Introduction
Disruption of intestinal epithelial barrier function
is a key characteristic of inflammatory bowel disease (IBD)
(1–4). An intact intestinal epithelial
barrier is essential for defending against the invasion of antigens
and for maintaining the internal environment, which separates
luminal contents within the body and prevents luminal bacterial and
microbial translocation. Intestinal barrier dysfunction leads to
entry of intestinal bacteria into the body, causing local or
systemic inflammatory responses (5,6).
Repair of the intestinal mucosa has become a standard criterion for
evaluating the efficacy of IBD treatments (7). Therefore, effective maintenance of
intestinal epithelial barrier function is of great importance in
IBD.
Integrity of the intestinal epithelial barrier
depends on a complex network of intercellular tight junctions (TJs)
and cytoskeletal structures, and disruption of this network results
in impaired barrier function (8–11).
TJ proteins are located at the apical ends of epithelial cells,
which contain both transmembrane (e.g., occludin, claudin and
junctional adhesion molecules) and cytoplasmic zonula occludens
(ZO) proteins. As the determinant of epithelial permeability, TJ
proteins are the most important component of the epithelial mucosal
barrier (12,13). Damage to intestinal epithelial TJs
leads to increased intercellular permeability, allowing for
bacteria and endotoxins to penetrate the mucosa and enter into
circulation, resulting in severe inflammatory responses (14,15). Rearrangement and redistribution of
cytoskeletal F-actin also leads to changes in intestinal epithelial
barrier permeability (16).
Tumor necrosis factor (TNF)-α is a multifunctional
proinflammatory cytokine which participates in the pathogenesis of
IBD (17). TNF-α is associated
with damage to intestinal TJs and cytoskeletal rearrangements,
which lead to increased intestinal permeability. It also promotes
release of the inflammatory factors interleukin (IL)-8, IL-17 and
interferon (IFN)-γ (18–20). Both the myosin light chain kinase
(MLCK)/p-MLC pathway and nuclear factor (NF)-κB activation have key
roles in TNF-α-mediated damage to the intestinal barrier (21,22). Activation of the MLCK/p-MLC
pathway leads to upregulation of the expression of TJ proteins,
alterations in cell adhesion and migration, and cytoskeletal
rearrangements (16,23,24).
Anterior gradient protein 2 homologue (AGR2) is a
member of the protein disulphide isomerase family that plays an
important role in maintaining intestinal homeostasis in IBD
(25). The prevalence of AGR2
gene mutations is higher in patients with IBD than in the general
population (26). In animal
models of IBD, AGR2 has been shown to maintain homeostatic
functions in intestinal Paneth and goblet cells (27), and AGR2-knockout mice have been
reported to be susceptible to the development of severe ileocolitis
(28). Thus, AGR2 is essential
for maintaining the intestinal epithelial cell function in IBD, but
it is unclear whether it regulates intestinal epithelial barrier
function.
Therefore, we investigated the roles of AGR2 in
epithelial permeability, TJ protein expression, and cytoskeletal
structure. Furthermore, we evaluated the mechanism by which AGR2
ameliorates TNF-α-mediated damage to the intestinal barrier.
Materials and methods
Cell culture and preparation
Caco-2 cells were purchased from the Chinese Academy
of Sciences (Shanghai, China). The cells were cultured in RPMI-1640
medium containing 10% fetal bovine serum (FBS; ScienCell Research
Laboratories, San Diego, CA, USA) at 37°C in 5% CO2. For
the monolayer model, cells were plated at 3×104/ml onto
Transwell filters with a 0.4-μm pore size (Millipore,
Billerica, MA, USA) and cultured for 21 to 28 days prior to the
experiments. The culture medium was changed every 2 days. The
experimental monolayers were pre-transfected with an AGR2 plasmid
and were then stimulated with 100 ng/ml rhTNF-α for 48 h.
Plasmid construction and
transfection
A pcDNA3.1 eukaryotic expression vector carrying the
human AGR2 gene was constructed. Cells were transfected with either
a pcDNA3.1-AGR2 plasmid or a pcDNA3.1 vector control plasmid. For
transfection, the cells were incubated in 100 μl Opti-MEM
medium containing 8 μl Lipofectamine 2000 (both from
Invitrogen Life Technologies, Carlsbad, CA, USA) for 5 min,
followed by the addition of 2 μg plasmids and incubation for
another 20 min. The mixtures were then transferred to 6-well plates
and cultured for 4 h. Subsequently, the transfection medium was
substituted with culture medium, and the transfected cells were
cultured at 37°C in 5% CO2.
