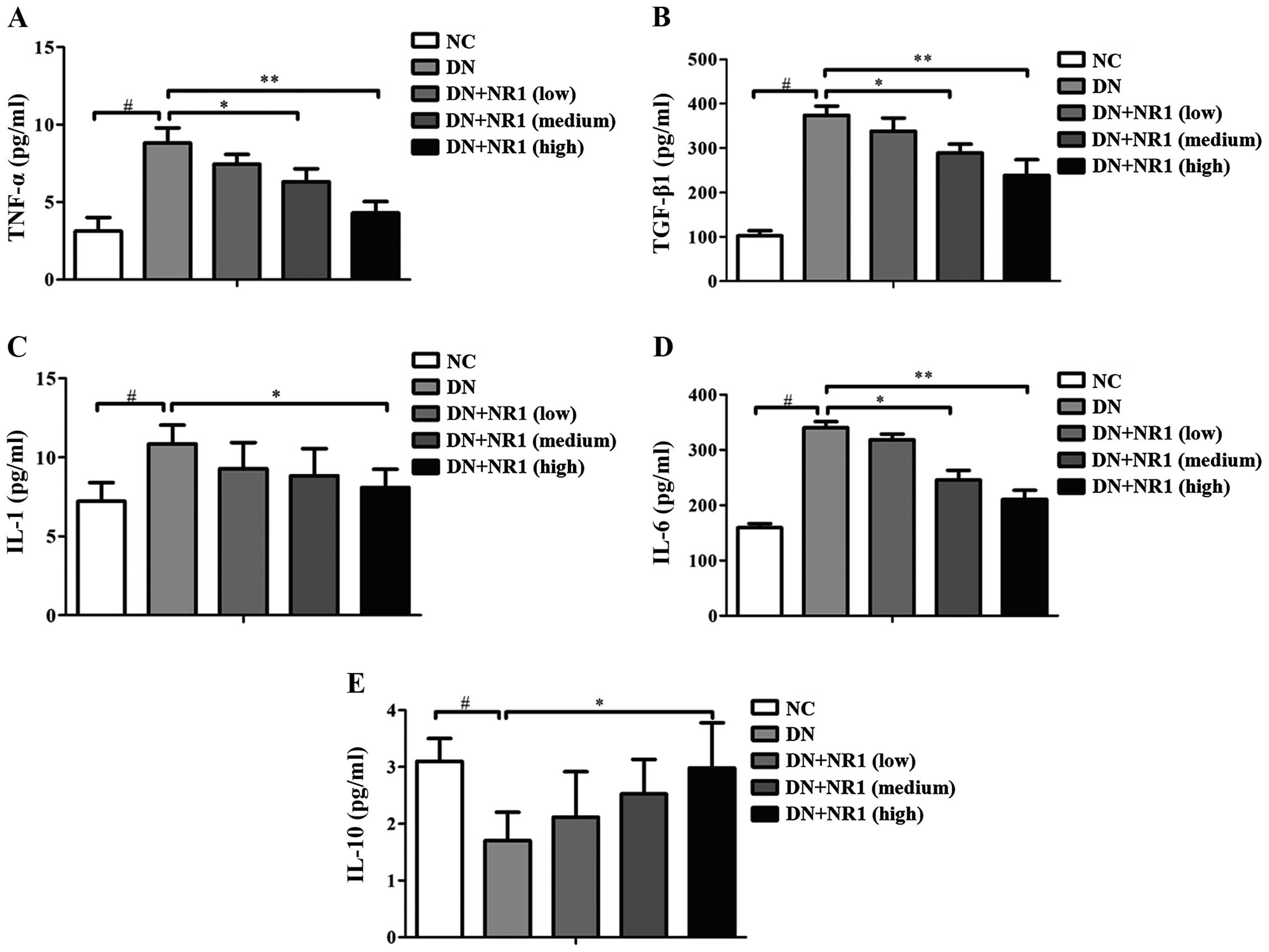
NR1 treatment inhibits the inflammatory
response in rats with DN
In order to evaluate the possible involvement of NR1
in the regulation of inflammatory cytokines in DN rats, we further
analyzed the secretion of inflammatory cytokines (TNF-α, TGF-β1,
IL-1 and IL-6) and an anti-inflammatory cytokine (IL-10) in the
serum. As shown in Fig. 4, the
secretion of TNF-α (Fig. 4A),
TGF-β1 (Fig. 4B), IL-1 (Fig. 4C) and IL-6 (Fig. 4D) was increased in the serum of
rats in the DN group, compared with the NC groups, and the
secretion of IL-10 (Fig. 4E) was
decreased in the serum of rats with DN compared with the
non-diabetic rats. However, following treatment with NR1, the
levels of the inflammatory cytokines were significantly suppressed
by NR1, whereas IL-10 levels were elevated. Taken together, our
data indicated that NR1 treatment significantly suppressed the
secretion of inflammatory cytokines and increased the secretion of
an anti-inflammatory cytokine in rats with DN.
