Oxidative stress is thought to play a significant
role in the development and progression of certain cell
degenerative diseases and aging (1,2).
Evidence supports the crucial role of mitochondrial dynamics in the
pathogenesis of various diseases. For example, mitochondria are
recognized to play an important role in neurodegenerative disorders
including multiple sclerosis, Parkinson's disease and Alzheimer's
disease, which are characterized by progressive and selective loss
of neuronal cell populations (3–5).
Aging and these neurodegenerative diseases are associated with a
chronic state of oxidative stress and inflammation mediated via
organized pathways (6,7). Mitochondria are dynamic organelles
and play an essential role in metabolic processes which are
prominent targets of oxidative stress. Alterations in mitochondrial
activity not only affect the function of individual cells but also
metabolism and the lifespan (8).
Thus, homeostasis is intensively regulated by a complex interplay
between mitochondrial biogenesis and mitochondrial autophagy
(mitophagy), an important control mechanism that clears damaged
mitochondria, critical for cell survival and for normal cellular
functions (9). Mitophagy may also
play a key role in mediating apoptosis and in determining their own
degradation. Phosphatase and tensin homolog (PTEN)-induced putative
kinase protein 1 (PINK1) is a mitochondrial-targeted
serine/threonine kinase, which is linked to autosomal recessive
familial Parkinson's disease (10). PINK1 has a protective role against
mitochondrial dysfunction and apoptosis with mitochondrial quality
control by activating a mitochondrial damage-response signaling
pathway (11). It is now clear
that mitochondrial oxidant production is controlled by redox
signaling under precise physiological conditions. The pathway
integrating environmental and genetic stimuli interacts with
crucial mitophagy-related effectors to stimulate cellular stress
response mechanisms modulating a healthy condition and long
lifespan (12). Needless to
state, there are many variables involved in how long we live and in
maintaining a healthy life. However, it may be possible to help
slow the aging process and/or avoid age-related diseases including
neurodegenerative disorders, diabetes and heart disease. As aging
may be the result of a number of factors, unraveling the regulatory
network that governs the crosstalk between mitochondrial biogenesis
and mitophagy may enhance our understanding of the molecular
mechanisms that regulate mitochondrial function to control aging
and age-related diseases. This review provides a concise overview
of the cellular functions of mitochondrial kinase PINK1 and the
relationship between aging/neurodegeneration and mitochondrial
dynamics.
Localization and stability of PINK1 require
catalytic activity of presenilin-associated rhomboid-like serine
protease (PARL) that can affect the proteolytic processing of PINK1
(13). In mitochondria, PARL
facilitates the cleavage of PINK1, and then mediates differential
cleavage of phosphoglycerate mutase family member 5 (PGAM5)
depending on the status of the mitochondria (14). In addition, PINK1 processing and
localization are indispensable in determining the interaction with
E3 ubiquitin ligase Parkin. PINK1 recruits Parkin, a Parkinson's
disease-related protein, to mitochondria in order to initiate
mitophagy (15,16). Therefore, PARL deficiency also
impairs Parkin recruitment to the mitochondria (15,16). Optineurin and NDP52 are the
primary receptors for PINK1-mediated mitophagy (17). In addition, several molecules
acting downstream of PINK1 are activated to maintain mitochondrial
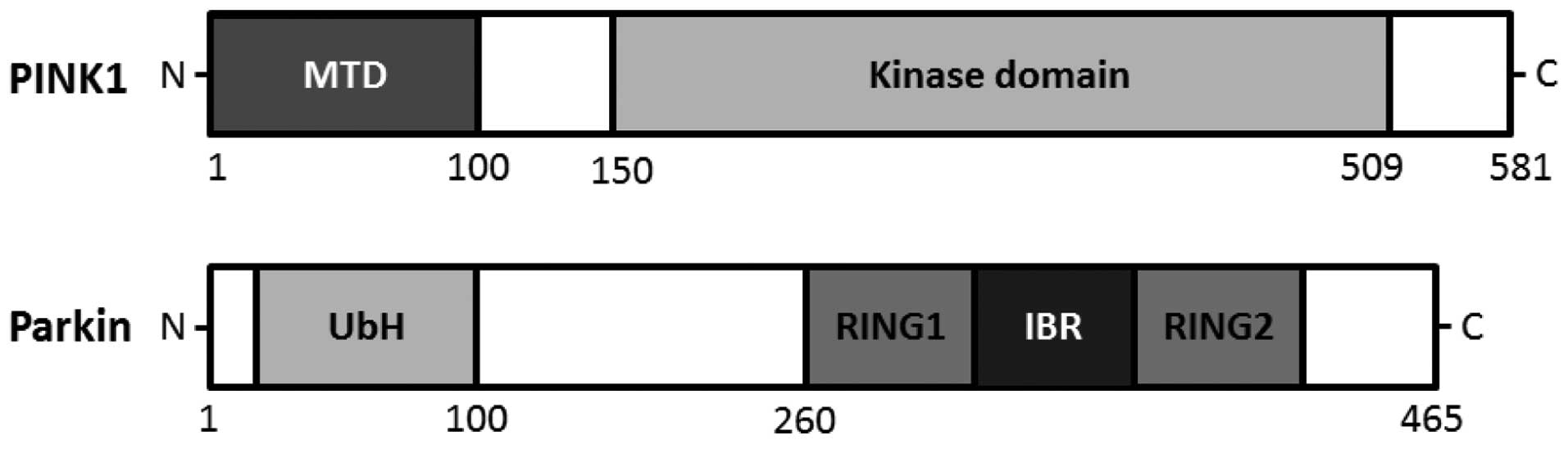
homeostasis. The PINK1 gene consists of eight exons, encoding a
581-amino acid protein with a calculated molecular mass of 66 kDa
(18). PINK1 mRNA is ubiquitously
expressed, and high expression is found in the brain, heart, testis
and skeletal muscle (19).
