Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic,
progressive interstitial lung disease (ILD), with the worst
prognosis among all types of ILD (1). In the US, 128,100 individuals are
affected by IPF, 48,000 new cases of IPF are diagnosed and 40,000
patients with IPF succumb to the disease annually (2,3).
Although there are some novel drugs available for the treatment IPF
which have been approved in the US and Europe, the effectiveness of
these drugs in the treatment of IPF remains limited (4). Pro- and anti-inflammatory cytokines,
growth factors, reactive oxygen species (ROS) and lipid mediators
have been implicated in the development of pulmonary fibrosis
(5–7). However, to date, the mechanisms
through which pathological changes progress in IPF remain unknown.
Thus, the complete understanding of the pathogenesis of IPF remains
to be achieved and this is a critical step in the development of
effective therapeutic approaches.
Heat-shock protein 27 (HSP27) is a member of the
small HSP family and is strongly induced by physiological and
pathological stresses (8). Hsp27
is found highly expressed (9) and
involved in tumorgenesis in various tissue and organs (10). Recent studies indicate that HSP27
is overexpressed in clusters between luminal epithelial cells and
myofibroblasts in IPF lung tissue (11). Furthermore, a previous in
vitro investigation demonstrated that HSP27 was directly
related to the process of epithelial-mesenchymal transition (EMT)
in lung mesothelial and epithelial cells (11). In addition, treatment with the
HSP27 specific inhibitor, OGX-427, significantly ameliorated
pleural/subpleural fibrosis in an animal model (11). However, the role and expression of
HSP27 in lung fibroblasts under fibrotic conditions has not yet
been investigated, at least to the best of our knowledge.
Bleomycin (BLM) has been extensively used to
construct models of lung fibrosis in both mice and rats (12). In the mouse model of BLM-induced
pulmonary fibrosis, transforming growth factor-β (TGF-β) plays
critical roles (7,12). On the one hand, the expression of
TGF-β leads to the disruption of the homeostatic microenvironment,
which is critical to promote fibroblast activation, migration,
invasion and hyperplastic changes in the progression of IPF
(7). In particular, lung
fibroblast differentiation and accumulation induced by TGF-β are
the major causes of pulmonary fibrogenesis (13). Recently, HSP27 was proven to be
essential for TGF-β-induced EMT features by in vitro study
using human epithelial cell lines (11). Therefore, we hypothesized that
HSP27 may be involved in the TGF-β-induced differentiation of lung
fibroblasts.
In this study, we detected the expression of HSP27
during pulmonary fibrosis. We found that the expression of HSP27
was markedly upregulated in lung tissue and fibroblasts from
BLM-challenged mice. Furthermore, we demonstrated that
TGF-β-induced HSP27 expression was Smad3- and nuclear factor-κB
(NF-κB)-dependent in vitro. Additionally, the expression of
HSP27 promoted TGF-β-induced fibroblast differentiation through the
activation of Smad3 and ERK. By contrast, the depletion of HSP27
partially inhibited the activation of Smad3 and ERK, and thus
prevented the differentiation of TGF-β-treated lung
fibroblasts.
Materials and methods
Mouse model of experimental pulmonary
fibrosis
As previously described (14,15), wild-type mice (C57BL/6J, male, 8
weeks old; n=32) were purchased from Vital River Laboratory Animal
Technology (Beijing, China) and housed in a pathogen-free and
light-controlled room (12 h light and 12 h dark) with free access
to food and water. BLM sulfate from Hospira, Inc. (Lake Forest, IL,
USA) was used to induce fibrosis. The mice were anesthetized (with
a 3 ml/kg mixture of 25 mg/kg of ketamine in 2.5 ml of xylazine),
followed by treatment with either saline (n=3–4 mice) or BLM (n=3–4
mice) (1.5 U/kg of body weight) in saline solution through an
intratracheal (IT) injection. Twenty-one days post-BLM
administration, the animals were sacrificed (by cervical
dislocation following CO2 inhalation), and the lungs
were removed for histological staining and the isolation of lung
fibroblasts. All animal experiments were performed in accordance
with protocols approved by the Animal Care and Use Committee of
Xuzhou Medical College, Xuzhou, China.
Fluorescence staining
The lung tissues were fixed in formalin and then
embedded in paraffin. Thereafter the samples were cut into
5-µm-thick sections. After removing the paraffin, the mouse
lung tissue sections (5-µM-thick) were hydrated and washed
in deionized water followed by block solution [Tris-buffered saline
(TBS) with 1% fetal bovine serum (FBS) 1 h] at room temperature,
incubated with goat anti-HSP27 antibody and rabbit anti-fibronectin
(FN) antibody (1:500 in the block solution; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) at 4°C overnight, and
incubated with Alexa Fluor secondary antibodies (1:200 dilutions in
blocking buffer) for 1 h, followed by mounting; and examined under
a Nikon Eclipse TE 2000-S fluorescence microscope with a 60X oil
immersion objective lens. For human lung fibroblasts, the treated
cells were fixed and permeabilized, then incubated with primary
antibodies (1:200 dilutions in blocking buffer) for 1 h and washed
3 times, thereafter incubated with Alexa Fluor secondary antibodies
(Alexa Fluor 594 for HSP27 and Alexa Fluor 488 for Fibronectin;
Biolegend, San Diego, CA, USA; 1:200 dilutions in blocking buffer)
for another 1 h. The nuclei were counterstained with DAPI (0.5
µg/ml; Sigma-Aldrich, Inc., St. Louis, MO, USA). After
washing, the slides were mounted on slides and examined under a
fluorescence microscope (Olympus IX83; Olympus, Tokyo, Japan), as
previously described (16,17).