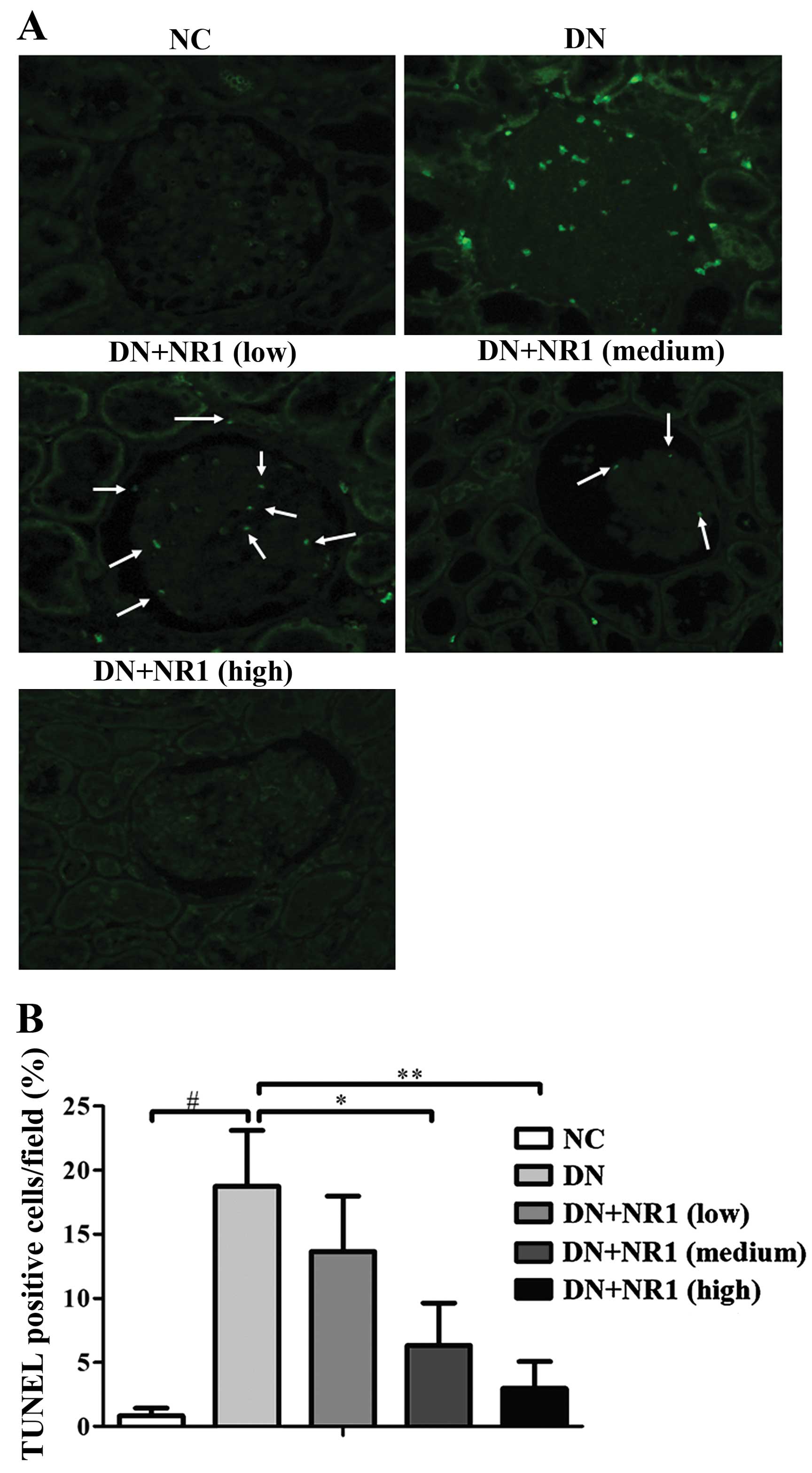
NR1 treatment ameliorates the
diabetes-induced apoptosis of podocytes
To determine whether the protective effect of NR1 in
podocytes was due to the amelioration of apoptosis, we performed
TUNEL staining in the kidneys of the experimental rats. As shown in
Fig. 5A, no positive signals were
present in the glomeruli of the normal control rats and the high
dose NR1-treated rats (Fig. 5A).
In addition, there were significantly reduced numbers of
TUNEL-positive cells in the kidneys of low and medium dose
NR1-treated rats. The percentages of TUNEL-positive cells indicated
that NR1 treatment decreased the diabetes-associated apoptosis of
renal cells as compared with the diabetic control (Fig. 5B). Thus, these results suggest
that NR1 treatment contributed to the podocyte protection by
inhibiting the apoptosis of podocytes under our experimental
conditions.