Determination of transepithelial
electrical resistance (TEER)
Caco-2 cells were seeded onto Transwell chambers,
and epithelial permeability was assessed by evaluating TEER using a
Millicell ERS-2 Voltohmmeter (Millipore). Two wells containing only
culture medium were used as blank controls. Measurements were
carried out at a constant temperature at three different locations
within each chamber. The mean value of three consecutive
measurements was used to calculate TEER.
TEER (Ω · cm2) = (measured TEER - control
TEER) × effective membrane area of the cell culture well.
Measurement of paracellular marker
fluorescein isothiocyanate (FITC)-dextran 40 (FD-40) (40 kDa)
flux
The monolayers were washed with HBSS, and the
RPMI-1640 medium in the apical chambers was replaced with
FITC-conjugated dextran (80 μg/ml, 40 kDa; Sigma-Aldrich,
St. Louis, MO, USA) in HBSS. The monolayers were incubated at 37°C
for 1 h. Then, the fluorescence of the culture medium in the lower
chambers was detected with a fluorescence plate reader (Varioskan
Flash; Thermo Electron Corporation, Vantaa, Finland) at excitation
and emission wavelengths of 427 and 536 nm, respectively.
Fluorescein concentrations were determined by comparison to a
standard curve.
Fluorescence transmittance (%) = FITC-dextran
concentration in the lower chamber/FITC-dextran concentration added
to the upper chamber × 100.
Quantitative (real-time) PCR (qPCR)
TRIzol was utilized to extract total RNA from the
cells, and reserve transcription was carried out using a
PrimeScript RT reagent kit (Takara Bio Inc., Shiga, Japan). qPCR
was performed using a SYBR Premix Ex kit (Takara Bio Inc.) and a
Bio-Rad iQ5 Real-Time system (Bio-Rad Laboratories, Inc., Hercules,
CA, USA). The primers for this study were as follows: β-actin
forward, 5′-CTTAGTTGCGTTACACCCTTTCTTG-3′ and reverse,
5′-CTGTCACCTTCACCGTTCCAGTTT-3′; AGR2 forward,
5′-GCATTCTTGCTCCTTGTGG-3′ and reverse, 5′-GACTGTGTGGGCACTCATCC-3′.
Expression levels were calculated using the 2−ΔΔCT
method.
Western blotting
Radioimmunoprecipitation assay (RIPA) and
phenylmethanesulfonyl fluoride (PMSF) lysate buffers were used to
extract total protein from the Caco-2 cells. The protein
concentrations of the extracted samples were determined by
bicinchoninic acid assay. These concentrations were adjusted by
addition of distilled water. Equal volumes of sample buffer and
boiling water were mixed, and the mixtures were boiled for 5 min to
denature the proteins. Equal amounts of proteins (40 μg) for
each sample were loaded onto SDS-PAGE gels for electrophoresis, and
then transferred to PVDF membranes and blocked by 5% non-fat milk
for 1 h. Next, incubation was carried out with the primary
antibodies, including anti-AGR2 (ab76473; 1:3,000), anti-ZO-1
(ab59720; 1:500), anti-claudin-1 (ab15098; 1:500), anti-occludin
(ab31721; 1:500) anti-MLCK (ab76092; 1:5,000) and anti-NF-κB
(ab207297; 1:500) (all from Abcam, Cambridge, MA, USA), anti-p-MLC
(#3671; 1:750) and anti-MLC (#8505; 1:750) (both from Cell
Signaling Technology, Inc., Danvers, MA, USA) and anti-β-actin
(20536-1-AP; 1:2,000; Proteintech, Wuhan, China) overnight at 4°C.
After that, incubation was carried out with horseradish peroxidase
(HRP)-conjugated secondary antibodies (SA00001-2; 1:5,000;
Proteintech) for 2 h and visualized by electrochemiluminescence
(ECL). Protein bands were detected using Gel-Pro Analyser 4.0
(Media Cybernetics, Inc., Rockville, MD, USA).