Isolation of primary mouse lung
fibroblasts and cell culture
Primary mouse lung fibroblasts were isolated from
the C57BL/6J mice treated with or without BLM as previously
described (16), and the isolated
cells were cultured in Dulbecco's modified Eagle's medium (DMEM)
containing 10% FBS for 14 days. The WI-38 cells (a human lung
fibroblast cell line) was obtained from the American Type Culture
Collection (ATCC, Manassas, VA, USA) and the cells were grown and
maintained in 6-well plates with DMEM medium containing 10%
FBS.
Treatment with TGF-β and NF-κB
inhibitor
Recombinant human TGF-β1 was obtained from
Peprotech, Inc. (Rocky Hill, NJ, USA) and dissolved in
phosphate-buffered saline (PBS) solution. Serum-starved lung
fibroblasts (~80% confluent) were treated with TGF-β (5 ng/ml) for
0–48 h. NF-κB inhibitor was used as previously described (16). In detail, NF-κB inhibitor (Bay
11-7082), which was obtained from Sigma-Aldrich, Inc., was applied
to the lung fibroblasts at a final concentration of 10 µM
for 1 h, as previously described (16). The NF-κB inhibitor was dissolved
in DMSO; DMSO was used as a parallel control to eliminate the
influence of DMSO itself. The cells were subsequently challenged
with TGF-β (5 ng/ml) for 48 h, and cell lysates (20 µg
protein) were subjected to western blot analysis.
Transfection with siRNA and shRNA
The scrambled RNA (scRNA), siRNA targeting human
NF-κB p65 subunit (si-p65) and siRNA targeting the human Smad3
(si-Smad3) smart pool, used in this study were from Santa Cruz
Biotechnology, Inc. Vehicle shRNA (Veh shRNA) and shRNA targeting
human HSP27 shRNA (HSP27 shRNA) were from Sigma-Aldrich, Inc. For
target gene depletion, the WI-38 cells cultured on 6-well plates
(50–60% confluence) were transiently transfected with 200 nmol/l of
siRNA (scRNA, si-p65 or si-Smad3), or transfected with 3
µg/well of shRNA (Veh shRNA or HSP27 shRNA) using Attractene
transfection reagent according to the manufacturer's instructions
(Qiagen, Inc., Gaithersburg, MD, USA). After 48 h, the cells were
further treated with TGF-β (5 ng/ml) for a further 0–48 h. Protein
expression was analyzed by western blot analysis or under a
fluorescence microscope, and the mRNA levels were analyzed by
reverse transcription-quantitative PCR (RT-qPCR).
Western blot analysis
Sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and western blot analysis were performed
as previously described (16,18). The protease inhibitor cocktail
tablets (EDTA-free complete) were from Roche Diagnostics
(Indianapolis, IN, USA). Mouse anti-α-smooth muscle actin (α-SMA)
and anti-β-actin antibodies were from Sigma-Aldrich, Inc. Cell
lysis buffer and rabbit anti-Smad3 antibody was from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Rabbit anti-FN and
anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibodies,
mouse anti-p65 antibody and goat anti-HSP27 antibody were purchased
from Santa Cruz Biotechnology, Inc. Horseradish peroxidase
(HRP)-linked anti-mouse IgG and anti-rabbit IgG antibodies were
obtained from Bio-Rad Laboratories, Inc. (Hercules, CA, USA).
Briefly, cell lysates were prepared in lysis buffer containing
EDTA-free complete protease inhibitors, followed by centrifugation
at 10,000 × g for 10 min, boiled with Laemmli sample buffer for 5
min, separated on 10 or 4–20% SDS-PAGE, transferred onto PVDF
membranes, and blocked with TBST containing 5% BSA prior to
incubation with primary antibodies (1:1,000 dilution) overnight,
and secondary antibodies (1:2,000 dilutions) for 2 h at room
temperature. The blots were developed and quantified using
ImageQuant 5.2 software (Molecular Dynamics, Sunnyvale, CA,
USA).
RNA isolation and real-time PCR
Total RNA was isolated from the cells using TRIzol
reagent (Life Technology, Rockville, MD, USA), and RNA (1
µg) was reversed transcribed using the cDNA synthesis kit
(Bio-Rad Laboratories, Inc.). Quantitative PCR (qPCR) was performed
as previously described (14,16). Briefly, qPCR detection was
performed using the ABI 7500 system (Life Technologies, Grand
Island, NY, USA) with the SYBR-Green supermix kit (Bio-Rad
Laboratories, Inc.). According to the manufacturer's instructions,
the running parameters were set as denaturation at 95°C for 3 min,
40 cycles of denaturation at 95°C for 15 sec and
annealing/extension at 60°C for 1 min, finally the melt curve
analysis was set as 95°C for 15 sec, 60°C for 1 min and 95°C for 15
sec. For human Smad3, the primer sequence was forward,
5′-TGGACGCAGGTTCTCCAAAC-3′ and reverse, 5′-CCGGCTCGCAGTAGGTAAC-3′;
for human GAPDH, the primer sequence was forward,
5′-TGTGGGCATCAATGGATTTGG-3′ and reverse,
5′-ACACCATGTATTCCGGGTCAAT-3′. GAPDH was used as a housekeeping gene
to normalize the expression levels as previously described
(14).