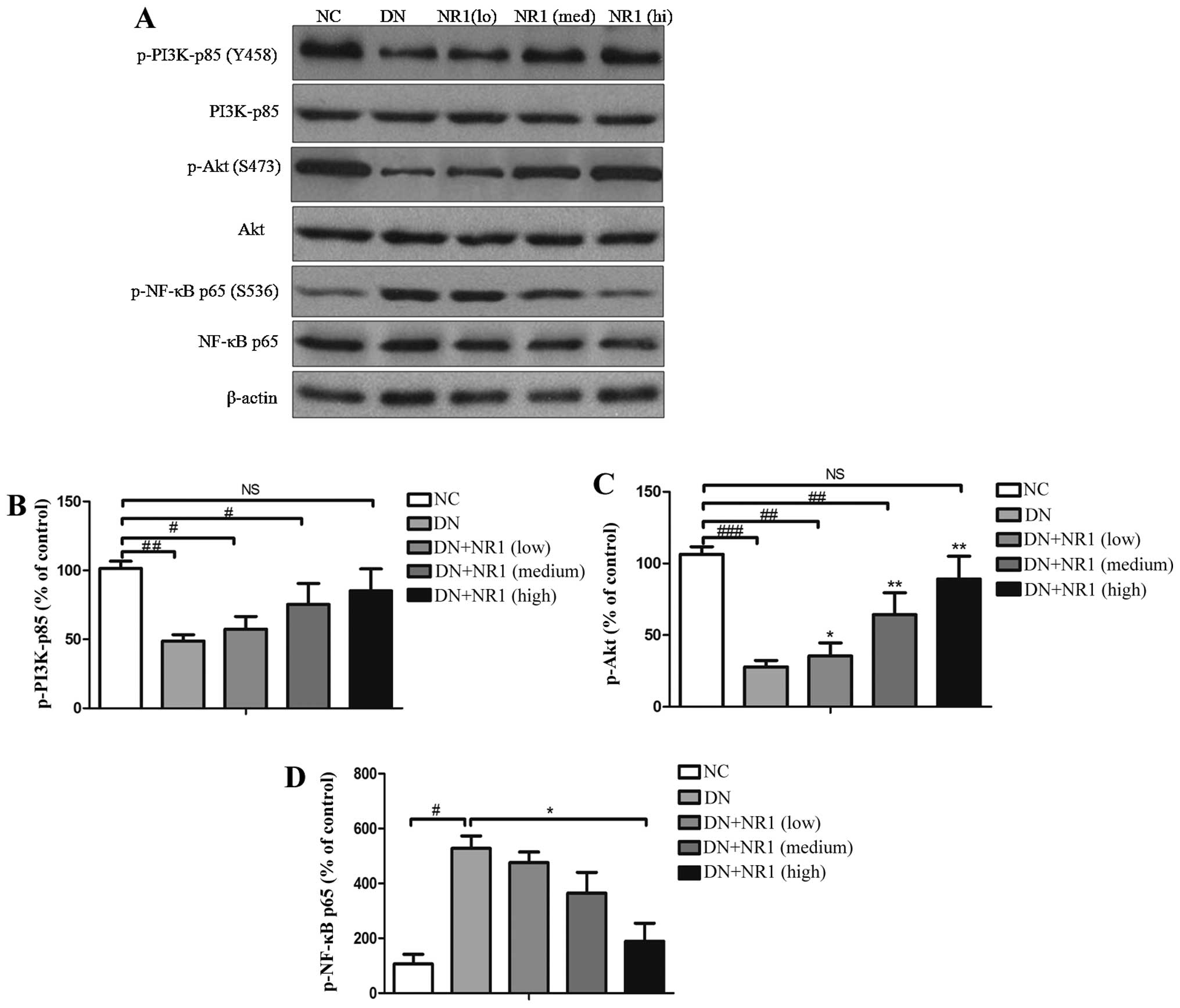
NR1 treatment induces activation of the
PI3K/Akt signaling pathway in rats with DN
In order to explore the mechanisms responsible for
the NR1-mediated inhibition of apoptosis and the suppressed
inflammatory response in rats with DN, we examined whether the
PI3K/Akt signaling pathway is suppressed in the podocytes of
STZ-exposed rats and whether NR1 is capable of reversing these
changes. Western blot analysis of p-PI3K-p85 (Tyr458), total
PI3K-p85, p-Akt (Ser473) and total Akt was performed, and it
indicated that the phosphorylation of both PI3K-p85 and Akt was
attenuated in the DN groups, compared with that of the NC control
group (Fig. 6A). Further
quantification of the grey scale of each blot was performed and the
ratio of p-p85 vs. total PI3K-p85 protein was calculated. As shown
in Fig. 6B, NR1 gradually
increased the phosphorylation of p85, in a dose-dependent manner.