Immunofluorescence staining
Caco-2 monolayers were pre-transfected with AGR2
gene plasmids or a control plasmid vector; after 24 h, 100 ng/ml
TNF-α was added. After 48 h of TNF-α exposure, the cultures were
fixed with 4% polyformaldehyde for 15 min and then incubated with
0.5% Triton X-100 for 30 min. Subsequently, the cultures were
incubated with goat serum for 15 min. After that, the cells were
incubated with primary antibodies including anti-F-actin (ab205;
1:100), anti-ZO-1 (1:200), anti-occludin (1:100) and anti-NF-κB p65
(1:200) (all from Abcam) overnight at 4°C. Next, they were
incubated with a fluorescent-labelled goat anti-rabbit IgG (H+L)
secondary antibody (SA00003-1; 1:200; Proteintech) for 1 h at room
temperature. After nuclear counterstaining of the cells with
4′,6-diamidino-2-phenylindole (DAPI) (Life Technologies, Thermo
Fisher Scientific, Inc., Waltham, MA, USA), images were captured by
laser scanning fluorescence microscopy (TCS SP5; Leica
Microsystems, Wetzlar, Germany) at ×400 magnification.
Statistical analysis
Assays were performed at least in triplicate, and
the results were reported as the mean ± SD. The significance of
differences was assessed by analysis of variance (ANOVA) or the