Statistical analysis
Primary data from at least 3 independent experiments
are expressed as the means ± SEM, and subjected to statistical
analysis using the two-tailed Student's t-test or one-way ANOVA.
Values of P<0.05 were considered to indicate statistically
significant differences (19,20).
Results
BLM challenge increases HSP27 expression
in mouse lung tissue
The role of HSP27 in the etiology of lung fibrosis
is elusive. In this study, we examined the expression of HSP27 in
lung tissue from C57BL/6J mice challenged with BLM by IT injection
(male, 8 weeks old) or treated with saline. At 21 days
post-challenge, lung tissues were isolated from the mice and and
used for immunofluorescence staining for HSP27 and FN as described
in the Materials and methods. As shown in Fig. 1A, the expression of HSP27 was
significantly increased and co-localized with FN, a marker of
fibrogenesis, in the lung fibrotic foci from the BLM-challenged
mice. The resutls of western blot analysis also indicated that
HSP27 expression was markedly increased in the lung tissue from the
BLM-challenged mice (Fig. 1B and
C).
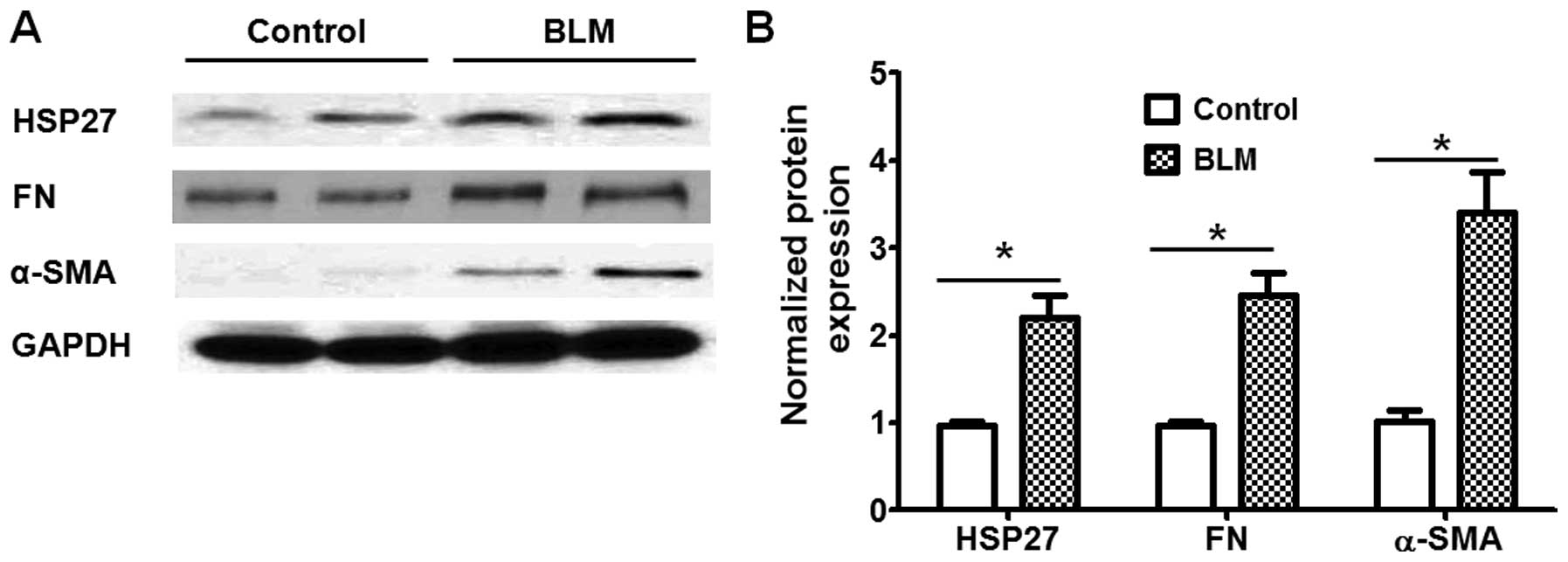
HSP27 expression in lung fibroblasts
We then examined the expression of HSP27 in lung
fibroblasts from the control and BLM-challenged mice. As shown in
Fig. 2, the expression levels of
HSP27, as well as that of bio-markers of fibroblast differentiation
(α-SMA and FN), were markedly higher in the lung fibroblasts from
BLM-challenged mice than in those from the control mice. These
findings suggest that HSP27 expression in lung fibroblasts may be
associated with fibroblast differentiation under fibrotic
conditions.