Mutations in the PINK1 gene are the most common causes of recessive
familial Parkinson's disease (20). An amino terminal targeting signal
domain is sufficient for mitochondrial introduction of PINK1, and
the carboxyl terminal tail holds regulatory motifs capable of
maintaining PINK1 kinase activity with homology to serine/threonine
kinases (21,22) (Fig.
1). PINK1 protein can be processed into at least two shorter
forms, which are distributed in both the mitochondrial and
cytosolic space. Generally, PINK1 is found on the outer and inner
mitochondrial membrane. PINK1 phosphorylates a mitochondrial
molecular chaperone heat shock protein 75 (Hsp75), also known as
mitochondrial heat shock protein 75 kDa [or tumor necrosis factor
receptor-associated protein 1 (TRAP1)], which increases neuronal
survival withstanding oxidative stress or heat shock by inhibiting
the release of cytochrome c (22,23). Serine protease high temperature
requirement protein A2 (HtrA2) is released from the inter-membrane
space of mitochondria to the cytosol during apoptosis, which may
also be regulated by PINK1 (24).
However, whether HtrA2 is a direct PINK1 substrate is fairly
uncertain. Targeted deletion of HtrA2 affects mitochondrial
dysfunction leading to a neurodegenerative disorder with
Parkinsonian phenotype in experimental mice (25). In addition, PINK1 may also
interact with Beclin1, a crucial autophagic protein implicated in
the pathogenesis of Alzheimer's disease and/or Huntington's disease
(26). Interaction of PINK1 with
Beclin1 was found to augment autophagy (27). Consequently, physiological PINK1
substrates may be localized in the outer membrane of mitochondria
or possibly in the cytosol adjacent to the mitochondrial surface.
Cytoplasmic PINK1 is degraded by proteasomes (28,29). It is feasible that differences in
cell viability initiated from PINK1 inactivation may affect several
substrates through other kinases such as p38 stress-activated
protein kinase (SAPK)/mitogen-activated protein kinase (MAPK)
(28,29).
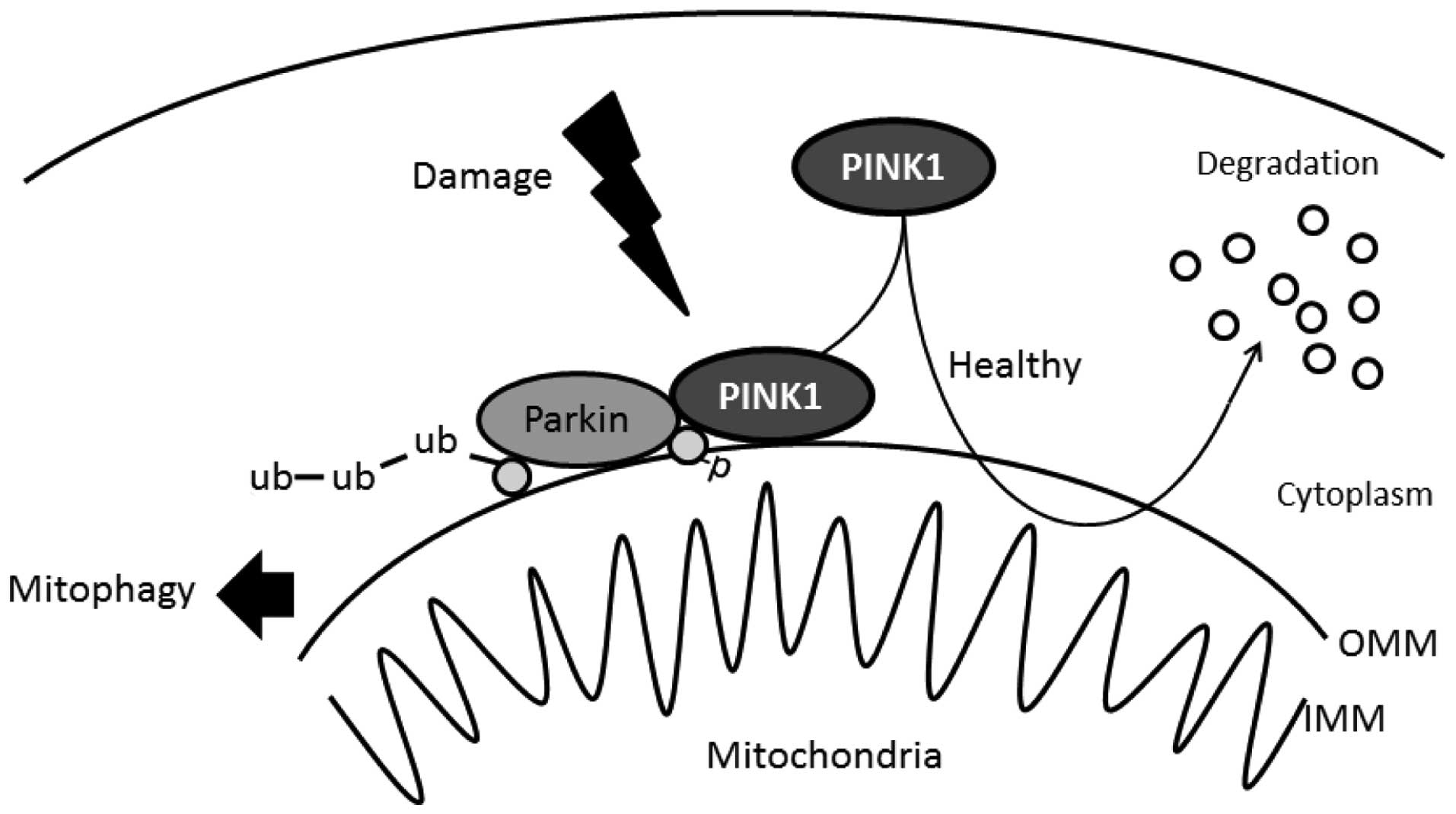
The importance of PINK1 in the mitochondria is
reflected by cell-protective properties for counteracting oxidative
stress. When mitochondria are compromised by cellular
depolarization, PINK1 accumulates on the mitochondrial surface
where it recruits the Parkin protein from the cytosol, which in
turn mediates the degradation of mitochondria termed mitophagy
(Fig. 2) (30,31). Knockdown of endogenous PINK1
and/or loss of PINK1 leads to alterations in mitochondrial
homeostasis as evidenced by increased mitochondrial reactive oxygen
species (ROS) carrying an escalation in mitophagy (32). Protective activity of PINK1 in
maintaining mitochondrial health depends on its mitochondrial
localization (33,34). Stable silencing of PINK1 may have
an incidental role in the activation of mitophagy (33,34). Thus, PINK1 plays a pivotal role in
mitochondrial quality control via the regulation of mitophagy
and/or mitochondrial maintenance. However, excessive rates of
mitophagy have been found to be harmful (35,36). In healthy mitochondria, PINK1 is
promptly degraded in a process comprising mitochondrial proteases
and proteasomes (37,38). As mentioned above, mitochondrial
protease PARL may mediate differential cleavage of PINK1 depending
on the health status of mitochondria (14). Stability of PINK1 requires
catalytic activity of PARL (14,15). PARL deficiency impairs Parkin
recruitment to mitochondria (15), suggesting that PINK1 localization
is important in defining the interaction with Parkin. Complete
PINK1 is free to be released into the mitochondrial inter-membrane
space or the cytosol. Blockage of PARL processing and the import of
PINK1 upon depolarization of the mitochondrial membrane, leads to
accumulation of PINK1 precursor (39). Targeting of this precursor protein