t-test using SPSS version 21.0 software (SPSS, Inc., Chicago, IL,
USA). A P<0.05 was considered statistically significant.
Results
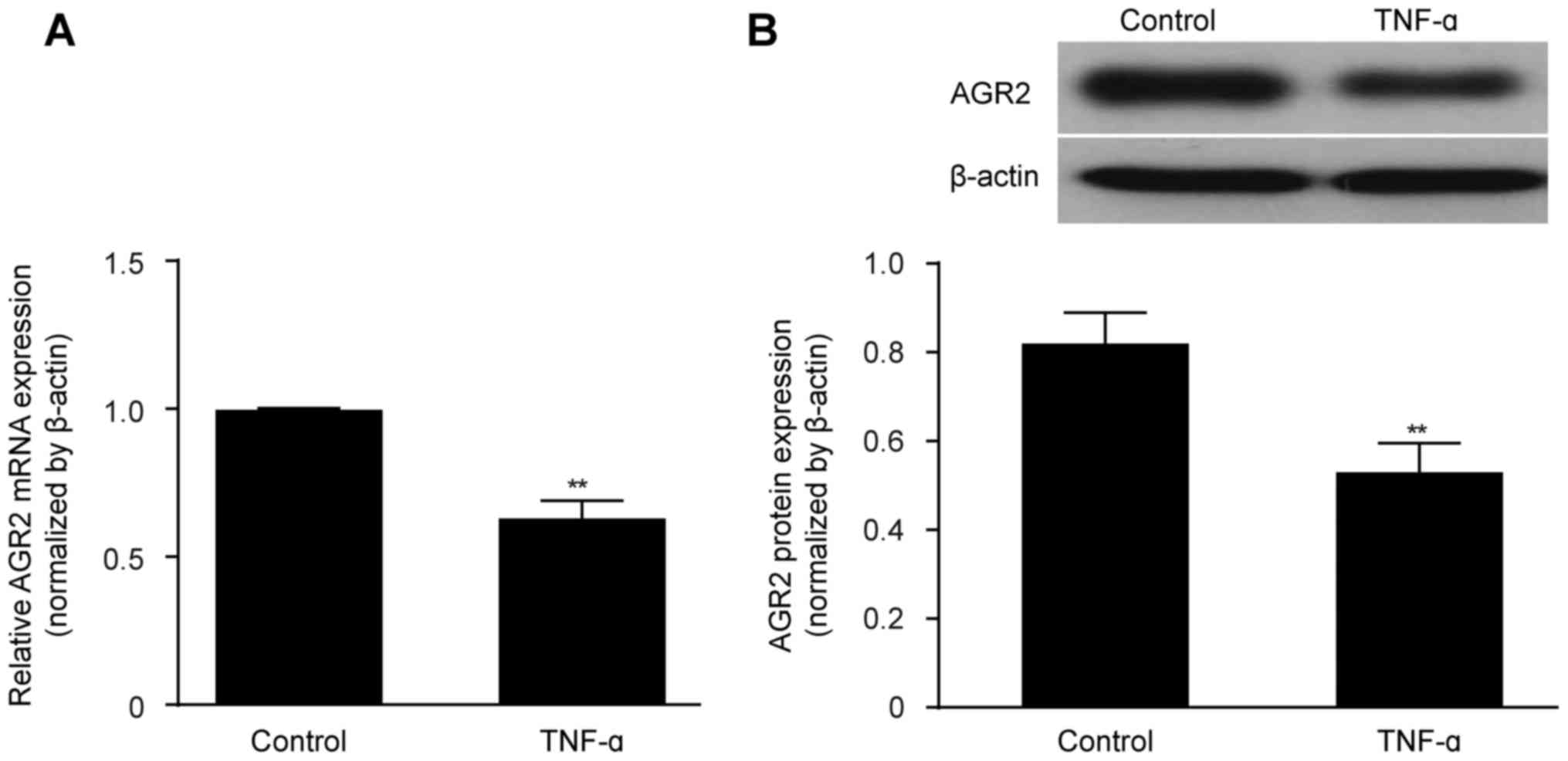
TNF-α decreases AGR2 expression in
confluent monolayers of cultured intestinal epithelial cells
Caco-2 cell cultures reached confluence by day 21,
forming monolayers consisting of irregular polygonal cells with a
cobblestone appearance and intercellular TJs, as observed by light
micro scopy. TEER remained stable at >600 Ω/cm2,
indicating the successful establishment of the in vitro
intestinal epithelial barrier model. TNF-α (100 ng/ml) was then
added to the model cultures, and after 48 h, the expression level
of AGR2 was detected. Both the AGR2 mRNA and protein expression
levels were obviously decreased by TNF-α exposure compared with the
levels in the untreated control monolayers (Fig. 1A and B).
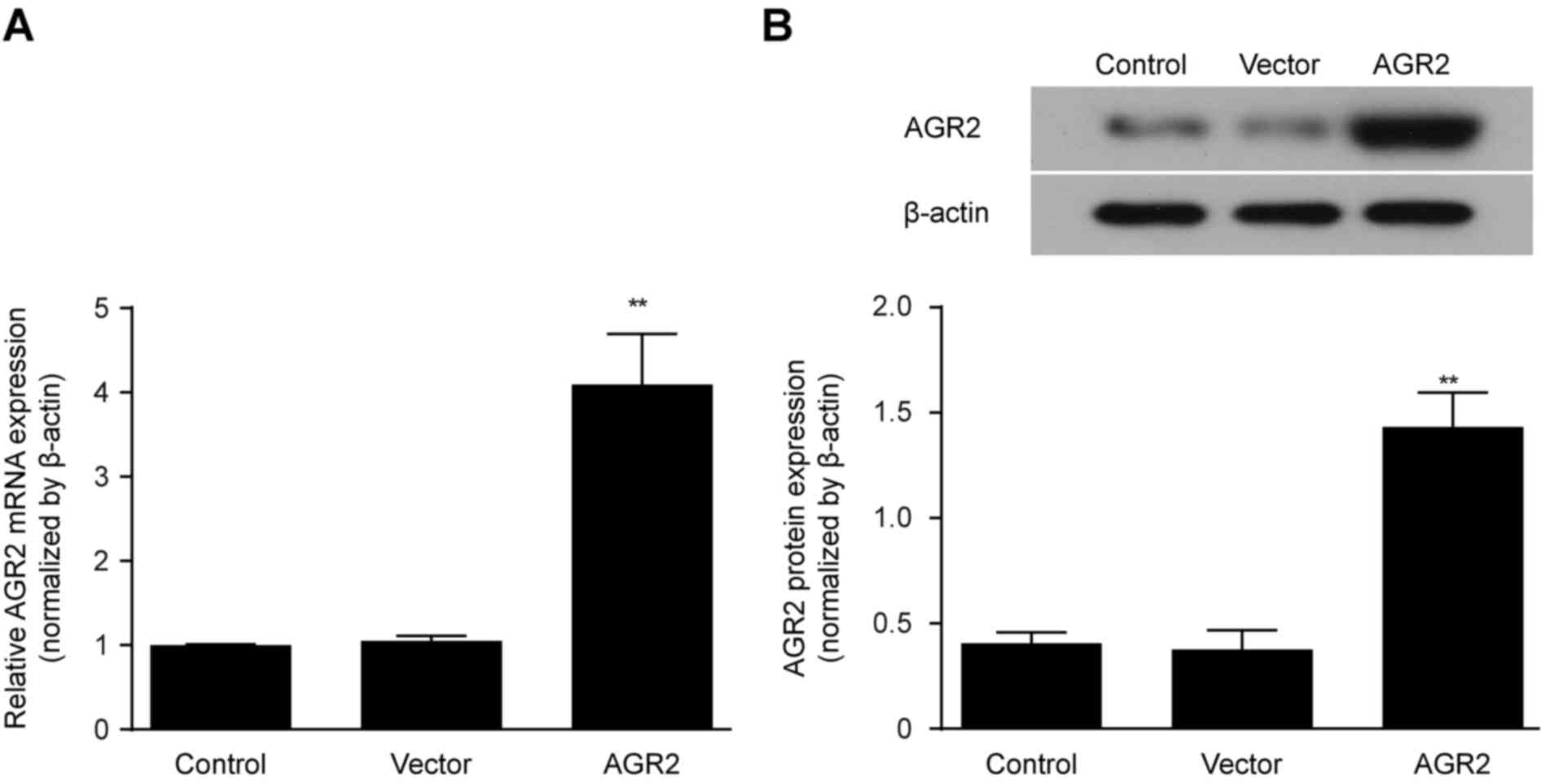
Construction of an AGR2 overexpression
model by transfection of an AGR2 or control plasmid
AGR2 or control plasmid vectors were transfected
in vitro into Caco-2 cell monolayers, and AGR2 mRNA
expression was determined by qRT-PCR after 24 h. After another 48 h