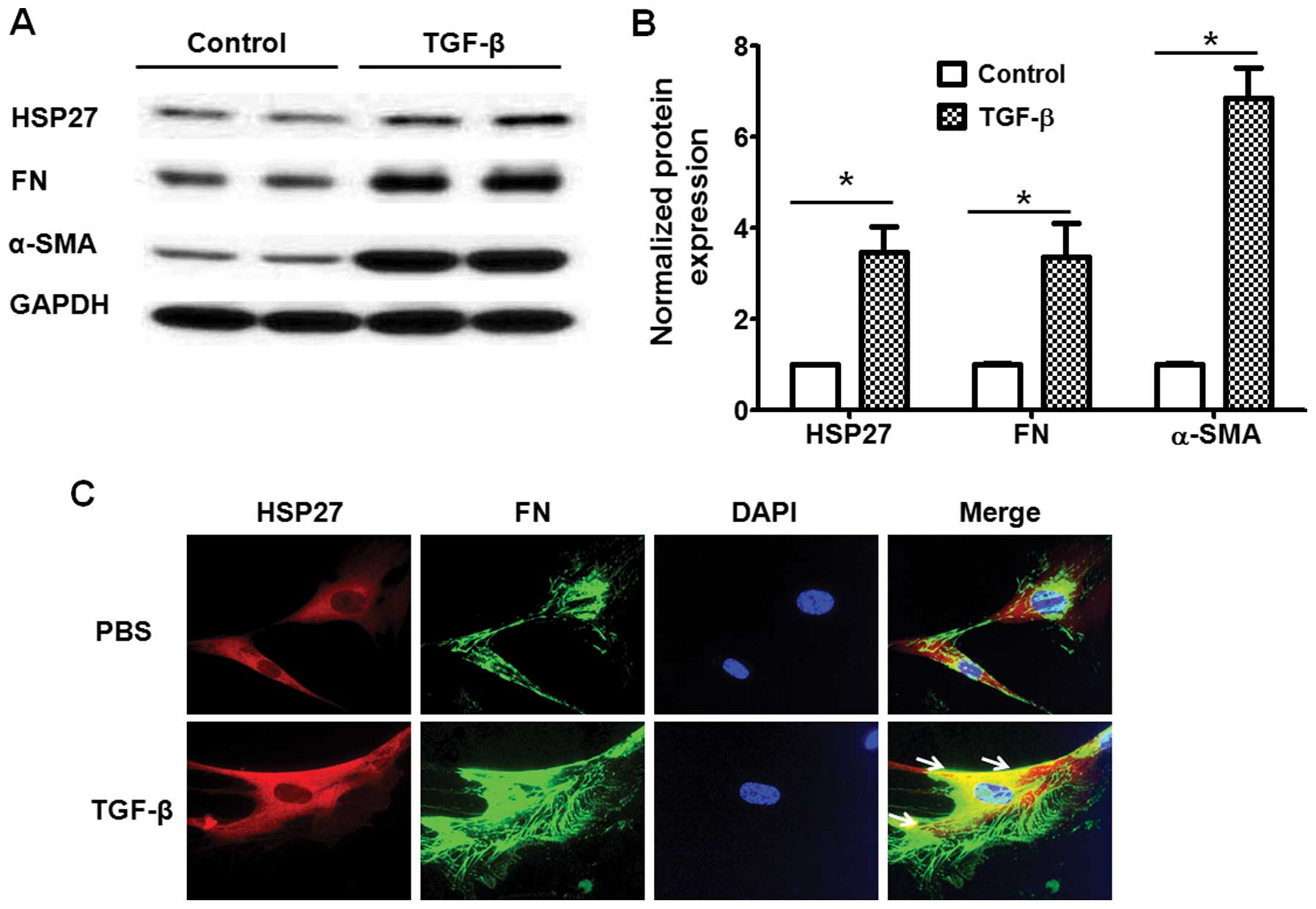
TGF-β promotes HSP27 expression in human
lung fibroblasts
TGF-β is a key factor for fibroblast differentiation
through the regulation of the expression of various genes during
pulmonary fibrosis (21). Thus,
we then investigated whether HSP27 expression is regulated by TGF-β
in lung fibroblasts. As shown in Fig.
3A and B, the TGF-β challenge (5 ng/ml, 48 h) markedly
increased HSP27 expression and promoted fibroblast differentiation.
Furthermore, we confirmed the expression of HSP27 by
immunofluorescence staining. Examination under a fluorescence
microscope also indicated that the TGF-β challenge upregulated the
expression of HSP27 and FN in human lung fibroblasts (Fig. 3C).