Furthermore, the enhanced phosphorylation of Akt was found in the
NR1-treated rats with DN (P<0.05, compared with DN group),
whereas the expression level of p-Akt in the rats with DN was
significantly suppressed (P<0.05, compared with NC group)
(Fig. 6C). The phosphorylation
data of both p85 and Akt demonstrated that the PI3K/Akt signaling
pathway was suppressed in the rats with DN, whereas NR1 protected
podocytes and improved proteinuria by activating the PI3K/Akt
signaling pathway.
NR1 treatment inhibits nuclear factor-κB
(NF-κB) p65 phosphorylation in rats with DN
Mounting evidence suggests that diabetes activates
NF-κB signaling, which regulates the expression of several genes
involved in the inflammatory response (28). Thus, to further evaluate the
mechanism of NR1-induced podocyte protection, western blot analysis
was employed to assess NF-κB activation by evaluating the
expression of total p65 and the extent of p65 activation. In the
kidneys of rats with STZ-induced diabetes, increased
phosphorylation of p65 was observed, compared with the non-diabetic
rats (Fig. 6A). However,
following treatment with NR1, p65 phosphorylation was significantly
decreased (Fig. 6D), indicating
the inactivation of NF-κB. Taken together, these findings suggest
that NR1 induced podocyte protection, apoptosis inhibition, and
inflammation suppression by activating the PI3K/Akt signaling
pathway and downregulating NF-κB.
Discussion
According to epidemiologic observations, the
prevalence of diabetes has increased significantly in recent
decades all over the world (29).
Of all diabetic complications, DN has drawn special focus, as the
incidence of end-stage renal failure in diabetic patients has
markedly increased in recent years (30). The high prevalence of the disease
calls for intense research into ways of protecting podocytes, which
have been recognized as critical regulators of glomerular injury
(14). As the available treatment
options for glomerular injury are limited to alkylating agents,
steroids, angiotensin-converting enzyme inhibitors and angiotensin
receptor blockers, exploring herbal medicine has attracted
particular attention (31). A
study by Gui et al (6)
demonstrated that NR1 was involved in the upregulation of α3β1
integrin expression and the inhibition of oxidative stress;
however, the mechanism responsible for NR1-induced podocyte
protection remains to be elucidated. In the present study, NR1
treatment significantly decreased proteinuria, plasma and urinary
biomarkers of renal function as well as blood glucose levels. The
plasma and urinary biochemical results indicate that NR1
ameliorates the diabetes-associated compromised renal function.
Furthermore, histological evaluation of the rat kidneys confirmed
that NR1 significantly ameliorated diabetes-induced structural
damage in a dose-dependent manner. More importantly, this is the
first study, to the best of our knowledge, to delineate the
protective role of NR1 against podocyte injury by activating the
PI3K/Akt signaling pathway in rats with DN.
Considerable evidence indicates that nephrin is a
podocyte protein crucial for the inter-podocyte slit membrane
structure and the maintenance of an intact filtration barrier
(32). It has been demonstrated
that the modulation of nephrin expression contributes to the loss
of glomerular filtration function, and is associated with the
extent of proteinuria in DN (33). Similarly, the injection of
anti-nephrin antibody in animals induced substructural alterations
of the slit diaphragm with reductions in permselectivity and
consequently proteinuria (34).
In addition, the inactivation of the nephrin gene in mice by
homologous recombination resulted in severe proteinuria, and
partial foot process effacement (35). Thus, therapies targeted at
correcting podocyte nephrin may be of value in the management of
diabetes (36). It was reported
that podocin is a member of the stomatin family, and it consists of
hairpin-like integral membrane proteins. Further studies have
demonstrated that podocin is expressed in glomerular podocytes, and
serves in the structural organization of the slit diaphragm and the
regulation of its filtration function (37,38). Thus, podocin serves as a
scaffolding molecule to localize nephrin, and the disruption of
slit diaphragm proteins such as nephrin and podocin plays a
significant role in the development and progression of DN. In the
present study, we observed that in rats with STZ-induced DN, the
expression of nephrin and podocin was decreased significantly,
which was indicative of the disruption of the slit diaphragm
proteins. However, when the rats with DN were treated with NR1, the
major component of P. notoginseng, the expression of both
nephrin and podocin was enhanced, which indicated that NR1 induced
podocyte protection by regulating the expression of nephrin and
podocin. In addition to nephrin and podocin, desmin, an
intermediate filament protein, is also regulated in response to
podocyte injury. Zou et al showed that the upregulation of
desmin may increase the mechanical stability of cells, thus
enabling podocytes to undergo morphological changes on the tensile
glomerular capillary wall (39).
Our results indicated that the expression level of desmin increased
significantly in the rats with DN, and NR1 treatment markedly
decreased desmin expression. These results suggest that NR1
protects podocytes from injury, by affecting the expression of slit
diaphragm proteins in the rats with DN.