to the outer mitochondrial membrane has been shown to initiate
mitophagy (22,40). Hence, the removal of PINK1 may act
as a cellular checkpoint for the control of mitochondrial
reliability, indicating that interactions of PINK1 with Parkin
promote healthy mitochondrial homeostasis. Expression of
full-length PINK1 is essential for mitochondrial Parkin
recruitment. Transient expression of Parkin further augments
mitochondrial mitophagy, resulting in cytoprotection of
mitochondrial networks (41).
Following severe mitochondrial damage, however, PINK1 facilitates
aggregation of depolarized mitochondria through interactions with
Parkin (27). Under conditions of
PINK1 deficiency, mitochondrial quality control is eventually
compromised. Parkin can be phosphorylated by PINK1 in its RING
finger domain, which may promote translocation of Parkin to
mitochondria (42). It has been
reported to facilitate the clearance of depolarized mitochondria
via mitophagy (43,44). Failure of this quality control
ultimately results in cell death. Therefore, PINK1 and Parkin
synergistically participate in a common mitochondrial complex
signaling pathway. Recently, it has been reported that PINK1
phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase
activity (45) (Fig. 2). This phosphorylation may be
required for the activation of Parkin for full activation to induce
selective autophagy of damaged mitochondria. Phosphorylation of
Parkin helps to prepare the ubiquitin ligase enzyme for activation
by ubiquitin phospho-Ser65 (46).
The phosphorylation-dependent interaction between ubiquitin and
Parkin suggests that phosphorylated ubiquitin unravels
auto-inhibition of this signaling (47). These findings demonstrate that
phosphorylated ubiquitin is a Parkin activator (47).
Dysfunction of mitochondria is a common feature in
aging and in neurodegeneration (48). In some cases, mitochondrial
abnormalities appear to be caused by decreased activation of Sirt1
initiated by hyperactivation of the DNA damage sensor PARP-1
[poly(ADP-ribose) polymerase 1] leading to mitochondrial membrane
depolarization, PINK1 cleavage, and impaired mitophagy (49). Notably, DNA repair is an essential
mechanism for cell survival, and various defects in the DNA repair
system lead to accelerated aging (49). Sirtuin 1 (silent mating type
information regulation 2, S. cerevisiae, homolog 1) (Sirt1)
is the most prominent member of the mammalian class III histone
deacetylase family implicated in healthy lifespan and longevity
(50). A nuclear-mitochondrial
crosstalk seems to be critical for the maintenance of mitochondrial
health (48), in which the
function of Sirt1 appears to be important (48,51). Sirt1 regulates the DNA repair and
the related metabolism by deacetylating target proteins such as p53
tumor suppressor. The mitochondrial abnormalities may be caused by
decreased activation of SIRT1 triggered by DNA damage. As PINK1
activates Parkin which translocates to depolarized mitochondria,
the PINK1/Parkin/Sirt1 pathway may also act synergistically to
promote mitochondrial degradation by mitophagy in order to protect
cells (52). In addition, several
members of transcription factors control PINK1 transcription. For
example, it is known that mitochondrial Sirt3 interacts and
regulates the activity of FoxO3a, the Forkhead box subgroup O
(FoxO), acting through the conserved FoxO binding elements in
mitochondria. Overexpression of the Sirt3 gene increases FoxO3a DNA
binding activity as well as FoxO3a-dependent gene expression
including PINK1 (53). Induction
of PINK1 by FoxO3a is crucial for critical survival signals in
normal cells, as depletion of PINK1 sensitizes those cells to cell
death (53). Furthermore,
increased FoxO3a expression reduces both ROS and thereby apoptotic
cell death (54).