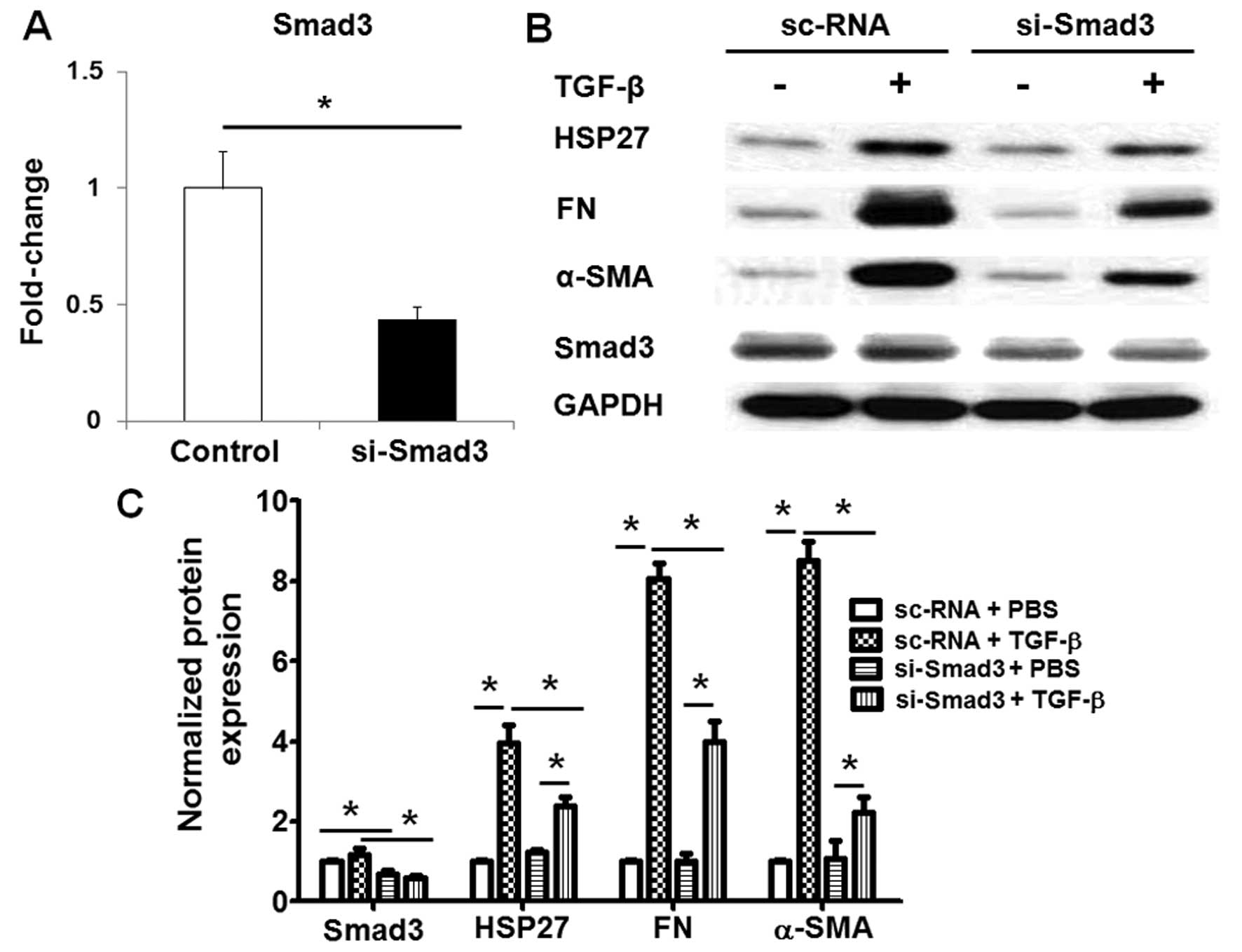
TGF-β induces HSP27 expression through
the Smad and NF-κB pathways
To further determine the molecular mechanisms
through which TGF-β regulates HSP27 expression in lung fibroblasts,
we knocked down the expression of Smad3 or the NF-κB p65 subunit,
which are the key transcription factors in TGF-β-challenged lung
fibroblasts. Transfection with si-Smad3 significantly decreased the
mRNA (Fig. 4A) and protein
expression (Fig. 4B and C) of
Smad3 in the human lung fibroblasts, and also inhibited the
TGF-β-induced HSP27 expression in the human lung fibroblasts
(Fig. 4B and C). Similarly, the
knockdown of the p65 subunit by transfection with si-p65 also
markedly inhibited the TGF-β-induced HSP27 expression and the
differentiation of human lung fibroblasts (Fig. 5A and B). Additionally, treatment
with NF-κB inhibited the TGF-β-induced increase in HSP27 expression
and the differentiation of human lung fibroblasts (Fig. 5C and D). Taken together, these
data indicated that TGF-β induced HSP27 expression through the
Smad3 and NF-κB pathways in human lung fibroblasts.
