Introduction
Diabetic nephropathy (DN) is the leading cause of
chronic kidney disease, which ultimately progresses to end-stage
renal failure. In early DN, glomerular abnormalities are observed,
including the increase in glomerular filtration rate and
albuminuria (1,2). Accumulating evidence indicates that
podocyte injury contributes to these pathological changes (3,4).
Clinical data also demonstrate that podocyte integrity is impaired
in individuals with type 1 and 2 diabetes (5,6).
It is widely acknowledged that the podocyte slit diaphragm plays a
critical role in establishing the selective permeability of the
glomerular filtration barrier (7). In our previous study (8), we demonstrated the in vivo
renal protective role of notoginsenoside R1 (NR1) through the
regulation of phosphoinositide 3-kinase (PI3K)/Akt signaling.
However, the mechanisms through which NR1 regulates this signaling
pathway remain unknown. Therefore in this study, we performed in
vitro experiments and focused on podocyte slit diaphragm
proteins that are important in maintaining podocyte integrity.
Podocyte decrement is also the leading cause of these pathological
changes. It has been reported that podocytes are highly
specialized, terminally differentiated cells with limited
replicative capacity (9). In both
type 1 and 2 diabetes, the decreased podocyte number is a major
pathophysiological precursor to the development of proteinuria
(10,11). Taken together, these data indicate
that the ability to maintain podocyte integrity and podocyte number
seems to be of pivotal importance in protecting against
diabetes.
It is acknowledged that the search for more
effective therapeutic drugs to prevent podocyte injury and to
maintain podocyte number is a great challenge. As herbal medicine
has been clinically used in the treatment of DN for centuries in
China, in this study, we investigated the role of NR1 (extracted
from Panax notoginseng) on podocytes and the underlying
mechanisms. It has been demonstrated that NR1 attenuates renal
ischemia-reperfusion injury in rats (12). It has also been demonstrated that
NR1 attenuates the glucose-induced impairement of podocyte adhesive
capacity and subsequent podocyte depopulation in rats through the
upregulation of α3β1 integrin (13). However, it is not clear whether
NR1 has any effect on the number of podocytes by affecting
apoptosis and inducing autophagy and podocyte integrity in addition
to its effect on podocyte adhesion (13).
Although a large body of evidence has indicated that
apoptosis contributes greatly to the podocyte depopulation,
autophagy has also been shown to be important in preservating the
number of podocytes (14–17). It has also been demonstrated that
in renal injury, autophagy plays a protective role by delaying the
progression of podocytopathies (18). In our previous in vivo
study (8), using mice, we
reported that NR1 protects podocytes from injury via the PI3K/Akt
pathway. However, which site activation/phosphorylation of the
PI3K/Akt pathway is regulated by NR1 remains unkown. Among several
signaling pathways that are functional in podocytes, it has been
reported that decreased the phosphorylation of Akt is associated
with podocyte loss in early DN (19). It has been demonstrated that in
rats with puromycin aminonucleoside (PAN) nephropathy, the
activation of the PI3K/Akt signaling inhibits podocyte apoptosis
(20). Furthermore, it has been
reported that Na+/H+ exchanger-1 (NHE-1) attenuates
podocyte injury via the PI3K/Akt pathway by inducing the activation
of autophagy (21). It is thus
possible that the PI3K/Akt pathway may be involved in both
apoptosis and autophagy during the process of podocyte injury.
Moreover, previous studies have demonstrated that the inhibition of
mammalian target of rapamycin (mTOR) protects podocytes from injury
and prevents DN through the regulation of autophagy (22,23). Due to the importance of the
PI3K/Akt/mTOR pathway in apoptosis and autophagy, in the present
study, we investigated the protective effects of NR1 on podocytes,
as well as whether and how the PI3K/Akt/mTOR signaling pathway is
regulated by NR1. Moreover, we wished to determine whether the
PI3K/Akt/mTOR signaling pathway is involved in the protective
effects of NR1 on podocytes, and to elucidate the mechanisms
through which Akt regulates both apoptosis and autophagy in
NR1-treated podocytes.
Materials and methods
Chemicals
NR1 was purchased from Sigma Chemical Co. (St.
Louis, MO, USA), and the purity of NR1 was >98%. The autophagy
inhibitor, 3-methyladenine (3-MA), monodansylcadaverine (MDC), and
2′,7′-dichlorofluorescein diacetate (DCF) were also purchased from
Sigma Chemical Co. Small interfering RNAs (siRNAs) against human
Beclin 1 and scramble siRNA were purchased from Cell Signaling
Technology, Inc. (Beverly, MA, USA). Hoechst 33342, the Click-iT
TUNEL Alexa Fluor 488 Imaging Assay kit for microscopy and
high-content screening (HCS), SYBR-Green PCR Master Mix, NuPage
Novex 10% Bis-Tris gel, NuPage transfer buffer, TRIzol, the High
Capacity cDNA Reverse Transcription kit, the Dead Cell Apoptosis
kit with Annexin V Alexa Fluor 488 and propidium iodide (PI) and
4′,6-diamidino-2-phenylindole (DAPI) were purchased from Invitrogen
Life Technologies (Carlsbad, CA, USA). Polyvinylidene fluoride
membranes were purchased from Pall Life Sciences (Port Washington,
NY, USA). Immobilon Western Chemiluminescent HRP Substrate was
purchased from Millipore Corp. (Billerica, MA, USA). The
CellTiter-Glo luminescent cell viability assay kit and terminal
deoxynucleotidyltransferase-mediated dUTP nick-end labeling (TUNEL)
assay kit were purchased from Promega Corp. (Madison, WI, USA)
The following antibodies were purchased from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA): nephrin (sc-377246),
desmin (sc-65983), pro-caspase-3 (sc-7148), pro-caspase-9
(sc-56073), cleaved caspase-9 (sc-22182), Bax (sc-493), Bid
(sc-135847), Bcl-2 (sc-492), Bcl-xL (sc-8392), poly(ADP-ribose)
polymerase (PARP; sc-7150) and β-actin (sc-47778). Besides, rat
anti-mouse podocalyxin (PCX) antibody (MAB1556) was purchased from
R&D Systems (Minneapolis, MN, USA). In addition, antibodies
against podocin (ab50339), CD2AP (ab84829), active caspase-3
(ab49822), Beclin (ab62557), PI3K p85 (ab189403), phosphorylated
(p-)Beclin 1 (S234; ab183335), mTOR (ab2732), p-mTOR (S2448;
ab109268), p-mTOR (S2481; ab137133), p-PI3K p85 (Y607; ab182651)
were from Abcam (Cambridge, UK). Antibodies to light chain 3 (LC3;
#12741), Akt (pan; #2920), cleaved caspase-9 (#9509), p-Akt (T308;
#9275), p-Akt (S473, #4060), p-p70S6K (Thr389; #9206) and
p-eukaryotic translation initiation factor 4E-binding protein 1
(4E-BP1; T37/46; #2855) were all purchased from Cell Signaling
Technology, Inc. Goat anti-rabbit IgG antibody conjugated with HRP
(#11-035-003), goat anti-mouse IgG antibody conjugated with HRP
(#115-035-003), and goat anti-rabbit IgG antibody conjugated with
Cy3 (#111-165-144) were purchased from Jackson ImmunoResearch
Laboratories, West Grove, PA, USA.