Multi-functional heat shock protein 40 kDa (HSP40) was found to
decrease PINK1 mRNA level by binding to FoxO3a that interacts with
the PINK1 promoter encouraging its transcriptional activity
(55). FoxO3a alteration is
determinedly linked to the progression of various types of cancer,
fibrosis, and other types of disease. Recently, several studies
have revealed the importance of Sirt3 along with FoxO3a in addition
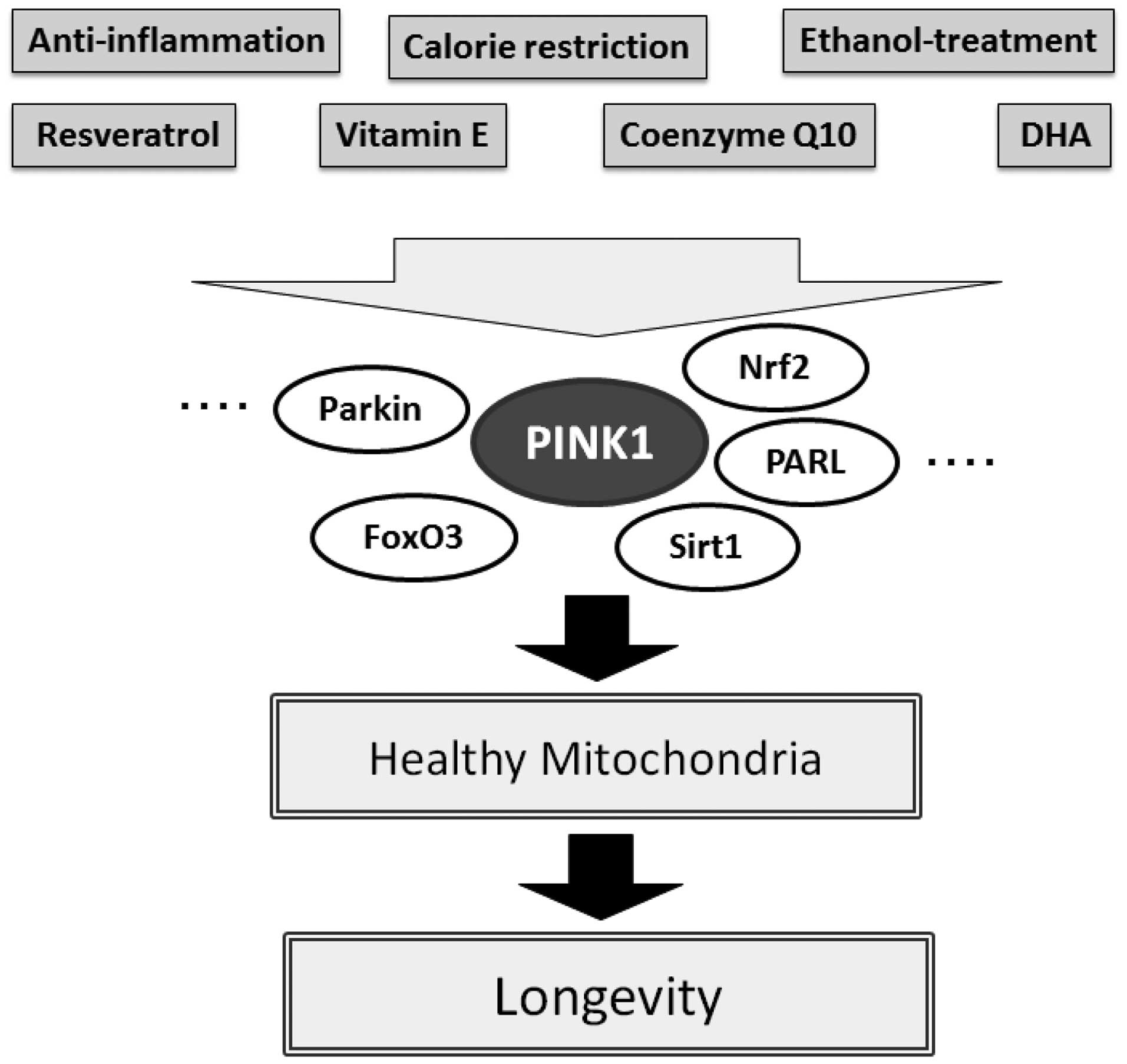
to Sirt1 with resveratrol in preventing premature aging (56,57). The favorable protective effect of
resveratrol has been shown to be diminished upon pharmacological
inhibition of Sirt1 by using sirtinol (56). Sirt1 in cooperation with Sirt3
activates FoxO3a thereby promoting the stimulation of the
PINK-1/Parkin pathway leading to mitophagy. The importance of
mitophagy has been revealed as it encourages a healthy pool of
mitochondria and prevents premature aging (49). In addition, the protein level of
nuclear factor erythroid 2-related factor (Nrf2) is also involved
in stress resistance and longevity (58,59). Notably, PINK1 expression is
positively regulated by NRF2 activity and the Nrf2/PINK1 signaling
axis is deeply involved in cell survival and longevity (60).
Various anti-inflammatory drugs could be used as a
supplement to scavenge ROS and hence improve the survival span of
several types of cells (61).