Mounting evidence, ranging from in vitro
experiments to pathological examinations, have highlighted that
inflammation plays a significant role in the development of DN
(40,41). Thus, the inhibition of
inflammatory processes is one of the cardinal mechanisms of
NR1-induced podocyte protection in DN. It has been reported that
the renal expression of IL-1 is increased in animal models of DN
(42). Moreover, researchers have
demonstrated the strong association between mesangial expansion and
the mRNA expression of IL-6 in podocytes, indicating that IL-6
affects the extracellular matrix dynamics of podocytes (43). A clinical study of IL-6 serum
levels demonstrated that IL-6 secretion was substantially higher in
patients with DN (44). Thus, the
expression of IL-1 and IL-6 was elevated in these models of DN,
which is in accordance with our results. Additionally, our results
demonstrated that the rats with DN receiving NR1 therapy showed
decreased serum levels of these two cytokines, indicating that NR1
exerts anti-inflammatory effects. TNF-α is a cytokine with
prominent inflammatory effects, and it is thought to play a pivotal
role in the pathogenesis of DN. It has been reported that TNF-α
levels are implicated in the development of renal hypertrophy and
hyperfunction during the initial stage of DN (45). Accumulating clinical studies have
revealed that the serum and urinary concentrations of TNF-α in
patients with DN are substantially higher than in non-diabetic
individuals (46). As expected,
in the present study, NR1-treated rats with DN exhibited decreased
serum TNF-α levels compared with the rats with DN treated with
vehicle. Moreover, it has been reported that accumulated TGF-β1
appears to play a pivotal role in the process of DN (47); apoptosis is increased in TGF-β
transgenic mice, which leads to a decrease in podocyte number and
glomerulosclerosis. Follow-up studies showed that TGF-β1-induced
apoptosis of podocytes was mediated by specific Smad pathways
(15,48). Thus, evaluating TGF-β1 secretion
levels in the rats with DN and the NR1-treated rats with DN is of
vital importance. In this study, TGF-β1 secretion levels were
enhanced significantly in the rats with DN, compared with those of
the normal control rats, and with the increased dosage of NR1, the
secretion levels of TGF-β1 decreased significantly. On the other
hand, IL-10 has been reported to block inflammation and improves
renal function in a model of chronic renal disease (49). We observed elevated serum IL-10
secretion in the NR1-treated rats with DN. Taken together, the
findings of the present study suggest that NR1 exerted protective
effects in the podocytes of rats with DN by regulating the
secretion of inflammatory cytokines as well as an anti-inflammatory
cytokine.
More importantly, the results suggest that NR1
exerts protective effects in DN by inhibiting apoptosis, activating
the PI3K/Akt signaling pathway, and downregulating NF-κB
expression. Many lines of evidence have shown that hyperglycemia
directly induces apoptosis in cultured podocytes, thereby providing
an additional possible explanation for the reduced podocyte numbers
in DN (50). It has also been
reported that a decrease in the podocyte number significantly
correlated with reduced renal function and global
glomerulosclerosis in diabetic patients (12,14). Thus, our observation regarding the
apoptosis of podocytes in the rats with DN was consistent with
these previous findings. Furthermore, NR1 treatment decreased
apoptosis and restored the podocyte number as revealed by TUNEL
staining and quantitative analysis of TUNEL-positive cell numbers,
which indicated that NR1 treatment contributed to the maintenance
of the podocyte number. To further explore the mechanism
responsible for NR1-induced inhibition of apoptosis, the PI3K/Akt
signaling pathway and the activation of NF-κB was explored.
Previous findings have demonstrated that PI3K was involved in
nephrin-mediated actin reorganization in podocytes, and that
disturbing nephrin-PI3K interactions may contribute to abnormal
podocyte morphology and proteinuria (17). In this study, we observed the
decreased expression of nephrin in the rats with DN, and NR1
treatment rescued those rats from DN by restoring the expression of
nephrin. Furthermore, it has been reported that Akt kinase is
activated by TGF-β in diabetic kidneys, and TGF-β may increase
FoxO3a phosphorylation and transcriptional inactivation through
PI3K/Akt, which also suggests the involvement of the PI3K/Akt
signaling pathway in the progression of DN (51,52). Huber et al revealed that
nephrin and CD2AP interacted with the p85 regulatory subunit of
PI3K in vivo, recruited PI3K to the plasma membrane, and,
together with podocin, stimulated PI3K-dependent Akt signaling in
podocytes (53). Thus, we
hypothesized that the PI3K/Akt signaling pathway may be involved.