Cell culture and treatment with NR1
Conditionally immortalized human podocytes were
kindly provided by Dr Guisen Li (Division of Nephrology, from the
Sichuan Provincial People's Hospital) and were cultured as
previously described (24). In
brief, in order to maintain an undifferentiated proliferative
state, podocytes were cultured at 33°C with RPMI-1640 medium (Life
Technologies) supplemented with 10% fetal bovine serum (Life
Technologies). Before use, when the cells had reached 70–80%
confluence, they were cultured at 37°C to reach differentiation.
The podocytes were divided into the following groups: i) the low
glucose (LG) group: the cells were incubated in RPMI-1640
containing 5 mM D-glucose; ii) the high glucose (HG) group: the
cells were incubated in RPMI-1640 containing 30 mM D-glucose; iii)
the NR1-treated (NR1) group: the cells were incubated in RPMI-1640
containing 30 mM D-glucose and then treated with 20 µM NR1
for 24 h.
Cell viability and cell morphology
To ensure that NR1 can abolish the growth inhibitory
effects of high glucose on podocytes, podocyte viability was
measured within 24 h of exposure to a series of NR1 concentrations
(0, 1, 5, 10, 20, 50 and 100 µM). The number of viable cells
was assessed by CellTiter-Glo luminescent cell viability assay kit
according to the manufacturer's instructions in triplicate. Cell
morphology was also observed under a light microscope (Leica
Microsystems GmbH, Wetzlar, Germany).
Hoechst staining
The podocytes were grown to 80% confluence on 6-well
plates under growth-restricted conditions and treated with low
glucose, high glucose, or a combination of both high glucose and 20
µM NR1 for 24 h. Hoechst 33342 was added to achieve a final
concentration of 10 µM and the cells were analyzed after 5
min of Hoechst staining. Apoptotic cells with condensed chromatin
were manually counted for a minimum of 20 high-powered fields per
experimental condition by two blinded technicians. The number of
apoptotic nuclei was expressed as a percentage of the total number
of apoptotic nuclei.
Annexin V-PI assay
Following the experimental treatments, the
differentiated human podocytes were dispersed from 6-well culture
plates, collected in PBS, washed with FACS buffer, and stained
using the Dead Cell Apoptosis kit with Annexin V-PI following the
manufacturer's instructions. Cells (104) were acquired
using a FACScan flow cytometer (Becton-Dickinson, San Jose, CA,
USA). The number of apoptotic cells was analyzed using CellQuest
software 3.3 (Becton-Dickinson) and was expressed as a percentage
of the number of total cells.
TUNEL assay
Podocyte apoptosis was analyzed by TUNEL assay
according to the manufacturer's recommendations. Following fixation
in 4% buffered paraformaldehyde (PFA), the cells were incubated
with terminal deoxynucleotidyl transferase using FITC-labeled
nucleotides. DAPI served as a nuclear counterstain (blue).
TUNEL-positive cells (green) in 15–20 randomly selected fields were
manually counted out of the total number of cells.
Caspase assay
Podocyte apoptosis was also assessed by measuring
the activity of caspase-3 using the caspase-3 activity kit
(Beyotime Institute of Biotechnology, Haimen, China) according to
the manufacturer's instructions. In brief, the cells were lysed,
and the supernatant was collected, quantified and incubated with
the caspase-3-specific color substrate Ac-DEVD-pNA. Caspase-3
activity was determination by measuring optical density at OD400
nm.
Immunofluorescence staining
The podocytes were grown in triplicate on type I
collagen-coated glass coverslips, fixed with 4% PFA, and
permeabilized with Triton X-100. Following the removal of the
remaining Triton X-100, the cells were incubated with anti-active
caspase-3 antibodies overnight, followed by Cy3-conjugated
secondary antibody. For nuclear staining, the cells were stained
with DAPI. For the negative contro, PBS was used instead of the
primary antibody. The total cell population and the number of
cleaved caspase-3-positive cells were manually counted.
MDC assay and Beclin knockdown
Following incubation with 20 µM NR1 for the
indicated periods of time, the podocytes were cultured with 0.05 mM
MDC at 37°C for 60 min. The fluorescence intensity of the cells was
analyzed by flow cytometry. The cells were respectively transfected
with Beclin 1 siRNA and scramble siRNA at 33 nM using DharmaFect
(#T-2004; Dharmacon, Lafayette, CO, USA) according to the
manufacturer's instructions (25). The transfected cells were used for
subsequent experiments 24 h later.
Western blot analysis
The cells were lysed and the protein lysates were
centrifuged at 12,000 rpm for 30 min. Supernatants were collected
and loaded on a SDS-PAGE gel (Bio-Rad Laboratories, Inc., Hercules,
CA, USA) for electrophoresis. Following electrophoresis, the
proteins were transferred onto PVDF membranes in transfer buffer.
The membranes were blocked with 5% non-fat dry milk in TBS for 1 h.
The blots were incubated overnight at 4°C with primary antibodies
at the dilutions recommended by the manufacturer, followed by
incubating with horseradish peroxidase (HRP)-conjugated secondary
antibody, and then visualized by Immobilon Western Chemiluminescent
HRP substrate (26).
Reverse transcription-quantitative PCR
(RT-qPCR)
RNA was extracted using the RNeasy micro kit
(Qiagen, Hilden, Germany) and reverse transcribed [1 µg
total RNA, 1 µl random primer (50 µmol/l; Applied
Biosystems, Foster City, CA, USA), 1X reverse transcriptase buffer,
and 10 units reverse transcriptase (Promega Corp.) in a total
volume of 20 µl]. The RNA and primer were heated to 72°C and
slowly cooled before reverse transcription at 42°C for 1 h. The
room temperature reaction was then diluted to 100 µl with
RNase-free water (Applied Biosystems) as previously described
(27). For quantitative PCR
(qPCR), 2.5% of the total room temperature reaction was used as
input for PCR using SYBR-Green Master Mix (Applied Biosystems) and
templates were subjected to 36 rounds of PCR (94°C for 30 sec; 56°C
for 30 sec; 72°C for 1 min) on an ABI 7900 Real-Time PCR system. As
previously described (28–30),
the following primers were used: podocin forward,
5′-GGCTGTGGAGGCTGAAGC-3′ and reverse 5′-CTCAGAAGCAGCCTTTTCCG-3′;
nephrin forward, 5′-CGGTACAGGATCTGGCTGTT-3′ and reverse
5′-CTCTCTCCACCTCGTCATACA-3′; podocalyxin forward,
5′-CTACGGACTCATCTAACAAA-3′ and reverse, 5′-AGATAACC GATGACGGTA-3′;
desmin forward, 5′-CAGAATTGAATCTCTCAACG-3′ and reverse,
5′-CTTCAGAAATGTTCTTAGCC-3′; CD2AP forward,
5′-AACAGATACCGAAGGTAAAA-3′ and reverse, 5′-ATCAAATGGATTATCTCCCC-3′
and GAPDH forward, 5′-TGGTCACCAGGGCTGCTT-3′ and reverse,
5′-AGCTTCCCGTTCTCAGCCTT-3′. The 2−ΔΔCt method was used
to calculate the relative amount of the target RNAs, as previously
described (31).