Antioxidants also act as inhibitors of radical production processes
by removing harmful ROS formed during normal cellular metabolism
(62,63). It is known that the polyphenolic
natural antioxidant resveratrol, present in red wine and grapes,
exhibits a number of pharmacological effects including
anti-inflammation, antioxidation, anti-apoptosis, subsequently
promoting longevity (64,65). Resveratrol can pass through the
blood-brain barrier and is water soluble (66). The anti-aging effects of
resveratrol are believed to be mediated by activation of Sirt1 and
the consequent reduced oxidative stress. As mentioned above,
several studies have revealed that Sirt3 along with FoxO3 in
addition to Sirt1 are of importance in promoting the anti-aging
function of resveratrol, which promotes the initial mitochondrial
signaling response to activate PINK1 thereby promoting mitophagy.
It is appealing to speculate that resveratrol promotes anti-aging
through this PINK1/Sirt1/FoxO3a signaling pathway comprising
mitophagy (Fig. 3). For example,
PINK1 is overexpressed in mitochondria of hepatocytes of
ethanol-treated rats, in which treatment with a small amount of
ethanol represent a possible protective mechanism via the
stimulation of mitophagy (67).
Dysfunctional mitophagy in response to gluco-lipotoxicities may
play an important role in several liver diseases including
hepatosteatosis (68). In
addition, metformin is known to improve hepatosteatosis by inducing
Sirt1-mediated mitophagy (68).
Dysregulation of the Parkin pathway by metabolic malregulation may
contribute to the pathogenesis of Parkinson's disease and metformin
may exert a neuroprotective effect on neuronal disease via the
restoration of Parkin (69).
Furthermore, DHA attenuates insulin resistance in obese mice
through the activation of Sirt1 (70). High fat diet-fed animals were
found to have reduced mRNA expression of PARL (71). Antioxidant vitamins and
vitamin-like substances, such as vitamin E and coenzyme Q10, have
been used in the treatment of neurodegenerative diseases with
noteworthy efficacy (72). It is
well known that a high intake of fruits and vegetables rich in
antioxidant vitamins is inversely related to the incidence of
neurodegenerative diseases. In contrast, treatment with an
isoflavonoid pesticide, which inhibits mitochondrial complex I
activity creating an environment of oxidative stress in cells, can
produce nigrastriatal neuronal cell-loss and it produces an
experimental animal model of Parkinson's disease with similar
symptoms (73,74). The level of mitochondrial PINK1
protein has been shown to be increased after pesticide exposure
(73,74). Cell protective proteins including
Parkin and heat shock proteins (HSPs) may play fundamental roles
even in slowing disease progression (73,74). Expression of Parkin and PINK1 was
found to be significantly attenuated in a rat group fed a
low-protein diet supplemented with various types of keto-acids
(75). Therefore, oxidative
damage can be reduced by adhering to certain diets with specific
vitamins and/or restriction of calorie intake controlling
hyperglycemia. Consequently, nutritional control related to
metabolic antioxidation may encourage longevity followed by healthy
mitochondria with minimal degeneration of cells.
Mitochondria are involved in cell stress-induced
programmed cell death, which also contributes to the regulation of
mitochondrial dynamics and mitophagy. In terms of the effect of
specific ROS on mitophagy, future research is needed to ascertain
how to maintain mitochondrial quality and ensure cellular
homeostasis. PINK1 protein may play key roles in the primary line
of mitochondrial quality control and in monitoring respiratory
function under conditions of oxidative stress. However, low levels
of redox signaling may be essential for normal mitophagy. Although
the detailed physiological substrate of PINK1 is not fully
determined, it is clear that kinase activity is important in many
aspects of mitochondrial function in addition to mitophagy
(76). Hence, reduction in PINK1
activity may have eventual lethal consequences on mitochondria
and/or cells. Enhancing pathways that promote dynamic mitophagy may
delay age-related diseases by promoting a healthy pool of viable
mitochondria in cells and by sustaining good energy metabolism.
Future experimental research may further clarify the mitochondrial
protective roles of PINK1 and Parkin.
This study was supported by grants from JSPS KAKENHI
(nos. 26-12035 and 24240098).
|
1
|
Prasad AS: Zinc: an antioxidant and
anti-inflammatory agent: role of zinc in degenerative disorders of
aging. J Trace Elem Med Biol. 28:364–371. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tebay LE, Robertson H, Durant ST, Vitale
SR, Penning TM, Dinkova-Kostova AT and Hayes JD: Mechanisms of
activation of the transcription factor Nrf2 by redox stressors,
nutrient cues, and energy status and the pathways through which it
attenuates degenerative disease. Free Radic Biol Med. 88:108–146.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Campbell GR, Worrall JT and Mahad DJ: The
central role of mitochondria in axonal degeneration in multiple
sclerosis. Mult Scler. 20:1806–1813. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Erpapazoglou Z and Corti O: The
endoplasmic reticulum/mitochondria interface: A subcellular
platform for the orchestration of the functions of the PINK1-Parkin
pathway? Biochem Soc Trans. 43:297–301. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kaminsky YG, Tikhonova LA and Kosenko EA:
Critical analysis of Alzheimer's amyloid-beta toxicity to
mitochondria. Front Biosci (Landmark Ed). 20:173–197. 2015.
View Article : Google Scholar
|
|
6
|
Oyewole AO and Birch-Machin MA:
Mitochondria-targeted antioxidants. FASEB J. 29:4766–4771. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tocchi A, Quarles EK, Basisty N, Gitari L
and Rabinovitch PS: Mitochondrial dysfunction in cardiac aging.
Biochim Biophys Acta. 1847:1424–1433. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pan Y, Nishida Y, Wang M and Verdin E:
Metabolic regulation, mitochondria and the life-prolonging effect
of rapamycin: A mini-review. Gerontology. 58:524–530. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Quirós PM, Langer T and López-Otín C: New
roles for mitochondrial proteases in health, ageing and disease.