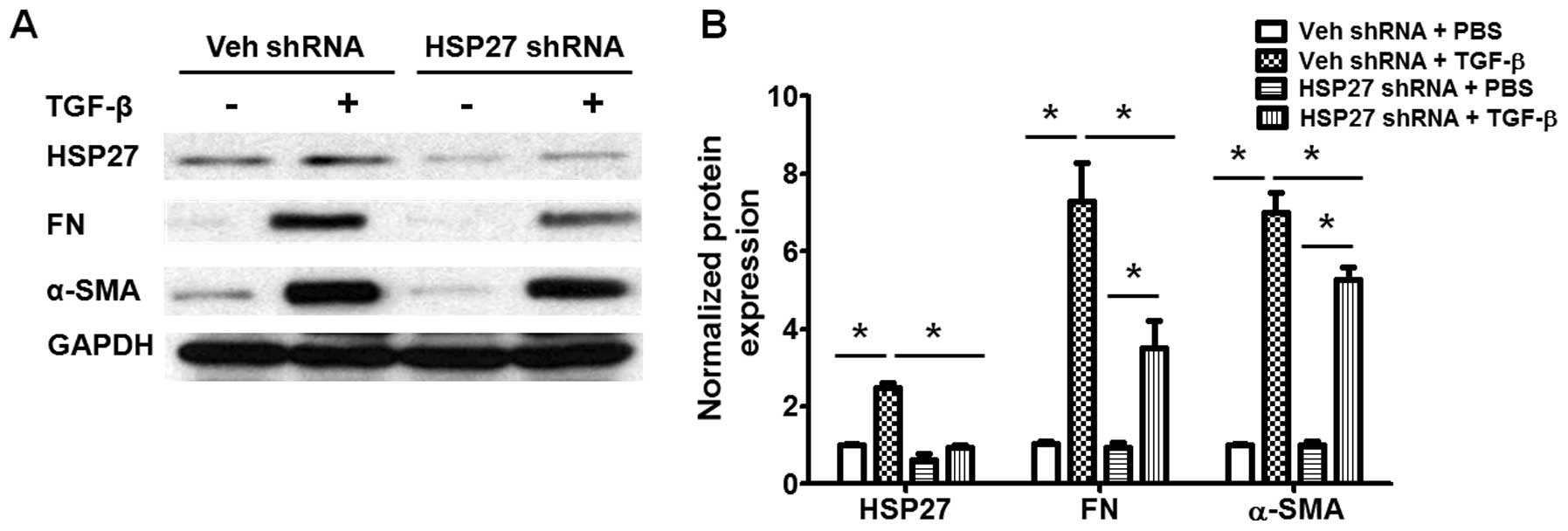
HSP27 regulates TGF-β-induced human lung
fibroblast differentiation via the regulation of Smad and ERK
activation
We also examined the role of HSP27 in TGF-β-induced
fibroblast differentiation. As shown in Fig. 6, transfection with HSP27 shRNA
significantly decreased the expression of HSP27 in human lung
fibroblasts, and also inhibited the TGF-β-induced differentiation
of lung fibroblasts (Fig. 6). The
TGF-β-induced activation of Smad3 and ERK are critical for lung
fibroblast differentiation during pulmonary fibrogenesis (6,14).
We then further examined whether HSP27 regulates the TGF-β-induced
activation of the Smad and ERK signaling pathways in human lung
fibroblasts. As shown in Fig. 7,
the knockdown of HSP27 markedly blocked the TGF-β-induced
activation of Smad3 and ERK. Additionally, the role of HSP27 in the
TGF-β-induced activation of Akt, p38 and JNK was examined studied.
However, the knockdown of HSP27 had shown no effect on the
TGF-β-induced activation of Akt, p38 and JNK (data not shown).
Taken together, these data indicate that HSP27 regulates
TGF-β-induced human lung fibroblast differentiation through the
regulation of Smad3 and ERK activation.
Discussion
IPF is a fatal fibrotic disease with a disconcerting
survival rate, and predominantly occurs in middle-aged and older
individuals (22). Some
therapeutic progress has been over the past few years; however,
this has been proven to be uneffective. Pirfenidone, an orally
available pyridine derivative, was approved for the treatment of
IPF in 2014 (23,24). In models of BLM-induced lung
fibrosis, treatment with pirfenidone has been shown to inhibit the
upregulation of HSP47 expression in lung epithelial cells and
fibroblasts (25–27). Similar to HSP27, HSP47 is also a
member of the small HSP family (28). Treatment with pirfenidone has been
shown to attenuate pulmonary fibrosis in animal models (27,29,30). Clinical trials with IPF patients
have also indicated that pirfenidone is a potent anti-fibrotic,
anti-inflammatory and antioxidant drug during pulmonary
fibrogenesis (4,24,31). These studies suggest that the HSP
family of proteins play critical roles in IPF, and thus,
understanding the role of the HSP family of proteins may be
essential to the identification and development of novel
therapeutic strategies for IPF.
HSP27 has been proven to participate in the
pathogenesis of pulmonary fibrosis, and treatment with HSP27
inhibitors has been shown to attenuate bleomycin-induced pulmonary
fibrosis by inhibiting epithelial-mesothelial transition in an
animal model (11). In in
vitro investigations with a human mesothelium cell line,
Met-5A, it was found that through interactions with Snail and via
the blocking of its proteasomal degradation, HSP27 accelerated EMT
(11). It is possible that the
HSP27-related EMT process is not limited to fibrogenesis, but may
also be involved in other diseases in which EMT occurs. However,
the expression and role of HSP27 in lung fibroblasts during
pulmonary fibrosis remain elusive. In the present study, we
isolated lung fibroblasts from mice with or without BLM-induced
pulmonary fibrosis, and investigated the expression and role of
HSP27 in lung fibroblasts during pulmonary fibrogenesis. Similar to
the findings of a previous study, our data also indicated that the
expression of HSP27 was markedly increased in experimental fibrotic
lung tissue (Fig. 1). Of note,
the expression of HSP27, as well as that of lung fibroblast
differentiation protein markers (FN and α-SMA), in the primary lung
fibroblasts from mice with pulmonary fibrosis was markedly higher
than that in fibroblasts from the control mice (Fig. 2). In human lung fibroblasts,
treatment with TGF-β also markedly increased the expression of
HSP27 in fibroblasts. Taken together, our data indicate that HSP27
may also be involve in fibroblast differentiation during lung
fibrosis.
Studies have proven that TGF-β is a key factor to
promote lung fibroblast differentiation through Smad-dependent and
-independent pathways (13).