Statistical analysis
Data are expressed as the means ± SEM from at least
3 independent experiments. Statistical significance was determined
by one-way ANOVA with Bonferroni correction for thye comparison of
3 groups or the Student's t-test for the comparison of 2
groups.
Results
NR1 protects against high glucose-induced
podocyte apoptosis
Accumulating evidence has indicated that podocyte
apoptosis is an early step in the progression of DN (15–17). We thus hypothesized that NR1 may
exert protective effects on podocytes by inhibiting apoptosis. Cell
viability was measured to determine the dose response of podocytes
to NR1 (Fig. 1A). NR1 at the
concentration of 20 µM was proven to be effective in
protecting the podocytes, as evidenced by a significant increase in
cell viability compared to the group exposed to high glucose.
Subsequent time-course experiments using 20 µM NR1 revealed
that NR1 treatment increased cell viability significantly after 8 h
and resulted in a 22.7% increase at 24 h (Fig. 1B). NR1 restored cell viability in
the HG-treated podocytes, indicating its protective effects on
podocytes. To induce significant changes in cell viability, NRI
treatment at 20 µM for 24 h of the HG-treated podocytes was
used in all subsequent experiments.
To determine whether NR1 protects podocytes from
apoptosis, cell morphology was assessed under a light miscroscope
(Fig. 1C). The podocytes in the
HG group exhibited marked apoptotic morphological changes,
including membrane blebbing, cell volume reduction and rounding.
These morphological changes were attenuated in the podocytes in the
NR1 group (Fig. 1C). Moreover,
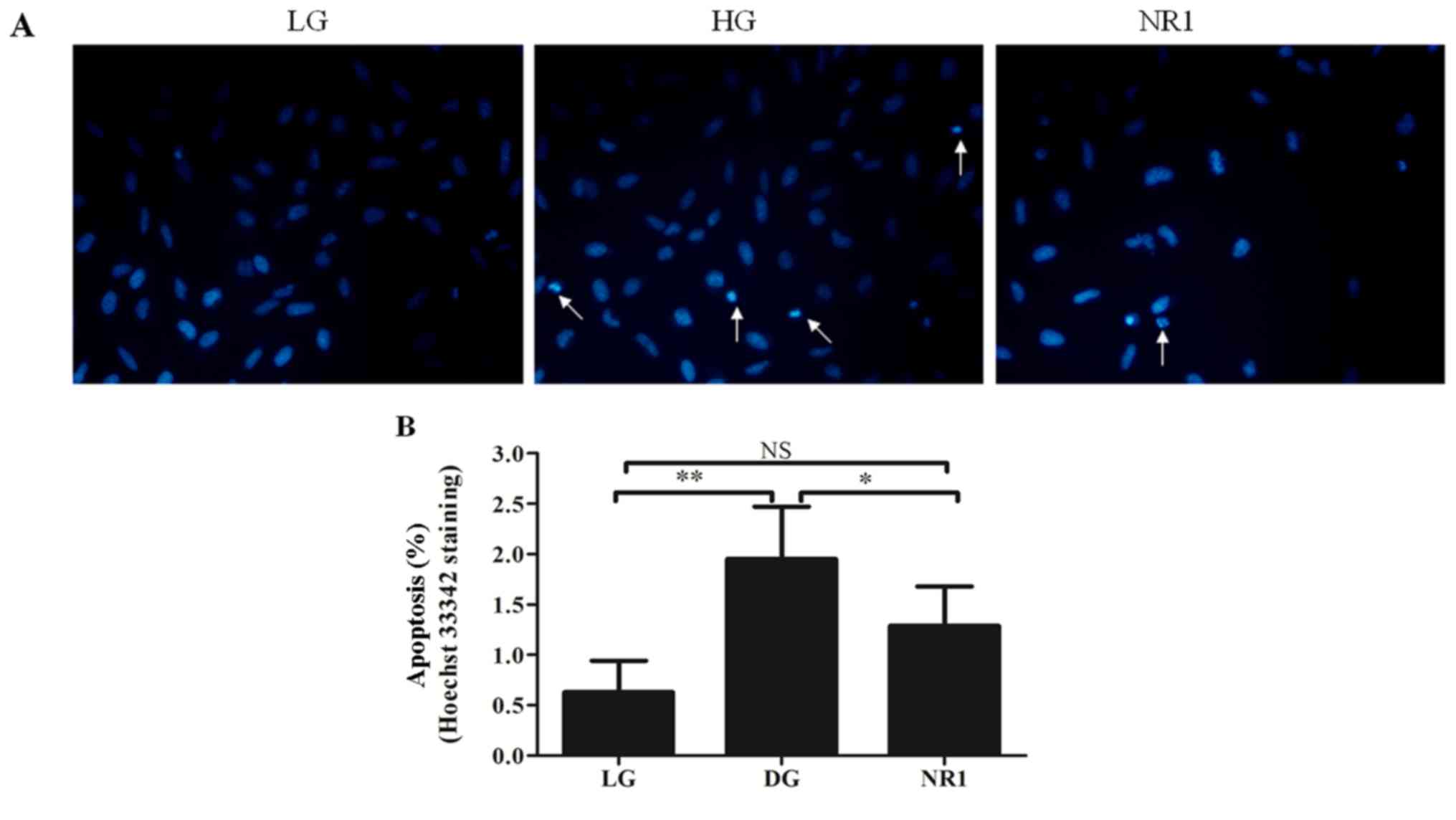
the percentage apoptosis was measured by Hoechst 33342 staining
(Fig. 2A and B). As shown in
Fig. 2A, considerably greater DNA
fragmentation in the nuclei was observed in the podocytes in the HG
group than in those in the LG group, and normal, round and
homogeneously stained nuclei were observed in the NR1-treated
podocytes. Statistical analysis also indicated that exposure to
high glucose resulted in increased apoptosis events that were
attenuated by NR1 (Fig. 2B).
There was no statistically significant difference in the percentage
apoptosis between the cells in the LG group and those in the NR1
group. In order to validate the results obtained by Hoechst 33342
staining, Annexin V-PI staining and FACS analysis were performed as
a second independent method to measure apoptosis (Fig. 3). The FACS data revealed similar
results as the Hoechst staining. In addition, TUNEL staining was
performed as a third independent method to measure apoptosis
(Fig. 4A), and the numbers of
TUNEL-positive cell percentages were manually calculated (Fig. 4B). Consistent with the other two
methods, the TUNEL staining results also demonstrated that high
glucose induced the apoptosis of podocytes and that this effect was
reversed by NR1. Taken together, these data convincingly
demonstrated that NR1 inhibits podocyte apoptosis in
vitro.