Nat Rev Mol Cell Biol. 16:345–359. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Durcan TM and Fon EA: The three ‘P's of
mitophagy: PARKIN, PINK1, and post-translational modifications.
Genes Dev. 29:989–999. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wilhelmus MM, van der Pol SM, Jansen Q,
Witte ME, van der Valk P, Rozemuller AJ, Drukarch B, de Vries HE
and Van Horssen J: Association of Parkinson disease-related protein
PINK1 with Alzheimer disease and multiple sclerosis brain lesions.
Free Radic Biol Med. 50:469–476. 2011. View Article : Google Scholar
|
|
12
|
Russell AP, Foletta VC, Snow RJ and Wadley
GD: Skeletal muscle mitochondria: A major player in exercise,
health and disease. Biochim Biophys Acta. 1840:1276–1284. 2014.
View Article : Google Scholar
|
|
13
|
Jin SM, Lazarou M, Wang C, Kane LA,
Narendra DP and Youle RJ: Mitochondrial membrane potential
regulates PINK1 import and proteolytic destabilization by PARL. J
Cell Biol. 191:933–942. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sekine S, Kanamaru Y, Koike M, Nishihara
A, Okada M, Kinoshita H, Kamiyama M, Maruyama J, Uchiyama Y,
Ishihara N, et al: Rhomboid protease PARL mediates the
mitochondrial membrane potential loss-induced cleavage of PGAM5. J
Biol Chem. 287:34635–34645. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shi G, Lee JR, Grimes DA, Racacho L, Ye D,
Yang H, Ross OA, Farrer M, McQuibban GA and Bulman DE: Functional
alteration of PARL contributes to mitochondrial dysregulation in
Parkinson's disease. Hum Mol Genet. 20:1966–1974. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pickrell AM and Youle RJ: The roles of
PINK1, parkin, and mitochondrial fidelity in Parkinson's disease.
Neuron. 85:257–273. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lazarou M, Sliter DA, Kane LA, Sarraf SA,
Wang C, Burman JL, Sideris DP, Fogel AI and Youle RJ: The ubiquitin
kinase PINK1 recruits autophagy receptors to induce mitophagy.
Nature. 524:309–314. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weihofen A, Ostaszewski B, Minami Y and
Selkoe DJ: Pink1 Parkinson mutations, the Cdc37/Hsp90 chaperones
and Parkin all influence the maturation or subcellular distribution
of Pink1. Hum Mol Genet. 17:602–616. 2008. View Article : Google Scholar
|
|
19
|
d'Amora M, Angelini C, Marcoli M, Cervetto
C, Kitada T and Vallarino M: Expression of PINK1 in the brain, eye
and ear of mouse during embryonic development. J Chem Neuroanat.
41:73–85. 2011. View Article : Google Scholar
|
|
20
|
Narendra D, Walker JE and Youle R:
Mitochondrial quality control mediated by PINK1 and Parkin: Links
to parkinsonism. Cold Spring Harb Perspect Biol. 4:a0113382012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sim CH, Lio DS, Mok SS, Masters CL, Hill
AF, Culvenor JG and Cheng HC: C-terminal truncation and Parkinson's
disease-associated mutations down-regulate the protein
serine/threonine kinase activity of PTEN-induced kinase-1. Hum Mol
Genet. 15:3251–3262. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Okatsu K, Oka T, Iguchi M, Imamura K,
Kosako H, Tani N, Kimura M, Go E, Koyano F, Funayama M, et al:
PINK1 autophosphorylation upon membrane potential dissipation is
essential for Parkin recruitment to damaged mitochondria. Nat
Commun. 3:10162012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pridgeon JW, Olzmann JA, Chin LS and Li L:
PINK1 protects against oxidative stress by phosphorylating
mitochondrial chaperone TRAP1. PLoS Biol. 5:e1722007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gaki GS and Papavassiliou AG: Oxidative
stress-induced signaling pathways implicated in the pathogenesis of
Parkinson's disease. Neuromolecular Med. 16:217–230. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Plun-Favreau H, Klupsch K, Moisoi N,
Gandhi S, Kjaer S, Frith D, Harvey K, Deas E, Harvey RJ, McDonald
N, et al: The mitochondrial protease HtrA2 is regulated by
Parkinson's disease-associated kinase PINK1. Nat Cell Biol.
9:1243–1252. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lenzi P, Marongiu R, Falleni A, Gelmetti
V, Busceti CL, Michiorri S, Valente EM and Fornai F: A subcellular
analysis of genetic modulation of PINK1 on mitochondrial
alterations, autophagy and cell death. Arch Ital Biol. 150:194–217.
2012.PubMed/NCBI
|
|
27
|
Chu CT: A pivotal role for PINK1 and
autophagy in mitochondrial quality control: Implications for
Parkinson disease. Hum Mol Genet. 19:R28–R37. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Choi I, Kim J, Jeong HK, Kim B, Jou I,
Park SM, Chen L, Kang UJ, Zhuang X and Joe EH: PINK1 deficiency
attenuates astrocyte proliferation through mitochondrial
dysfunction, reduced AKT and increased p38 MAPK activation, and
down-regulation of EGFR. Glia. 61:800–812. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lin W and Kang UJ: Characterization of
PINK1 processing, stability, and subcellular localization. J
Neurochem. 106:464–474. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wei H, Liu L and Chen Q: Selective removal
of mitochondria via mitophagy: Distinct pathways for different
mitochondrial stresses. Biochim Biophys Acta. 1853:2784–2790. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Eiyama A and Okamoto K:
PINK1/Parkin-mediated mitophagy in mammalian cells. Curr Opin Cell
Biol. 33:95–101. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dias V, Junn E and Mouradian MM: The role
of oxidative stress in Parkinson's disease. J Parkinsons Dis.