Additionally, TGF-β is also involved in a crosstalk with
wnt/β-catenin, sphingosine 1 phospate, lysophosphatidic acid and
NF-κB pathways to induce pro- or anti-fibrotic gene expression
during pulmonary fibrogenesis (5,14,15,32,33). To investigate the mechanisms
through which TGF-β induces HSP27 expression in lung fibroblasts,
we knocked down the expression of Smad3 or NF-κB p65 subunit and
examined the expression of HSP27 in TGF-β-treated cells. A previous
study suggested that the depletion of HSP27 in epithelial cells
significantly inhibited TGF-β-induced EMT and suppressed the
expression of Snail, which is a subfamily of the zinc finger
transcription factor that represses the transcription of the
adhesion molecule, E-cadherin, and thus inhibits EMT (34). Similarly, this study indicated
that the knockdown of HSP27 markedly blocked the TGF-β-induced
activation of Smad3 and ERK, and inhibited the expression of FN and
α-SMA in human lung fibroblasts (Figs. 6 and 7). Our findings indicated that TGF-β
induced HSP27 expression through the activation of the Smad3 and
NF-κB pathways; furthermore, HSP27 expression also synergistically
regulated the TGF-β-induced differentiation of human lung
fibroblasts through the regulation of the Smad3 and ERK signaling
pathways.
In conclusion, in this study, we demonstrated that
HSP27 expression increased in lung fibrotic foci, and TGF-β
increased HSP27 expression through the activation of the Smad3 and
NF-κB pathways. HSP27 regulated the TGF-β-induced activation of
lung fibroblasts during pulmonary fibrogenesis.
Acknowledgments
This study was supported by the China National
Natural Science Foundation (grant nos. 81400047 and 81400055) and
the Natural Science Foundation of Jiangsu Province (grant nos.
BK20150213 and BK20140242). The funders had no role in the study
design, data collection and analysis, decision to publish, or
preparation of the manuscript.
References
|
1
|
Zibrak JD and Price D: Interstitial lung
disease: Raising the index of suspicion in primary care. NPJ Prim
Care Respir Med. 24:140542014.PubMed/NCBI
|
|
2
|
Raghu G, Weycker D, Edelsberg J, Bradford
WZ and Oster G: Incidence and prevalence of idiopathic pulmonary
fibrosis. Am J Respir Crit Care Med. 174:810–816. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Esposito DB, Lanes S, Donneyong M, Holick
CN, Lasky JA, Lederer D, Nathan SD, O'Quinn S, Parker J and Tran
TN: Idiopathic pulmonary fibrosis in US automated claims:
Incidence, prevalence and algorithm validation. Am J Respir Crit
Care Med. 192:1200–1207. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Azuma A: Pirfenidone: Antifibrotic agent
for idiopathic pulmonary fibrosis. Expert Rev Respir Med.
4:301–310. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rancoule C, Pradère JP, Gonzalez J, Klein
J, Valet P, Bascands JL, Schanstra JP and Saulnier-Blache JS:
Lysophosphatidic acid-1-receptor targeting agents for fibrosis.
Expert Opin Investig Drugs. 20:657–667. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang LS and Natarajan V: Sphingolipids in
pulmonary fibrosis. Adv Biol Regul. 57:55–63. 2015. View Article : Google Scholar :
|
|
7
|
Wynn TA: Integrating mechanisms of
pulmonary fibrosis. J Exp Med. 208:1339–1350. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shashidharamurthy R, Koteiche HA, Dong J
and McHaourab HS: Mechanism of chaperone function in small heat
shock proteins: Dissociation of the HSP27 oligomer is required for
recognition and binding of destabilized T4 lysozyme. J Biol Chem.
280:5281–5289. 2005. View Article : Google Scholar
|
|
9
|
Hadaschik BA, Jackson J, Fazli L, Zoubeidi
A, Burt HM, Gleave ME and So AI: Intravesically administered
antisense oligonucleotides targeting heat-shock protein-27 inhibit
the growth of non-muscle-invasive bladder cancer. BJU Int.
102:610–616. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kamada M, So A, Muramaki M, Rocchi P,
Beraldi E and Gleave M: Hsp27 knockdown using nucleotide-based
therapies inhibit tumor growth and enhance chemotherapy in human
bladder cancer cells. Mol Cancer Ther. 6:299–308. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wettstein G, Bellaye PS, Kolb M, Hammann
A, Crestani B, Soler P, Marchal-Somme J, Hazoume A, Gauldie J,
Gunther A, et al: Inhibition of HSP27 blocks fibrosis development
and EMT features by promoting Snail degradation. FASEB J.
27:1549–1560. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Della Latta V, Cecchettini A, Del Ry S and
Morales MA: Bleomycin in the setting of lung fibrosis induction:
From biological mechanisms to counteractions. Pharmacol Res.
97:122–130. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wolters PJ, Collard HR and Jones KD:
Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol.
9:157–179. 2014. View Article : Google Scholar :
|
|
14
|
Huang LS, Fu P, Patel P, Harijith A, Sun
T, Zhao Y, Garcia JG, Chun J and Natarajan V: Lysophosphatidic acid
receptor-2 deficiency confers protection against bleomycin-induced
lung injury and fibrosis in mice. Am J Respir Cell Mol Biol.