Effects of NR1 on foot process
effacement
It has been previously demonstrated that the slit
diaphragm bridges adjacent foot processes and functions as the
ultimate molecular size filter (32). Given the clinical significance of
the slit diaphragm proteins, we further analyzed the expression
levels of podocin, nephrin and CD2AP. As shown in Fig. 5A, the expression levels of these
proteins were significantly decreased under high glucose conditions
and NR1 reversed this effect. Consistent with the results of
western blot analysis, NR1 also restored the mRNA levels of
podocin, nephrin and CD2AP in the HG-treated podocytes (Fig. 5B). As PCX inhibits cell-matrix
interactions (33), and desmin is
a marker of podocyte injury (34,35), the protein expression of PCX and
desmin was assessed by western blot analysis. Exposure to high
glucose enhanced the expression levels of PCX and desmin in
podocytes, while NR1 inhibited these effects (Fig. 5).
NR1 inhibits apoptosis via the
caspase-dependent pathway
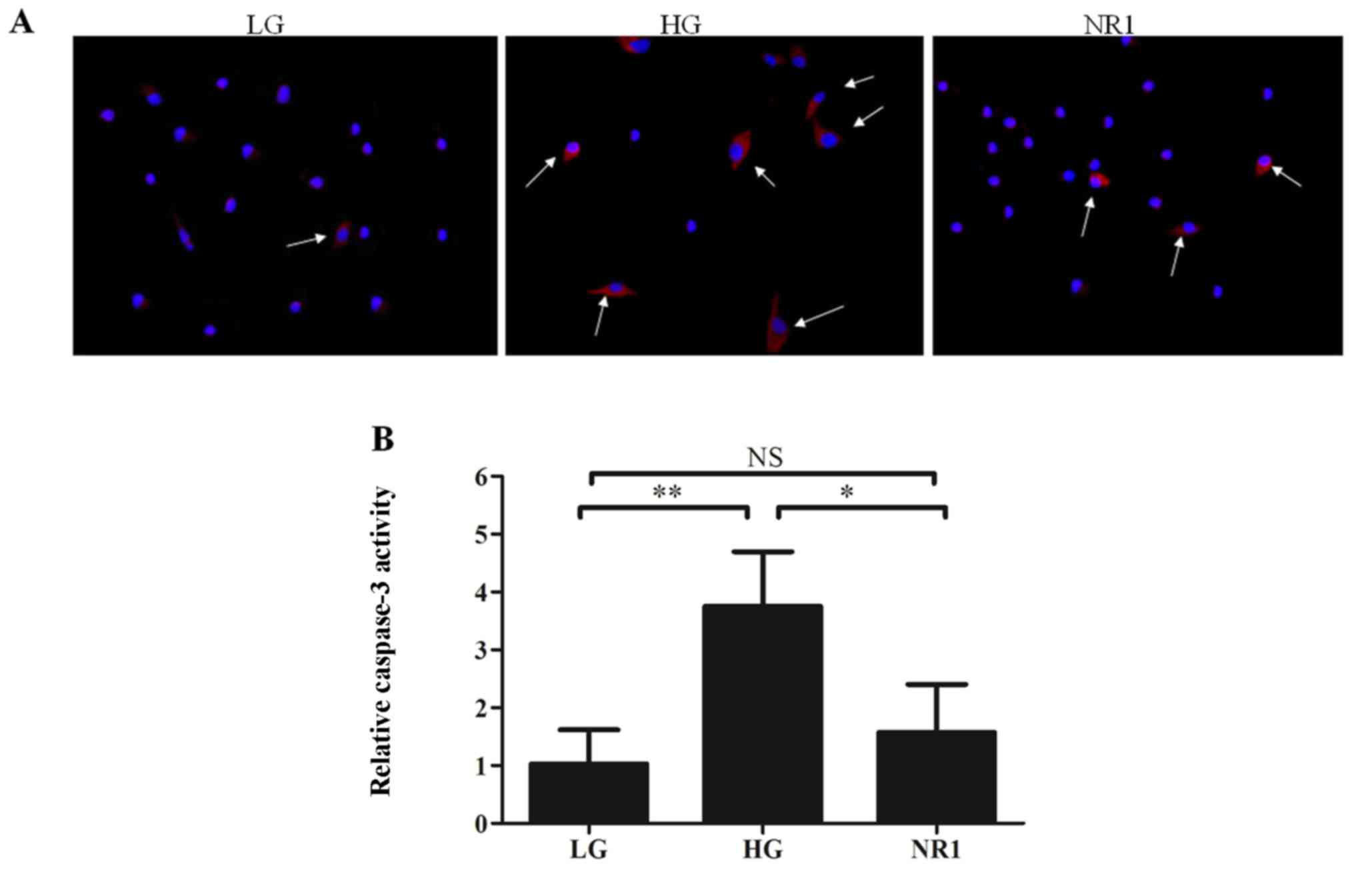
To further determine how NR1 inhibits podocyte
apoptosis, podocytes in the LG, HG and NR1 groups were stained with
cleaved caspase-3 antibody. The results revealed that high glucose
contributed to podocyte apoptosis by increasing cleaved caspase-3
expression, while NR1 significantly inhibited the expression of
cleaved caspase-3 (Fig. 6A).