3:461–491. 2013.PubMed/NCBI
|
|
33
|
McCoy MK, Kaganovich A, Rudenko IN, Ding J
and Cookson MR: Hexokinase activity is required for recruitment of
parkin to depolarized mitochondria. Hum Mol Genet. 23:145–156.
2014. View Article : Google Scholar
|
|
34
|
Michiorri S, Gelmetti V, Giarda E,
Lombardi F, Romano F, Marongiu R, Nerini-Molteni S, Sale P, Vago R,
Arena G, et al: The Parkinson-associated protein PINK1 interacts
with Beclin1 and promotes autophagy. Cell Death Differ. 17:962–974.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cherra SJ III and Chu CT: Autophagy in
neuroprotection and neurodegeneration: A question of balance.
Future Neurol. 3:309–323. 2008.PubMed/NCBI
|
|
36
|
Cherra SJ III, Dagda RK and Chu CT:
Review: autophagy and neurodegeneration: survival at a cost?
Neuropathol Appl Neurobiol. 36:125–132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Moriwaki Y, Kim YJ, Ido Y, Misawa H,
Kawashima K, Endo S and Takahashi R: L347P PINK1 mutant that fails
to bind to Hsp90/Cdc37 chaperones is rapidly degraded in a
proteasome-dependent manner. Neurosci Res. 61:43–48. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sun F, Kanthasamy A, Anantharam V and
Kanthasamy AG: Environmental neurotoxic chemical-induced ubiquitin
proteasome system dysfunction in the pathogenesis and progression
of Parkinson's disease. Pharmacol Ther. 114:327–344. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Meissner C, Lorenz H, Weihofen A, Selkoe
DJ and Lemberg MK: The mitochondrial intramembrane protease PARL
cleaves human Pink1 to regulate Pink1 trafficking. J Neurochem.
117:856–867. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Matsuda S, Kitagishi Y and Kobayashi M:
Function and characteristics of PINK1 in mitochondria. Oxid Med
Cell Longev. 2013:6015872013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dagda RK and Chu CT: Mitochondrial quality
control: Insights on how Parkinson's disease related genes PINK1,
parkin, and Omi/HtrA2 interact to maintain mitochondrial
homeostasis. J Bioenerg Biomembr. 41:473–479. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kim Y, Park J, Kim S, Song S, Kwon SK, Lee
SH, Kitada T, Kim JM and Chung J: PINK1 controls mitochondrial
localization of Parkin through direct phosphorylation. Biochem
Biophys Res Commun. 377:975–980. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Narendra D, Tanaka A, Suen DF and Youle
RJ: Parkin is recruited selectively to impaired mitochondria and
promotes their autophagy. J Cell Biol. 183:795–803. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Narendra DP and Youle RJ: Targeting
mitochondrial dysfunction: Role for PINK1 and Parkin in
mitochondrial quality control. Antioxid Redox Signal. 14:1929–1938.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kane LA, Lazarou M, Fogel AI, Li Y, Yamano
K, Sarraf SA, Banerjee S and Youle RJ: PINK1 phosphorylates
ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell
Biol. 205:143–153. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kazlauskaite A, Kondapalli C, Gourlay R,
Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M
and Muqit MM: Parkin is activated by PINK1-dependent
phosphorylation of ubiquitin at Ser65. Biochem J. 460:127–139.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Koyano F, Okatsu K, Kosako H, Tamura Y, Go
E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, et al:
Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature.
510:162–166. 2014.PubMed/NCBI
|
|
48
|
Fang EF, Scheibye-Knudsen M, Brace LE,
Kassahun H, SenGupta T, Nilsen H, Mitchell JR, Croteau DL and Bohr
VA: Defective mitophagy in XPA via PARP-1 hyperactivation and
NAD(+)/SIRT1 reduction. Cell. 157:882–896. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Scheibye-Knudsen M, Fang EF, Croteau DL
and Bohr VA: Contribution of defective mitophagy to the
neurodegeneration in DNA repair-deficient disorders. Autophagy.
10:1468–1469. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tang BL: Sirt1 and the mitochondria. Mol
Cells. 39:87–95. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Greene AW, Grenier K, Aguileta MA, Muise
S, Farazifard R, Haque ME, McBride HM, Park DS and Fon EA:
Mitochondrial processing peptidase regulates PINK1 processing,
import and Parkin recruitment. EMBO Rep. 13:378–385. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Das S, Mitrovsky G, Vasanthi HR and Das
DK: Antiaging properties of a grape-derived antioxidant are
regulated by mitochondrial balance of fusion and fission leading to
mitophagy triggered by a signaling network of
Sirt1-Sirt3-Foxo3-PINK1-PARKIN. Oxid Med Cell Longev.
2014:3451052014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mei Y, Zhang Y, Yamamoto K, Xie W, Mak TW
and You H: FOXO3a-dependent regulation of Pink1 (Park6) mediates
survival signaling in response to cytokine deprivation. Proc Natl
Acad Sci USA. 106:5153–5158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sengupta A, Molkentin JD, Paik JH, DePinho
RA and Yutzey KE: FoxO transcription factors promote cardiomyocyte
survival upon induction of oxidative stress. J Biol Chem.