of culturing, AGR2 protein expression was measured by western
blotting and compared with that in the controls. The results showed
that both the AGR2 mRNA and protein expression levels were
significantly increased in the Caco-2 cells transfected with the
AGR2 plasmids (Fig. 2A and B).
These findings indicate the successful construction of the AGR2
plasmid.
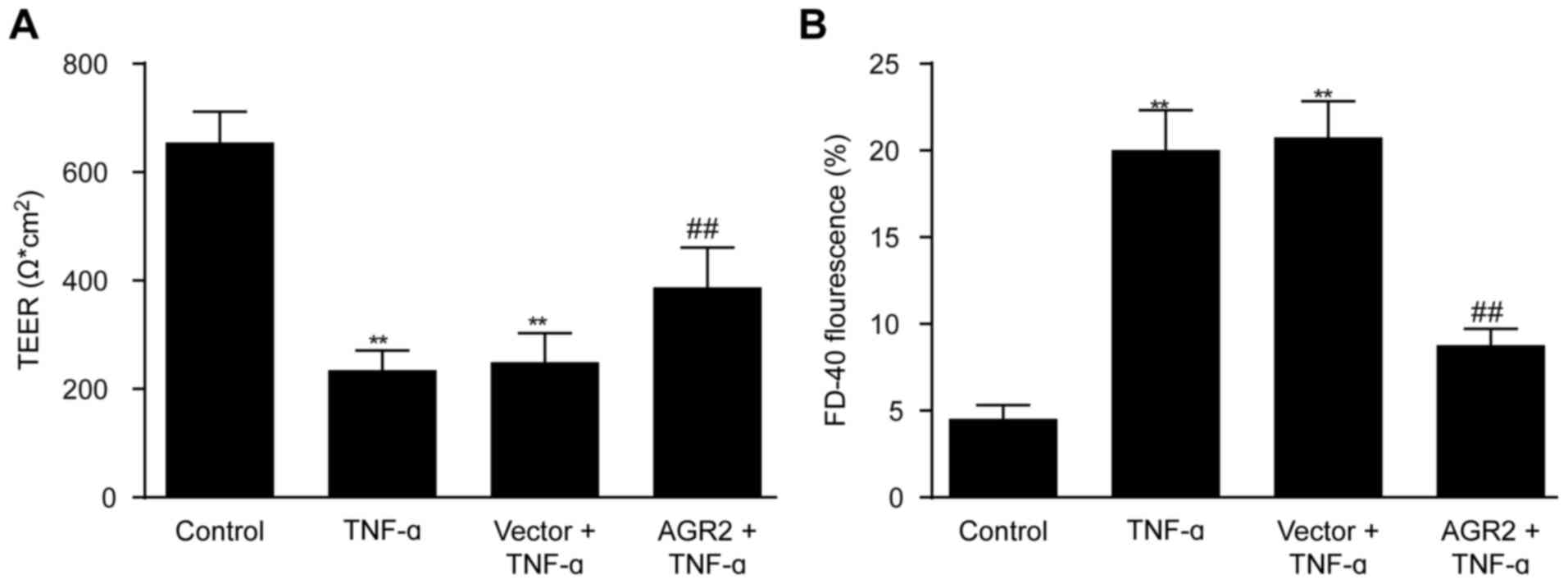
AGR2 reduces TNF-α-induced increase in
permeability of intestinal epithelial cell monolayers
Caco-2 cell monolayers were transfected with AGR2 or
control plasmids, and after incubation for 24 h, 100 ng/ml TNF-α
was added. Permeability of the intestinal epithelial cell
monolayers was assessed by measuring TEER and FD-40 flux after 48 h
of incubation with TNF-α. The results showed that TEER was
significantly lower and FD-40 flux was significantly higher in the
TNF-α-stimulated monolayers compared with that noted in the control
monolayers (Fig. 3A and B). AGR2
plasmid transfection significantly inhibited the decrease in TEER
as well as the increase in FD-40 flux induced by TNF-α (Fig. 3A and B), indicating that AGR2
ameliorated the TNF-α-induced increase in permeability of the model
system.