Furthermore, caspase-3 activity assay (Fig. 6B) revealed that high glucose
enhanced caspase-3 activity in the podocytes, while NR1
significantly attenuated this effect. Western blot analysis of
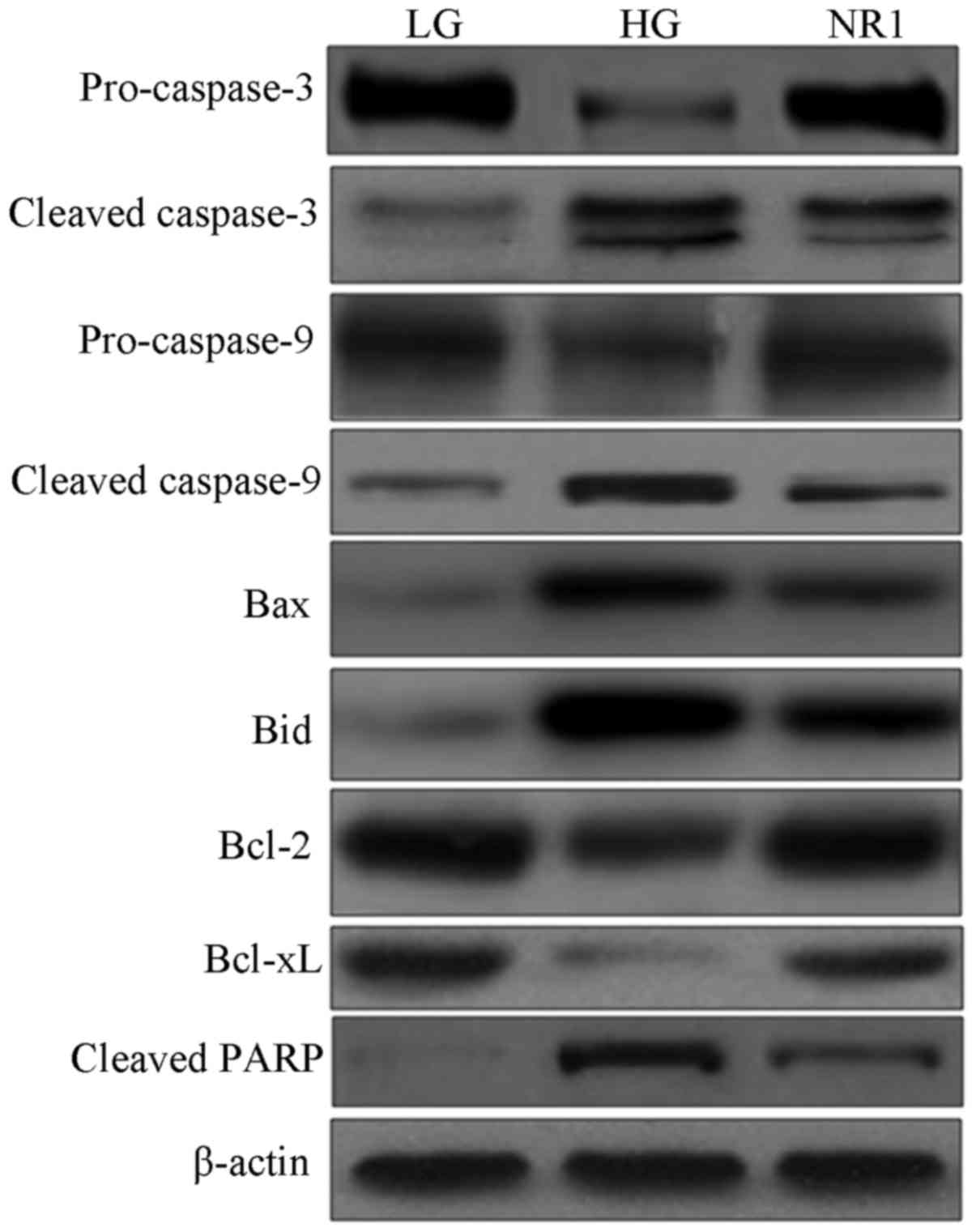
proteins involved in the apoptotic pathway (Fig. 7) revealed decreased expression
levels of pro-caspase-3 and -9, and increased expression levels of
cleaved caspase-3 and -9 in the HG group podocytes, indicating the
activation of caspase-9 and caspase-3. However the cleavage of
caspase-3 and -9 was partially inhibited by NR1. In addition, the
upregulation of the levels of pro-apoptotic Bax and Bid proteins,
as well as the downregulation of the anti-apoptotic Bcl-2 and
Bcl-xL proteins were observed in the podocytes in the HG group;
however, NR1 downregulated the expression levels of Bax and Bid,
and upregulated the expression levels of Bcl-2 and Bcl-xL in the
HG-treated podocytes. Further experiments of the PARP cleavage
product indicated that the exposure of the podocytes to HG
increased the cleavage of PARP, and treatment with NR1 resulted in
a substantial decrease in the cleavage of PARP. Taken together, our
data demonstrate that NR1 significantly attenuates HG-induced
podocyte apoptosis via the caspase-dependent pathway.
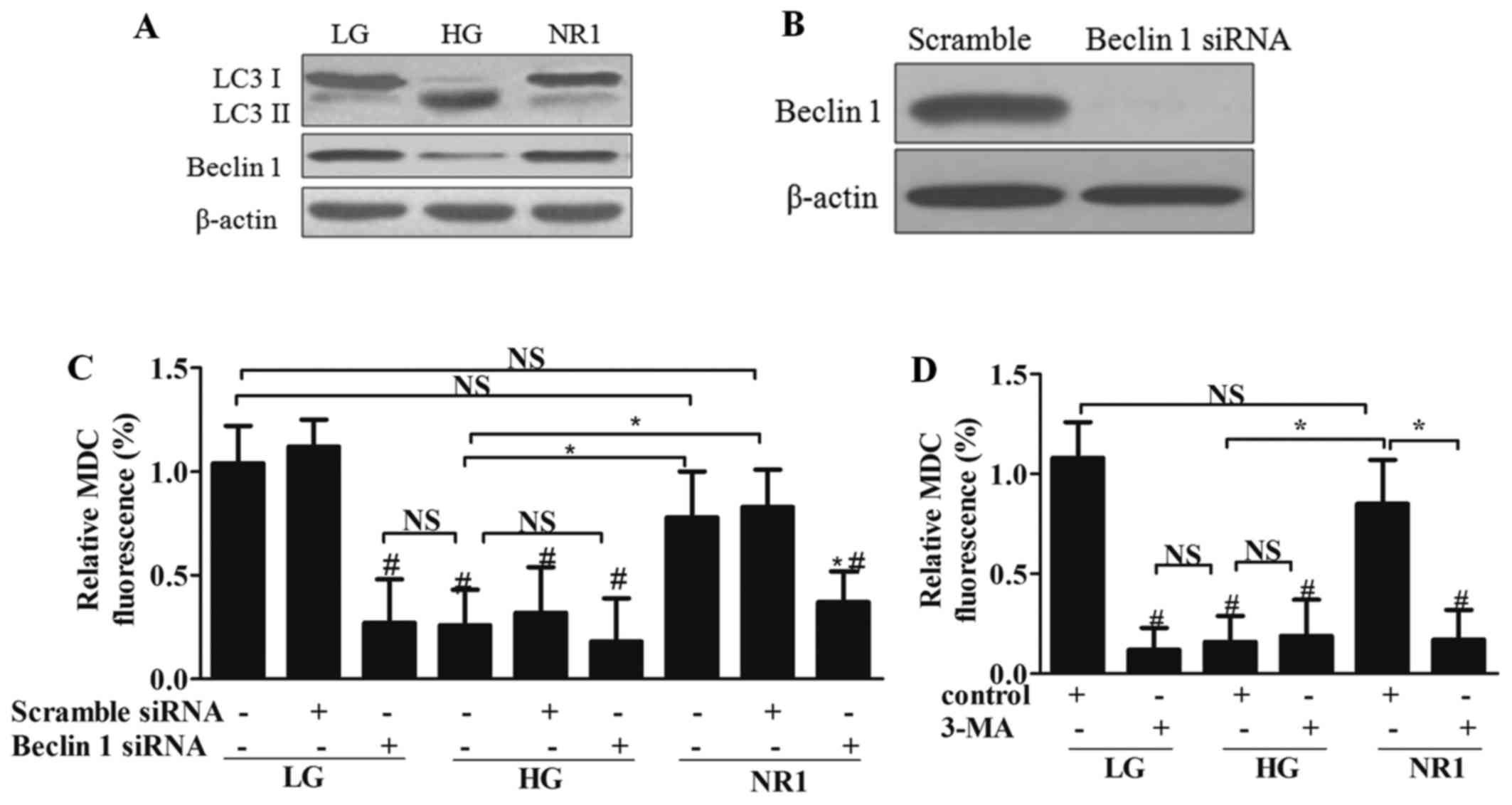
NR1 induces autophagy in podocytes
Increasing evidence indicates that podocytes have a
higher level of constitutive autophagy than other intrinsic renal
cells do, and we thus investigated the expression levels of
autophagy-related proteins. Western blot ananlysis (Fig. 8A) revealed that HG increased the
expression of LC3-II, and decreased the expression of LC3-I and
Beclin 1 in podocytes. However, NR1 significantly reversed these
effects on these proteins in the HG-treated cells to levels
comparable to those of the LG group. To further investigate the
role of autophagy in NR1 treatment, Beclin 1 was knocked down by
siRNA (Fig. 8B). By measuring the
MDC fluorescence intensity in the presence of Beclin 1 siRNA, we
found that the silencing of Beclin 1 resulted in reduced autophagy
in the NR1-treated podocytes (Fig.