In this study, we observed a significant decrease in the
phosphorylation of PI3K (p85) and Akt in the rats with STZ-induced
DN. However, this inactivation of the PI3K/Akt signaling pathway
was abrogated by NR1 administration. To further prove our
hypothesis, our next study aims to provide in vitro evidence
of NR1-induced PI3K/Akt signaling pathway activation. As NF-κB
regulates the expression of numerous genes that play key roles in
the inflammatory response during human and experimental kidney
injury (54), we postulated that
the inactivation of NF-κB was also involved in NR1-induced podocyte
protection. In the present study, NR1 treatment significantly
decreased the diabetes-induced activation of NF-κB by the
phosphorylation of NF-κB at serine 536, whereas the expression of
total NF-κB was unchanged in the different groups. Collectively,
these findings suggest that NR1 treatment protects against
diabetes-associated renal damage through the suppression of
apoptosis, the inactivation of NF-κB and the induction of PI3K/Akt
activation.
In conclusion, to the best of our knowledge, this is
the first study to examine the protective effects of NR1 treatment
on podocytes which occur through the regulation of the PI3K/Akt and
the NF-κB signaling pathway. The present study clearly demonstrated
that NR1 ameliorates podocyte and renal injuries as well as
apoptosis and inflammation by activating the PI3K/Akt signaling
pathway and inactivating NF-κB. The present results provide new
evidence for the further potential applications of NR1 in
maintaining podocyte numbers and glomerular filtration barrier
integrity as well as preventing proteinuria. However, further in
vitro studies of the molecular mechanism responsible for the
NR1-mediated protective effects on podocytes are warranted prior to
conducting clinical investigations.
Acknowledgments
The present study was funded by the National Science
Foundation of China (no. 81160434), the Guangxi Science Foundation
(no. 2013GXNSFDA019016), and the Sichuan Science Foundation (no.
2015JY0183).
References
|
1
|
Wiggins RC: The spectrum of
podocytopathies: a unifying view of glomerular diseases. Kidney
Int. 71:1205–1214. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shankland SJ: The podocyte's response to
injury: role in proteinuria and glomerulosclerosis. Kidney Int.
69:2131–2147. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mundel P and Shankland SJ: Podocyte
biology and response to injury. J Am Soc Nephrol. 13:3005–3015.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kumar PA, Brosius FC III and Menon RK: The
glomerular podocyte as a target of growth hormone action:
implications for the pathogenesis of diabetic nephropathy. Curr
Diabetes Rev. 7:50–55. 2011. View Article : Google Scholar
|
|
5
|
Liu WJ, Tang HT, Jia YT, Ma B, Fu JF, Wang
Y, Lv KY and Xia ZF: Notoginsenoside R1 attenuates renal
ischemia-reperfusion injury in rats. Shock. 34:314–320. 2010.
View Article : Google Scholar
|
|
6
|
Gui D, Wei L, Jian G, Guo Y, Yang J and
Wang N: Notoginsenoside R1 ameliorates podocyte adhesion under
diabetic condition through alpha3beta1 integrin upregulation in
vitro and in vivo:. Cell Physiol Biochem. 34:1849–1862. 2014.
View Article : Google Scholar
|
|
7
|
Tryggvason K: Unraveling the mechanisms of
glomerular ultra-filtration: nephrin, a key component of the slit
diaphragm. J Am Soc Nephrol. 10:2440–2445. 1999.PubMed/NCBI
|
|
8
|
Kestilä M, Lenkkeri U, Männikkö M,
Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T,
Nissinen M, Herva R, et al: Positionally cloned gene for a novel
glomerular protein -nephrin-is mutated in congenital nephrotic
syndrome. Mol Cell. 1:575–582. 1998. View Article : Google Scholar
|
|
9
|
Khoshnoodi J, Sigmundsson K, Ofverstedt
LG, Skoglund U, Obrink B, Wartiovaara J and Tryggvason K: Nephrin
promotes cell-cell adhesion through homophilic interactions. Am J
Pathol. 163:2337–2346. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McLaughlin JK, Lipworth L, Chow WH and
Blot WJ: Analgesic use and chronic renal failure: a critical review
of the epidemiologic literature. Kidney Int. 54:679–686. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lemley KV, Lafayette RA, Safai M, Derby G,
Blouch K, Squarer A and Myers BD: Podocytopenia and disease
severity in IgA nephropathy. Kidney Int. 61:1475–1485. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Steffes MW, Schmidt D, McCrery R and
Basgen JM; International Diabetic Nephropathy Study Group:
Glomerular cell number in normal subjects and in type 1 diabetic
patients. Kidney Int. 59:2104–2113. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
White KE and Bilous RW; Diabiopsies Study
Group: Structural alterations to the podocyte are related to
proteinuria in type 2 diabetic patients. Nephrol Dial Transplant.
19:1437–1440. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pagtalunan ME, Miller PL, Jumping-Eagle S,
Nelson RG, Myers BD, Rennke HG, Coplon NS, Sun L and Meyer TW:
Podocyte loss and progressive glomerular injury in type II
diabetes. J Clin Invest. 99:342–348. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schiffer M, Bitzer M, Roberts IS, Kopp JB,
ten Dijke P, Mundel P and Böttinger EP: Apoptosis in podocytes
induced by TGF-beta and Smad7. J Clin Invest. 108:807–816. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhu J, Sun N, Aoudjit L, Li H, Kawachi H,
Lemay S and Takano T: Nephrin mediates actin reorganization via
phosphoinositide 3-kinase in podocytes. Kidney Int. 73:556–566.