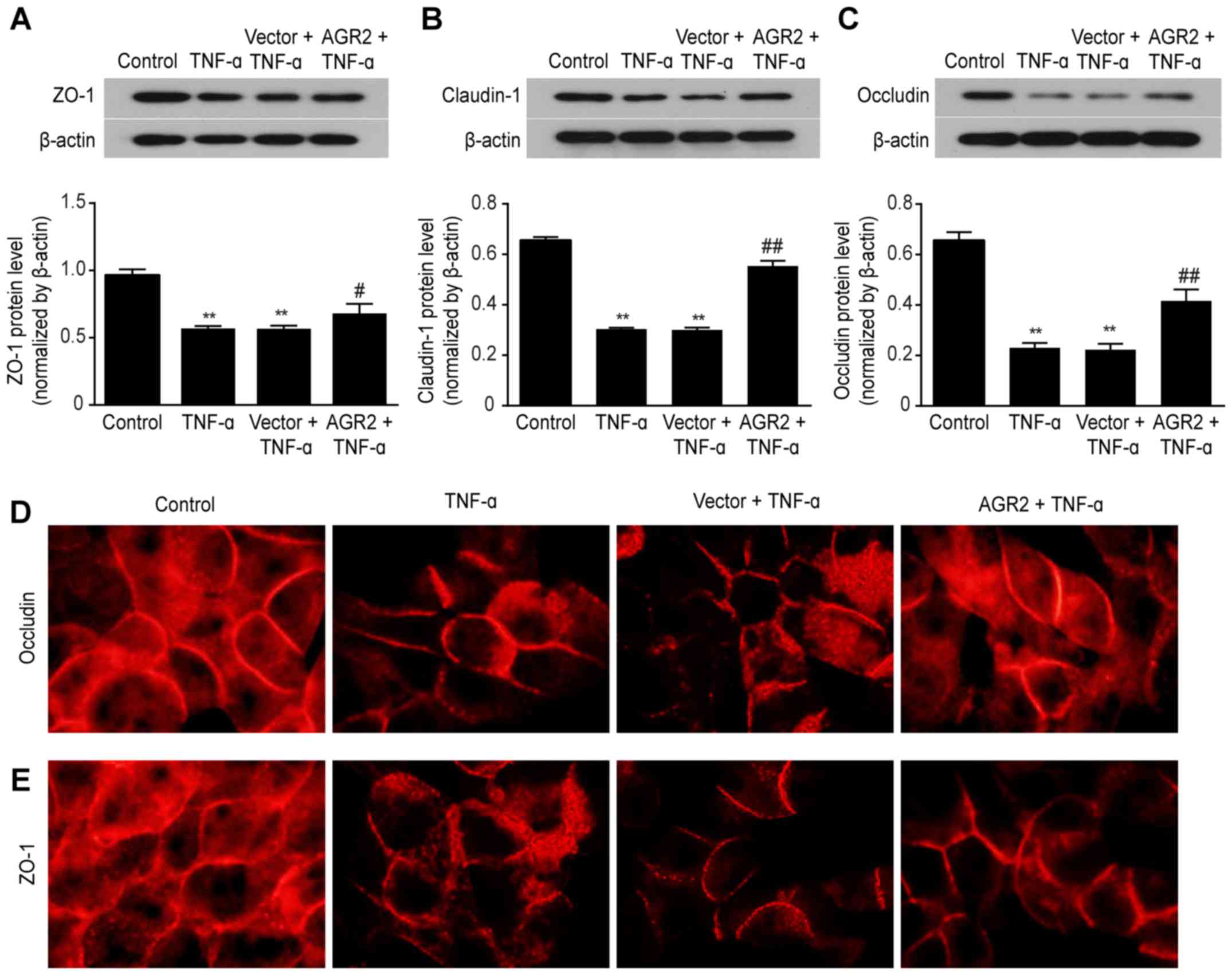
AGR2 inhibits the decreased expression of
ZO-1, occludin, and claudin-1 TJ proteins induced by TNF-α
Disruption of TJs is an important component of
altered intestinal epithelial barrier function (29). We then examined the role of AGR2
and TNF-α in regulating TJ proteins. Western blotting confirmed
that TNF-α stimulation decreased the expression of ZO-1, claudin-1
and occludin (Fig. 4A–C) and that
AGR2 plasmid transfection attenuated these decreases in expression
(Fig. 4A–C). Immunofluorescence
staining showed that in the control group, occludin and ZO-1
appeared as a continuous band surrounding the cell boundary, with
no obvious gap. After 48 h of incubation with TNF-α, these proteins
had rough edges and an interrupted distribution; there were visible
irregular openings, with a honeycomb appearance between the cells
of the monolayers. AGR2 plasmid transfection significantly reduced
the TNF-α-induced damage to the TJs, causing the surfaces of the