286:7468–7478. 2011. View Article : Google Scholar :
|
|
55
|
Requejo-Aguilar R, Lopez-Fabuel I,
Jimenez-Blasco D, Fernandez E, Almeida A and Bolaños JP: DJ1
represses glycolysis and cell proliferation by transcriptionally
upregulating Pink1. Biochem J. 467:303–310. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sin TK, Yung BY, Yip SP, Chan LW, Wong CS,
Tam EW and Siu PM: SIRT1-dependent myoprotective effects of
resveratrol on muscle injury induced by compression. Front Physiol.
6:2932015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lin CH, Lin CC, Ting WJ, Pai PY, Kuo CH,
Ho TJ, Kuo WW, Chang CH, Huang CY and Lin WT: Resveratrol enhanced
FOXO3 phosphorylation via synergetic activation of SIRT1 and
PI3K/Akt signaling to improve the effects of exercise in elderly
rat hearts. Age (Dordr). 36:97052014. View Article : Google Scholar
|
|
58
|
Castillo-Quan JI, Li L, Kinghorn KJ,
Ivanov DK, Tain LS, Slack C, Kerr F, Nespital T, Thornton J, Hardy
J, et al: Lithium promotes longevity through GSK3/NRF2-dependent
hormesis. Cell Rep. 15:638–650. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lewis KN, Wason E, Edrey YH, Kristan DM,
Nevo E and Buffenstein R: Regulation of Nrf2 signaling and
longevity in naturally long-lived rodents. Proc Natl Acad Sci USA.
112:3722–3727. 2015.PubMed/NCBI
|
|
60
|
Murata H, Takamatsu H, Liu S, Kataoka K,
Huh NH and Sakaguchi M: NRF2 regulates PINK1 expression under
oxidative stress conditions. PLoS One. 10:e01424382015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Huang ST, Ho CS, Lin CM, Fang HW and Peng
YX: Development and biological evaluation of C(60)
fulleropyrrolidine-thalidomide dyad as a new anti-inflammation
agent. Bioorg Med Chem. 16:8619–8626. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Amorati R, Valgimigli L, Panzella L,
Napolitano A and d'Ischia M: 5-S-lipoylhydroxytyrosol, a
multidefense antioxidant featuring a solvent-tunable peroxyl
radical-scavenging 3-thio-1,2-dihydroxybenzene motif. J Org Chem.
78:9857–9864. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kenneth NS, Hucks GE Jr, Kocab AJ,
McCollom AL and Duckett CS: Copper is a potent inhibitor of both
the canonical and non-canonical NFκB pathways. Cell Cycle.
13:1006–1014. 2014. View Article : Google Scholar :
|
|
64
|
Deng ZY, Hu MM, Xin YF and Gang C:
Resveratrol alleviates vascular inflammatory injury by inhibiting
inflammasome activation in rats with hypercholesterolemia and
vitamin D2 treatment. Inflamm Res. 64:321–332. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Baolin L, Inami Y, Tanaka H, Inagaki N,
Iinuma M and Nagai H: Resveratrol inhibits the release of mediators
from bone marrow-derived mouse mast cells in vitro. Planta Med.
70:305–309. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wang Q, Xu J, Rottinghaus GE, Simonyi A,
Lubahn D, Sun GY and Sun AY: Resveratrol protects against global
cerebral ischemic injury in gerbils. Brain Res. 958:439–447. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Eid N, Ito Y, Maemura K and Otsuki Y:
Elevated autophagic sequestration of mitochondria and lipid
droplets in steatotic hepatocytes of chronic ethanol-treated rats:
An immunohistochemical and electron microscopic study. J Mol
Histol. 44:311–326. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Song YM, Lee WK, Lee YH, Kang ES, Cha BS
and Lee BW: Metformin restores Parkin-mediated mitophagy,
suppressed by cytosolic p53. Int J Mol Sci. 17:E1222016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Khang R, Park C and Shin JH: Dysregulation
of parkin in the substantia nigra of db/db and high-fat diet mice.
Neuroscience. 294:182–192. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Luo X, Jia R, Yao Q, Xu Y, Luo Z, Luo X
and Wang N: Docosahexaenoic acid attenuates adipose tissue
angiogenesis and insulin resistance in high fat diet-fed
middle-aged mice via a sirt1-dependent mechanism. Mol Nutr Food
Res. 60:871–885. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Borengasser SJ, Faske J, Kang P, Blackburn
ML, Badger TM and Shankar K: In utero exposure to prepregnancy
maternal obesity and postweaning high-fat diet impair regulators of
mitochondrial dynamics in rat placenta and offspring. Physiol
Genomics. 46:841–850. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ono K and Yamada M: Vitamin A potently
destabilizes preformed alpha-synuclein fibrils in vitro:
Implications for Lewy body diseases. Neurobiol Dis. 25:446–454.
2007. View Article : Google Scholar
|
|
73
|
Casarejos MJ, Menéndez J, Solano RM,
Rodríguez-Navarro JA, García de Yébenes J and Mena MA:
Susceptibility to rotenone is increased in neurons from parkin null
mice and is reduced by minocycline. J Neurochem. 97:934–946. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Sonia Angeline M, Chaterjee P, Anand K,
Ambasta RK and Kumar P: Rotenone-induced parkinsonism elicits
behavioral impairments and differential expression of parkin, heat
shock proteins and caspases in the rat. Neuroscience. 220:291–301.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zhang YY, Huang J, Yang M, Gu LJ, Ji JY,
Wang LJ and Yuan WJ: Effect of a low-protein diet supplemented with
keto-acids on autophagy and inflammation in 5/6 nephrectomized
rats. Biosci Rep. 35:e002632015. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Koh H and Chung J: PINK1 as a molecular
checkpoint in the maintenance of mitochondrial function and
integrity. Mol Cells. 34:7–13. 2012. View Article : Google Scholar : PubMed/NCBI
|