49:912–922. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang LS, Berdyshev E, Mathew B, Fu P,
Gorshkova IA, He D, Ma W, Noth I, Ma SF, Pendyala S, et al:
Targeting sphingosine kinase 1 attenuates bleomycin-induced
pulmonary fibrosis. FASEB J. 27:1749–1760. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sun X, Chen E, Dong R, Chen W and Hu Y:
Nuclear factor (NF)-κB p65 regulates differentiation of human and
mouse lung fibroblasts mediated by TGF-β. Life Sci. 122:8–14. 2015.
View Article : Google Scholar
|
|
17
|
Huang LS, Mathew B, Li H, Zhao Y, Ma SF,
Noth I, Reddy SP, Harijith A, Usatyuk PV, Berdyshev EV, et al: The
mitochondrial cardiolipin remodeling enzyme lysocardiolipin
acyltransferase is a novel target in pulmonary fibrosis. Am J
Respir Crit Care Med. 189:1402–1415. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Usatyuk PV, Burns M, Mohan V, Pendyala S,
He D, Ebenezer DL, Harijith A, Fu P, Huang LS, Bear JE, et al:
Coronin 1B regulates S1P-induced human lung endothelial cell
chemotaxis: Role of PLD2, protein kinase C and Rac1 signal
transduction. PLoS One. 8:e630072013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang LS, Kim MR and Sok D-E: Enzymatic
reduction of polyunsaturated lysophosphatidylcholine hydroperoxides
by glutathione peroxidase-1. Eur J Lipid Sci Technol. 111:584–592.
2009. View Article : Google Scholar
|
|
20
|
Huang LS, Hung ND, Sok DE and Kim MR:
Lysophosphatidylcholine containing docosahexaenoic acid at the sn-1
position is anti-inflammatory. Lipids. 45:225–236. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Midgley AC, Rogers M, Hallett MB, Clayton
A, Bowen T, Phillips AO and Steadman R: Transforming growth
factor-β1 (TGF-β1)-stimulated fibroblast to myofibroblast
differentiation is mediated by hyaluronan (HA)-facilitated
epidermal growth factor receptor (EGFR) and CD44 co-localization in
lipid rafts. J Biol Chem. 288:14824–14838. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Morell F: Idiopathic pulmonary fibrosis:
Importance of accurate diagnosis and treatment. Arch Bronconeumol.
49:319–320. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Antoniou KM, Tomassetti S, Tsitoura E and
Vancheri C: Idiopathic pulmonary fibrosis and lung cancer: A
clinical and pathogenesis update. Curr Opin Pulm Med. 21:626–633.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Aravena C, Labarca G, Venegas C, Arenas A
and Rada G: Pirfenidone for idiopathic pulmonary fibrosis: A
systematic review and meta-analysis. PLoS One. 10:e01361602015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hisatomi K, Mukae H, Sakamoto N, Ishimatsu
Y, Kakugawa T, Hara S, Fujita H, Nakamichi S, Oku H, Urata Y, et
al: Pirfenidone inhibits TGF-β1-induced over-expression of collagen
type I and heat shock protein 47 in A549 cells. BMC Pulm Med.
12:242012. View Article : Google Scholar
|
|
26
|
Nakayama S, Mukae H, Sakamoto N, Kakugawa
T, Yoshioka S, Soda H, Oku H, Urata Y, Kondo T, Kubota H, et al:
Pirfenidone inhibits the expression of HSP47 in
TGF-beta1-stimulated human lung fibroblasts. Life Sci. 82:210–217.
2008. View Article : Google Scholar
|
|
27
|
Kakugawa T, Mukae H, Hayashi T, Ishii H,
Abe K, Fujii T, Oku H, Miyazaki M, Kadota J and Kohno S:
Pirfenidone attenuates expression of HSP47 in murine
bleomycin-induced pulmonary fibrosis. Eur Respir J. 24:57–65. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Razzaque MS, Le VT and Taguchi T: Heat
shock protein 47 and renal fibrogenesis. Contrib Nephrol.
148:57–69. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schaefer CJ, Ruhrmund DW, Pan L, Seiwert
SD and Kossen K: Antifibrotic activities of pirfenidone in animal
models. Eur Respir Rev. 20:85–97. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Seifirad S, Keshavarz A, Taslimi S, Aran
S, Abbasi H and Ghaffari A: Effect of pirfenidone on pulmonary
fibrosis due to paraquat poisoning in rats. Clin Toxicol (Phila).
50:754–758. 2012. View Article : Google Scholar
|
|
31
|
Raghu G, Johnson WC, Lockhart D and Mageto
Y: Treatment of idiopathic pulmonary fibrosis with a new
antifibrotic agent, pirfenidone: Results of a prospective,
open-label phase II study. Am J Respir Crit Care Med.
159:1061–1069. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chilosi M, Poletti V, Zamò A, Lestani M,
Montagna L, Piccoli P, Pedron S, Bertaso M, Scarpa A, Murer B, et
al: Aberrant Wnt/beta-catenin pathway activation in idiopathic
pulmonary fibrosis. Am J Pathol. 162:1495–1502. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tager AM, LaCamera P, Shea BS, Campanella
GS, Selman M, Zhao Z, Polosukhin V, Wain J, Karimi-Shah BA, Kim ND,
et al: The lysophosphatidic acid receptor LPA1 links pulmonary
fibrosis to lung injury by mediating fibroblast recruitment and
vascular leak. Nat Med. 14:45–54. 2008. View Article : Google Scholar
|
|
34
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|