2008. View Article : Google Scholar
|
|
18
|
Yu S and Li Y: Dexamethasone inhibits
podocyte apoptosis by stabilizing the PI3K/Akt signal pathway.
BioMed Res Int. 2013:3269862013.
|
|
19
|
Wang Y, Liu Y, Wang H, Li C, Qi P and Bao
J: Agaricus bisporus lectins mediate islet β-cell proliferation
through regulation of cell cycle proteins. Exp Biol Med (Maywood).
237:287–296. 2012. View Article : Google Scholar
|
|
20
|
Sugimoto H, LeBleu VS, Bosukonda D, Keck
P, Taduri G, Bechtel W, Okada H, Carlson W Jr, Bey P, Rusckowski M,
et al: Activin-like kinase 3 is important for kidney regeneration
and reversal of fibrosis. Nat Med. 18:396–404. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Y, Wang H, Liu Y, Li C, Qi P and Bao
J: Antihyperglycemic effect of ginsenoside Rh2 by inducing islet
beta-cell regeneration in mice. Horm Metab Res. 44:33–40. 2012.
View Article : Google Scholar
|
|
22
|
Jiagang D, Li C, Wang H, Hao E, Du Z, Bao
C, Lv J and Wang Y: Amygdalin mediates relieved atherosclerosis in
apolipoprotein E deficient mice through the induction of regulatory
T cells. Biochem Biophys Res Commun. 411:523–529. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rantanen M, Palmén T, Pätäri A, Ahola H,
Lehtonen S, Aström E, Floss T, Vauti F, Wurst W, Ruiz P, et al:
Nephrin TRAP mice lack slit diaphragms and show fibrotic glomeruli
and cystic tubular lesions. J Am Soc Nephrol. 13:1586–1594. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kawachi H, Koike H, Kurihara H, Sakai T
and Shimizu F: Cloning of rat homologue of podocin: expression in
proteinuric states and in developing glomeruli. J Am Soc Nephrol.
14:46–56. 2003. View Article : Google Scholar
|
|
25
|
Wiggins JE, Goyal M, Sanden SK, Wharram
BL, Shedden KA, Misek DE, Kuick RD and Wiggins RC: Podocyte
hypertrophy, 'adaptation' and 'decompensation' associated with
glomerular enlargement and glomerulosclerosis in the aging rat:
prevention by calorie restriction. J Am Soc Nephrol. 16:2953–2966.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huber TB and Benzing T: The slit
diaphragm: a signaling platform to regulate podocyte function. Curr
Opin Nephrol Hypertens. 14:211–216. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li Q and Verma IM: NF-kappaB regulation in
the immune system. Nat Rev Immunol. 2:725–734. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Whiting DR, Guariguata L, Weil C and Shaw
J: IDF diabetes atlas: global estimates of the prevalence of
diabetes for 2011 and 2030. Diabetes Res Clin Pract. 94:311–321.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ritz E and Stefanski A: Diabetic
nephropathy in type II diabetes. Am J Kidney Dis. 27:167–194. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
de Jong PE and Brenner BM: From secondary
to primary prevention of progressive renal disease: the case for
screening for albuminuria. Kidney Int. 66:2109–2118. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Aaltonen P, Luimula P, Aström E, Palmen T,
Grönholm T, Palojoki E, Jaakkola I, Ahola H, Tikkanen I and
Holthöfer H: Changes in the expression of nephrin gene and protein
in experimental diabetic nephropathy. Lab Invest. 81:1185–1190.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Langham RG, Kelly DJ, Cox AJ, Thomson NM,
Holthöfer H, Zaoui P, Pinel N, Cordonnier DJ and Gilbert RE:
Proteinuria and the expression of the podocyte slit diaphragm
protein, nephrin, in diabetic nephropathy: effects of angiotensin
converting enzyme inhibition. Diabetologia. 45:1572–1576. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Orikasa M, Matsui K, Oite T and Shimizu F:
Massive proteinuria induced in rats by a single intravenous
injection of a monoclonal antibody. J Immunol. 141:807–814.