proteins to become completely adherent (Fig. 4D and E). These results indicated
that AGR2 inhibited the TNF-α-induced decreases in expression of TJ
proteins.
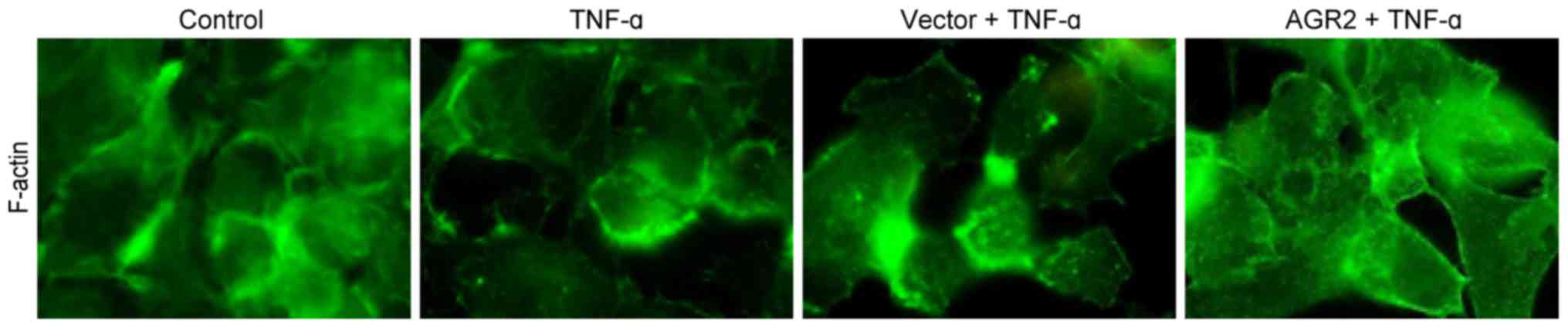
AGR2 inhibits TNF-α-induced cytoskeletal
F-actin rearrangement
Immunofluorescence staining showed that in the
control group, F-actin was arranged in the cytoplasm in neat rows
close to the cell membrane, forming a dense, smooth, continuous
ring with a uniform distribution. After 48 h of incubation with
TNF-α, cytoskeletal F-actin was rearranged, forming a fuzzy,
peripheral actin ribbon with a decrease in the cortical F-actin
fibre macula densa. Some cells had obvious transcellular stress
fibre formation. AGR2 plasmid transfection inhibited the
rearrangement of cytoskeletal F-actin, which appeared as a clear,
peripheral actin ribbon with a decrease in cytoplasmic stress
fibres (Fig. 5). The results
indicated that AGR2 inhibited the TNF-α-induced rearrangement of
cytoskeletal F-actin.