8C). To further evaluate whether NR1 induces autophagy, the
chemical autphagy inhibitor, 3-MA, was added to the medium of
podocytes in each group. The MDC fluorescence intensity of the
podocytes treated with 3-MA also revealed that 3-MA inhibited
NR1-induced autophagy, while 3-MA had no effect on the HG-treated
podocytes (Fig. 8D). These
observations suggested that NR1 increased the basal autophagy that
was inhibited by HG.
NR1 protects podocytes from injury via
the PI3K/Akt/mTOR pathway
In order to further explore the mechanisms through
which NR1 inhibits apoptosis and increases autophagy, we
investigated whether the PI3K/Akt/mTOR signaling pathway is
involved in these mechanisms. As shown in Fig. 9, the phosphorylation levels of
PI3K (p85), Akt, mTOR and p70S6K and 4E-BP1, which are downstream
genes of mTOR, were markedly decreased in the podocytes in the HG
group. However, NR1 increased the phosphorylation levels of these
proteins, indicating that the activation of PI3K/Akt/mTOR pathway
may be involved in NR1 induced podocyte protection. These data
suggested that the NR1-induced protective effects on podocytes were
dependent on the phosphorylation and activation of the
PI3K/Akt/mTOR pathway.
Discussion
In the present study, we demonstrated that NR1
preserved the podocyte number by inhibiting apoptosis and inducing
autography. NR1 increased cell viability in a dose- and
time-dependent manner (Fig. 1A and
B). NR1 significantly attenuated high glucose induced podocyte
apoptosis (Fig. 1), mainly
through the caspase-dependent pathway (Figs. 6 and 7). NR1 also protected podocytes from
high glucose-induced damage by restoring the expression of proteins
involved in podocyte slit diaphragm (Fig. 5), which was dependent on the
activation of autophagy by NR1 (Fig.
8). Additionally, we found that NR1 activated the PI3K/Akt/mTOR
signaling pathway, which in turn inhibited apoptosis and enhanced
autography (Fig. 9). Taken
together, our studies suggest that NR1 protects podocytes from high
glucose-induced injury by inhibiting apoptosis and promoting
autophagy via the PI3K/Akt/mTOR pathway.
Podocytes, terminally differentiated cells, have a
very limited capacity for division and replacement. Therefore,
podocyte damage and decreased podocyte number contribute to
proteinuria and to the development of glomerulosclerosis. Emerging
evidence indicates that the podocyte slit diaphragm contributes
significantly to the size-selective filtration barrier in the
kidneys (36,37). Moreover, the disruption of the
cytoskeleton leads to foot process effacement in podocytes
(38). Herein, to elucidate the
mechanisms responsible for the protective effects of NR1 against
high glucose-induced podocyte damage, we performed western blot
analysis of slit diaphragm proteins and found that NR1 restored the
expression levels of these proteins (Fig. 5A). In addition, the results of
RT-qPCR (Fig. 5B) also
demonstrated that NR1 reduced the expression of desmin and
increased the expression of slit diaphragm proteins, which are
closely linked to the pathogenesis of foot process effacement under
hyperglycemia.
The inhibition of basal autophagy has also been
reported to be detrimental to the podocyte architectural structure
(22). Maintaining podocyte
number plays a pivotal role in the treatment of DN; therefore, in
our study, we focused on the anti-apoptotic and autophagy-inducing
effects of NR1. Accumulating clinical and experimental data have
demonstrated that increased apoptosis and the inhibition of
autophagy are important causes of decreased podocyte number
(14,39). Therefore, therapies targeting
apoptosis and autophagy pathways may stabilize podocyte number and
delay the progression of chronic glomerular disease. Our study
demonstrated that NR1 preserves the podocyte number by inhibiting
apoptosis and inducing autophagy. Firstly, we found that NR1
increased cell viability of HG-treated podocytes in a dose- and
time-dependent manner. Furthermore, NR1 protected the HG-treated
podocytes from apoptosis, verified by 3 independent methods.
Secondly, we found that NR1 was capable of inducing autophagy in
HG-treated podocytes, and this effect was inhibited by Beclin siRNA
or 3-MA (an autophagy inhibitor). Moreover, the enhancement of
autophagy induced by NR1 resulted in elevated expression levels of
slit diaphragm proteins and restored the cytoskeleton structure.
These data are consistent with the hypothesis that autophagy plays
an important role in maintaining podocyte number and function.
To explore the mechanisms underlying the
anti-apoptotic effects of NR1, we focused on the caspase pathway.
There is evidence to indicate that caspase-3 is involved in many
forms of apoptosis (40). It has
been demonstrated that caspase-3 is dispensable for certain forms
of apoptosis (41). Therefore,
whether caspase pathway is involved in the NR1-induced inhibition
of podocyte apoptosis gained our attention. It has been
demonstrated that in podocytes, PA caused activation of caspase-3
(42,43). Furthermore, previous studies have
indicated that caspase-3 is activated in podocytes under high
glucose conditions, and the activation of caspase-3 is caused by
Bcl-2 insufficiency (44,45). However, to the best of our
knowledge, no studies to date have focused on whether Bcl-xL and
other proteins in the caspase pathway are involved in the effects
of NR1. One of the major findings of our study was that NR1
suppressed apoptosis in a caspase-dependent manner. The results of
western blot analysis revealed that Bcl2, Bcl-xL and other proteins
in the caspase pathway wre involved in the NR1-induced inhibition
of apoptosis. In addition, immunofluorescence staining of cleaved
caspase-3 and caspase-3 activity assay also demonstrated that
caspase-3 activity was inhibited in the NR1-treated podocytes.
Taken together, by three independent methods, our study
demonstrates that the caspase pathway is involved in the
NR1-induced inhibition of the apoptosis of podocytes.