1988.PubMed/NCBI
|
|
35
|
Putaala H, Soininen R, Kilpeläinen P,
Wartiovaara J and Tryggvason K: The murine nephrin gene is
specifically expressed in kidney, brain and pancreas: inactivation
of the gene leads to massive proteinuria and neonatal death. Hum
Mol Genet. 10:1–8. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Benigni A, Gagliardini E, Tomasoni S,
Abbate M, Ruggenenti P, Kalluri R and Remuzzi G: Selective
impairment of gene expression and assembly of nephrin in human
diabetic nephropathy. Kidney Int. 65:2193–2200. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schwarz K, Simons M, Reiser J, Saleem MA,
Faul C, Kriz W, Shaw AS, Holzman LB and Mundel P: Podocin, a
raft-associated component of the glomerular slit diaphragm,
interacts with CD2AP and nephrin. J Clin Invest. 108:1621–1629.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Roselli S, Gribouval O, Boute N, Sich M,
Benessy F, Attié T, Gubler MC and Antignac C: Podocin localizes in
the kidney to the slit diaphragm area. Am J Pathol. 160:131–139.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zou J, Yaoita E, Watanabe Y, Yoshida Y,
Nameta M, Li H, Qu Z and Yamamoto T: Upregulation of nestin,
vimentin, and desmin in rat podocytes in response to injury.
Virchows Arch. 448:485–492. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Navarro-González JF and Mora-Fernández C:
The role of inflammatory cytokines in diabetic nephropathy. J Am
Soc Nephrol. 19:433–442. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Navarro-González JF, Mora-Fernández C,
Muros de Fuentes M and García-Pérez J: Inflammatory molecules and
pathways in the pathogenesis of diabetic nephropathy. Nat Rev
Nephrol. 7:327–340. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sassy-Prigent C, Heudes D, Mandet C,
Bélair MF, Michel O, Perdereau B, Bariéty J and Bruneval P: Early
glomerular macrophage recruitment in streptozotocin-induced
diabetic rats. Diabetes. 49:466–475. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Suzuki D, Miyazaki M, Naka R, Koji T,
Yagame M, Jinde K, Endoh M, Nomoto Y and Sakai H: In situ
hybridization of interleukin 6 in diabetic nephropathy. Diabetes.
44:1233–1238. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sekizuka K, Tomino Y, Sei C, Kurusu A,
Tashiro K, Yamaguchi Y, Kodera S, Hishiki T, Shirato I and Koide H:
Detection of serum IL-6 in patients with diabetic nephropathy.
Nephron. 68:284–285. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
DiPetrillo K, Coutermarsh B and Gesek FA:
Urinary tumor necrosis factor contributes to sodium retention and
renal hypertrophy during diabetes. Am J Physiol Renal Physiol.
284:F113–F121. 2003. View Article : Google Scholar
|
|
46
|
Navarro JF and Mora-Fernández C: The role
of TNF-alpha in diabetic nephropathy: pathogenic and therapeutic
implications. Cytokine Growth Factor Rev. 17:441–450. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mizuno S and Nakamura T: Suppressions of
chronic glomerular injuries and TGF-beta 1 production by HGF in
attenuation of murine diabetic nephropathy. Am J Physiol Renal
Physiol. 286:F134–F143. 2004. View Article : Google Scholar
|
|
48
|
Schiffer M, Schiffer LE, Gupta A, Shaw AS,
Roberts IS, Mundel P and Böttinger EP: Inhibitory smads and TGF-β
signaling in glomerular cells. J Am Soc Nephrol. 13:2657–2666.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mu W, Ouyang X, Agarwal A, Zhang L, Long
DA, Cruz PE, Roncal CA, Glushakova OY, Chiodo VA, Atkinson MA, et
al: IL-10 suppresses chemokines, inflammation, and fibrosis in a
model of chronic renal disease. J Am Soc Nephrol. 16:3651–3660.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lewko B and Stepinski J: Hyperglycemia and
mechanical stress: Targeting the renal podocyte. J Cell Physiol.
221:288–295. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kato M, Putta S, Wang M, Yuan H, Lanting
L, Nair I, Gunn A, Nakagawa Y, Shimano H, Todorov I, et al:
TGF-beta activates Akt kinase through a microRNA-dependent
amplifying circuit targeting PTEN. Nat Cell Biol. 11:881–889. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kato M, Yuan H, Xu ZG, Lanting L, Li SL,
Wang M, Hu MC, Reddy MA and Natarajan R: Role of the Akt/FoxO3a
pathway in TGF-beta1-mediated mesangial cell dysfunction: a novel
mechanism related to diabetic kidney disease. J Am Soc Nephrol.
17:3325–3335. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Huber TB, Hartleben B, Kim J, Schmidts M,
Schermer B, Keil A, Egger L, Lecha RL, Borner C, Pavenstädt H, et
al: Nephrin and CD2AP associate with phosphoinositide 3-OH kinase
and stimulate AKT-dependent signaling. Mol Cell Biol. 23:4917–4928.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sanz AB, Sanchez-Niño MD, Ramos AM, Moreno
JA, Santamaria B, Ruiz-Ortega M, Egido J and Ortiz A: NF-kappaB in
renal inflammation. J Am Soc Nephrol. 21:1254–1262. 2010.
View Article : Google Scholar : PubMed/NCBI
|