Among a wealth of signaling pathways in podocyte
protection, the PI3K/Akt signaling pathway is one of the main
anti-apoptotic pathways. A growing body of evidence supports the
notion that the PI3K/Akt pathway controls the remodeling of the
actin cytoskeleton and cell viability (46,47). Studies on podocytes, isolated from
diabetic mice, have demonstrated that the insulin-dependent
phosphorylation of Akt is impaired, and the dysregulation of Akt
phosphorylation is associated with podocyte apoptosis (19). Moreover, in vivo studies
have indicated that PI3K p85 interacts with nephrin and CD2AP to
form the slit diaphragm protein complex that was associated with a
strong activation of Akt (48).
Therefore, the PI3K/Akt pathway is involved in both the control of
cell number and the maintenance of podocyte biology. In this study,
we provide evidence showing that the PI3K/Akt pathway is
inactivated after podocyte injury by HG, and is then activated
following treatment with NR1. We also proved that NR1 induced
podocyte autophagy by the phosphorylation of Akt and mTOR.
In conclusion, our study suggests that NR1 protects
podocytes from injury, whose damage and decrement account for
proteinuria. NR1 plays an anti-apoptotic and autophagy-inducing
role in cultured podocytes through the PI3K/Akt/mTOR signaling.
Moreover, NR1 protects slit diaphragm proteins in podocytes under
high glucose conditions. Taken together, these findings provide
insight into the possible mechanisms through which NR1 treatment
may preserve the overall podocyte number and prevent podocyte
injury, thereby attenuating the development of glomerulosclerosis
and many forms of chronic kidney disease.
Acknowledgments
The present study was supported by grant no.
81160434 from the National Science Foundation of China, grant no.
2013GXNSFDA019016 from the Guangxi Science Foundation, grant no.
2015JY0183 from the Sichuan Science Foundation, a grant from the
Chengdu Science Foundation, grant no. 16ZD0253 from the Sichuan
Health and Family Planning Commission Funding, and Sichuan
Scientific Research Foundation of the Returned Overseas Chinese
Scholars. Funding was also provided from Sichuan Provincial
People's Hospital.
References
|
1
|
Mogensen CE: Microalbuminuria as a
predictor of clinical diabetic nephropathy. Kidney Int. 31:673–689.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rudberg S, Persson B and Dahlquist G:
Increased glomerular filtration rate as a predictor of diabetic
nephropathy - an 8-year prospective study. Kidney Int. 41:822–828.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
White KE, Bilous RW and Diabiopsies Study
Group: Structural alterations to the podocyte are related to
proteinuria in type 2 diabetic patients. Nephrol Dial Transplant.
19:1437–1440. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kriz W, Gretz N and Lemley KV: Progression
of glomerular diseases: is the podocyte the culprit. Kidney Int.
54:687–697. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dalla Vestra M, Masiero A, Roiter AM,
Saller A, Crepaldi G and Fioretto P: Is podocyte injury relevant in
diabetic nephropathy? Studies in patients with type 2 diabetes.
Diabetes. 52:1031–1035. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meyer TW, Bennett PH and Nelson RG:
Podocyte number predicts long-term urinary albumin excretion in
Pima Indians with Type II diabetes and microalbuminuria.
Diabetologia. 42:1341–1344. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pavenstadt H, Kriz W and Kretzler M: Cell
biology of the glomerular podocyte. Physiol Rev. 83:253–307. 2003.
View Article : Google Scholar
|
|
8
|
Huang G, Lv J, Li T, Huai G, Li X, Xiang
S, Wang L, Qin Z, Pang J, Zou B and Wang Y: Notoginsenoside R1
ameliorates podocyte injury in rats with diabetic nephropathy by
activating the PI3K/Akt signaling pathway. Int J Mol Med.
38:1179–1189. 2016.PubMed/NCBI
|
|
9
|
Quaggin SE: Transcriptional regulation of
podocyte specification and differentiation. Microsc Res Tech.
57:208–211. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
White KE, Bilous RW, Marshall SM, El Nahas
M, Remuzzi G, Piras G, De Cosmo S and Viberti G: Podocyte number in
normotensive type 1 diabetic patients with albuminuria. Diabetes.
51:3083–3089. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Verzola D, Gandolfo MT, Ferrario F,
Rastaldi MP, Villaggio B, Gianiorio F, Giannoni M, Rimoldi L,
Lauria F, Miji M, et al: Apoptosis in the kidneys of patients with
type II diabetic nephropathy. Kidney Int. 72:1262–1272. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu WJ, Tang HT, Jia YT, Ma B, Fu JF, Wang
Y, Lv KY and Xia ZF: Notoginsenoside R1 attenuates renal
ischemia-reperfusion injury in rats. Shock. 34:314–320. 2010.
View Article : Google Scholar
|
|
13
|
Gui D, Wei L, Jian G, Guo Y, Yang J and
Wang N: Notoginsenoside R1 ameliorates podocyte adhesion under
diabetic condition through α3β1 integrin upregulation in vitro and
in vivo. Cell Physiol Biochem. 34:1849–1862. 2014. View Article : Google Scholar
|
|
14
|
Hartleben B, Gödel M, Meyer-Schwesinger C,
Liu S, Ulrich T, Köbler S, Wiech T, Grahammer F, Arnold SJ,
Lindenmeyer MT, et al: Autophagy influences glomerular disease
susceptibility and maintains podocyte homeostasis in aging mice. J
Clin Invest. 120:1084–1096. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schiffer M, Bitzer M, Roberts IS, Kopp JB,
ten Dijke P, Mundel P and Böttinger EP: Apoptosis in podocytes
induced by TGF-beta and Smad7. J Clin Invest. 108:807–816. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Isermann B, Vinnikov IA, Madhusudhan T,
Herzog S, Kashif M, Blautzik J, Corat MA, Zeier M, Blessing E, Oh
J, et al: Activated protein C protects against diabetic nephropathy
by inhibiting endothelial and podocyte apoptosis. Nat Med.
13:1349–1358. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mundel P and Shankland SJ: Podocyte
biology and response to injury. J Am Soc Nephrol. 13:3005–3015.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zeng C, Fan Y, Wu J, Shi S, Chen Z, Zhong
Y, Zhang C, Zen K and Liu Z: Podocyte autophagic activity plays a
protective role in renal injury and delays the progression of
podocytopathies. J Pathol. 234:203–213. 2014.PubMed/NCBI
|
|
19
|
Tejada T, Catanuto P, Ijaz A, Santos JV,
Xia X, Sanchez P, Sanabria N, Lenz O, Elliot SJ and Fornoni A:
Failure to phosphorylate AKT in podocytes from mice with early
diabetic nephropathy promotes cell death. Kidney Int. 73:1385–1393.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xiao H, Shi W, Liu S, Wang W, Zhang B,
Zhang Y, Xu L, Liang X and Liang Y: 1,25-Dihydroxyvitamin D(3)
prevents puromycin aminonucleoside-induced apoptosis of glomerular
podocytes by activating the phosphatidylinositol
3-kinase/Akt-signaling pathway. Am J Nephrol. 30:34–43. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Feng Z, Tang L, Wu L, Cui S, Hong Q, Cai
G, Wu D, Fu B, Wei R and Chen X: Na+/H+
exchanger-1 reduces podocyte injury caused by endoplasmic reticulum
stress via autophagy activation. Lab Invest. 94:439–454. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cinà DP, Onay T, Paltoo A, Li C, Maezawa
Y, De Arteaga J, Jurisicova A and Quaggin SE: Inhibition of MTOR
disrupts autophagic flux in podocytes. J Am Soc Nephrol.
23:412–420. 2012. View Article : Google Scholar :
|
|
23
|
Gödel M, Hartleben B, Herbach N, Liu S,
Zschiedrich S, Lu S, Debreczeni-Mór A, Lindenmeyer MT, Rastaldi MP,
Hartleben G, et al: Role of mTOR in podocyte function and diabetic
nephropathy in humans and mice. J Clin Invest. 121:2197–2209. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Saleem MA, O'Hare MJ, Reiser J, Coward RJ,
Inward CD, Farren T, Xing CY, Ni L, Mathieson PW and Mundel P: A
conditionally immortalized human podocyte cell line demonstrating
nephrin and podocin expression. J Am Soc Nephrol. 13:630–638.
2002.PubMed/NCBI
|
|
25
|
Li C, Chen J, Lu B, Shi Z, Wang H, Zhang
B, Zhao K, Qi W, Bao J and Wang Y: Molecular switch role of Akt in
Polygonatum odoratum lectin-induced apoptosis and autophagy in
human non-small cell lung cancer A549 cells. PLoS One.
9:e1015262014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Y, Liu Y, Wang H, Li C, Qi P and Bao
J: Agaricus bisporus lectins mediates islet β-cell proliferation
through regulation of cell cycle proteins. Exp Biol Med (Maywood).
237:287–296. 2012. View Article : Google Scholar
|
|
27
|
Wang Y, Wang H, Liu Y, Li C, Qi P and Bao
J: Antihyperglycemic effect of ginsenoside Rh2 by inducing islet
β-cell regeneration in mice. Horm Metab Res. 44:33–40. 2012.
View Article : Google Scholar
|
|
28
|
Rantanen M, Palmén T, Pätäri A, Ahola H,
Lehtonen S, Aström E, Floss T, Vauti F, Wurst W and Ruiz P: Nephrin
TRAP mice lack slit diaphragms and show fibrotic glomeruli and
cystic tubular lesions. J Am Soc Nephrol. 13:1586–1594. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kawachi H, Koike H, Kurihara H, Sakai T
and Shimizu F: Cloning of rat homologue of podocin: expression in
proteinuric states and in developing glomeruli. J Am Soc Nephrol.
14:46–56. 2003. View Article : Google Scholar
|
|
30
|
Wiggins JE, Goyal M, Sanden SK, Wharram
BL, Shedden KA, Misek DE, Kuick RD and Wiggins RC: Podocyte
hypertrophy, 'adaptation,' and 'decompensation' associated with
glomerular enlargement and glomerulosclerosis in the aging rat:
prevention by calorie restriction. J Am Soc Nephrol. 16:2953–2966.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Reiser J, Kriz W, Kretzler M and Mundel P:
The glomerular slit diaphragm is a modified adherens junction. J Am
Soc Nephrol. 11:1–8. 2000.PubMed/NCBI
|
|
33
|
Nielsen JS and McNagny KM: The role of
podocalyxin in health and disease. J Am Soc Nephrol. 20:1669–1676.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yaoita E, Kawasaki K, Yamamoto T and
Kihara I: Variable expression of desmin in rat glomerular
epithelial cells. The Am J Pathol. 136:899–908. 1990.PubMed/NCBI
|
|
35
|
Floege J, Alpers CE, Sage EH, Pritzl P,
Gordon K, Johnson RJ and Couser WG: Markers of complement-dependent
and complement-independent glomerular visceral epithelial cell
injury in vivo. Expression of antiadhesive proteins and
cytoskeletal changes. Lab Invest. 67:486–497. 1992.PubMed/NCBI
|
|
36
|
Somlo S and Mundel P: Getting a foothold
in nephrotic syndrome. Nat Genet. 24:333–335. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kerjaschki D: Caught flat-footed: podocyte
damage and the molecular bases of focal glomerulosclerosis. J Clin
Invest. 108:1583–1587. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Saleem MA, Ni L, Witherden I, Tryggvason
K, Ruotsalainen V, Mundel P and Mathieson PW: Co-localization of
nephrin, podocin, and the actin cytoskeleton: evidence for a role
in podocyte foot process formation. Am J Pathol. 161:1459–1466.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Asanuma K, Tanida I, Shirato I, Ueno T,
Takahara H, Nishitani T, Kominami E and Tomino Y: MAP-LC3, a
promising autophagosomal marker, is processed during the
differentiation and recovery of podocytes from PAN nephrosis. FASEB
J. 17:1165–1167. 2003.PubMed/NCBI
|
|
40
|
Nunez G, Benedict MA, Hu Y and Inohara N:
Caspases: the proteases of the apoptotic pathway. Oncogene.
17:3237–3245. 1998. View Article : Google Scholar
|
|
41
|
Mohr S, McCormick TS and Lapetina EG:
Macrophages resistant to endogenously generated nitric
oxide-mediated apoptosis are hypersensitive to exogenously added
nitric oxide donors: dichotomous apoptotic response independent of
caspase 3 and reversal by the mitogen-activated protein kinase
kinase (MEK) inhibitor PD 098059. Proc Natl Acad Sci USA.
95:5045–5050. 1998. View Article : Google Scholar
|
|
42
|
Wada T, Pippin JW, Marshall CB, Griffin SV
and Shankland SJ: Dexamethasone prevents podocyte apoptosis induced
by puromycin aminonucleoside: role of p53 and Bcl-2-related family
proteins. J Am Soc Nephrol. 16:2615–2625. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li X, Zhang X, Li X, Ding F and Ding J:
The role of survivin in podocyte injury induced by puromycin
aminonucleoside. Int J Mol Sci. 15:6657–6673. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen YQ, Wang XX, Yao XM, Zhang DL, Yang
XF, Tian SF and Wang NS: MicroRNA-195 promotes apoptosis in mouse
podocytes via enhanced caspase activity driven by BCL2
insufficiency. Am J Nephrol. 34:549–559. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gui D, Guo Y, Wang F, Liu W, Chen J, Chen
Y, Huang J and Wang N: Astragaloside IV, a novel antioxidant,
prevents glucose-induced podocyte apoptosis in vitro and in vivo.
PLoS One. 7:e398242012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Franke TF, Hornik CP, Segev L, Shostak GA
and Sugimoto C: PI3K/Akt and apoptosis: size matters. Oncogene.
22:8983–8998. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Huber TB, Hartleben B, Kim J, Schmidts M,
Schermer B, Keil A, Egger L, Lecha RL, Borner C, Pavenstädt H, et
al: Nephrin and CD2AP associate with phosphoinositide 3-OH kinase
and stimulate AKT-dependent signaling. Mol Cell Biol. 23:4917–4928.
2003. View Article : Google Scholar : PubMed/NCBI
|