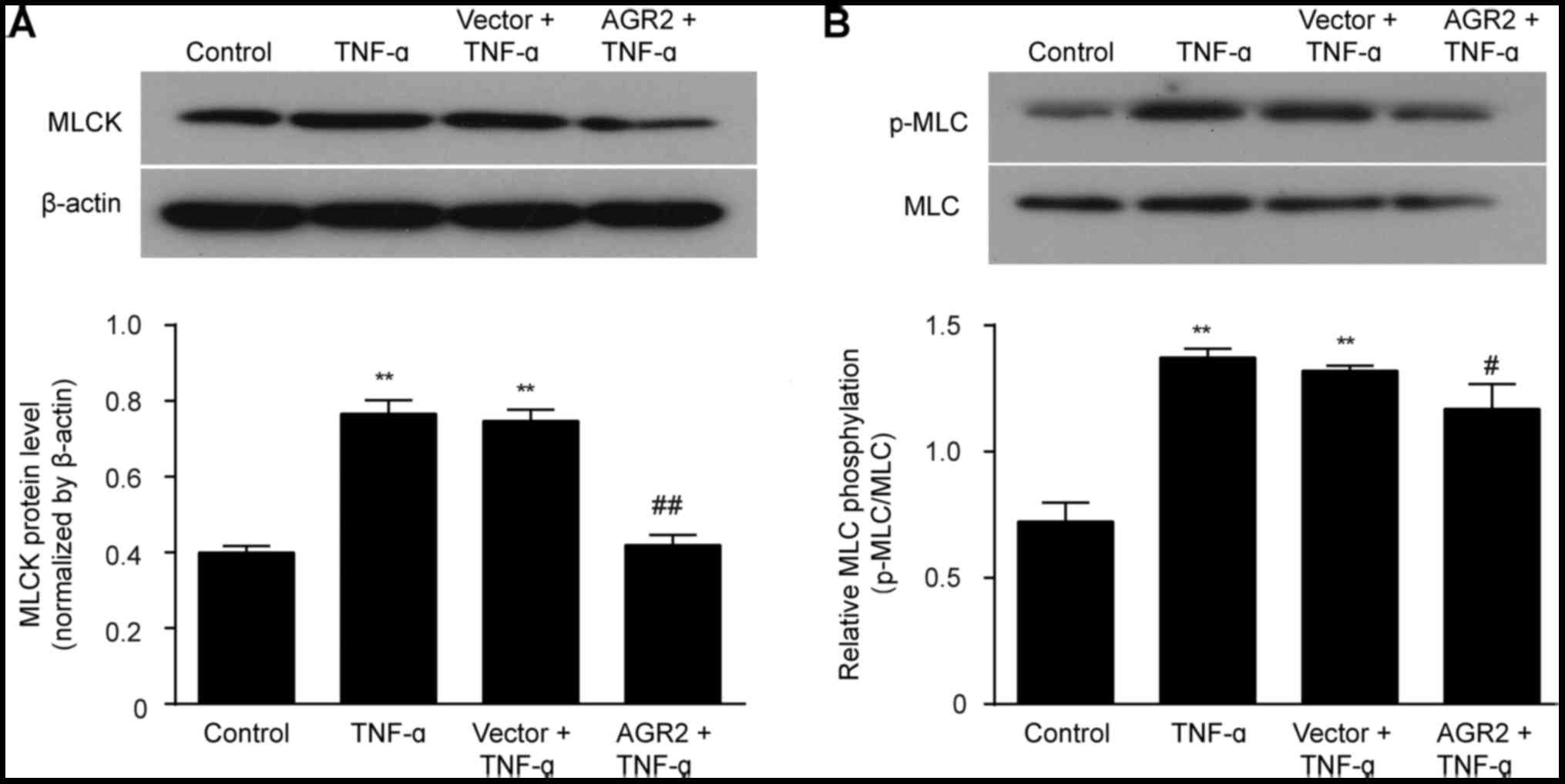
AGR2 inhibits TNF-α activation of the
MLCK/p-MLC pathway
The western blotting results showed that the
expression of MLCK and p-MLC was significantly higher in cells
incubated with TNF-α than levels noted in the control cells.
Transfection with the AGR2 gene plasmid inhibited the TNF-α-induced
elevations in MLCK and p-MLC expression (Fig. 6A and B). These results indicated
that AGR2 attenuated the TNF-α-induced intestinal mucosal barrier
injury by regulating the MLCK/p-MLC pathway.
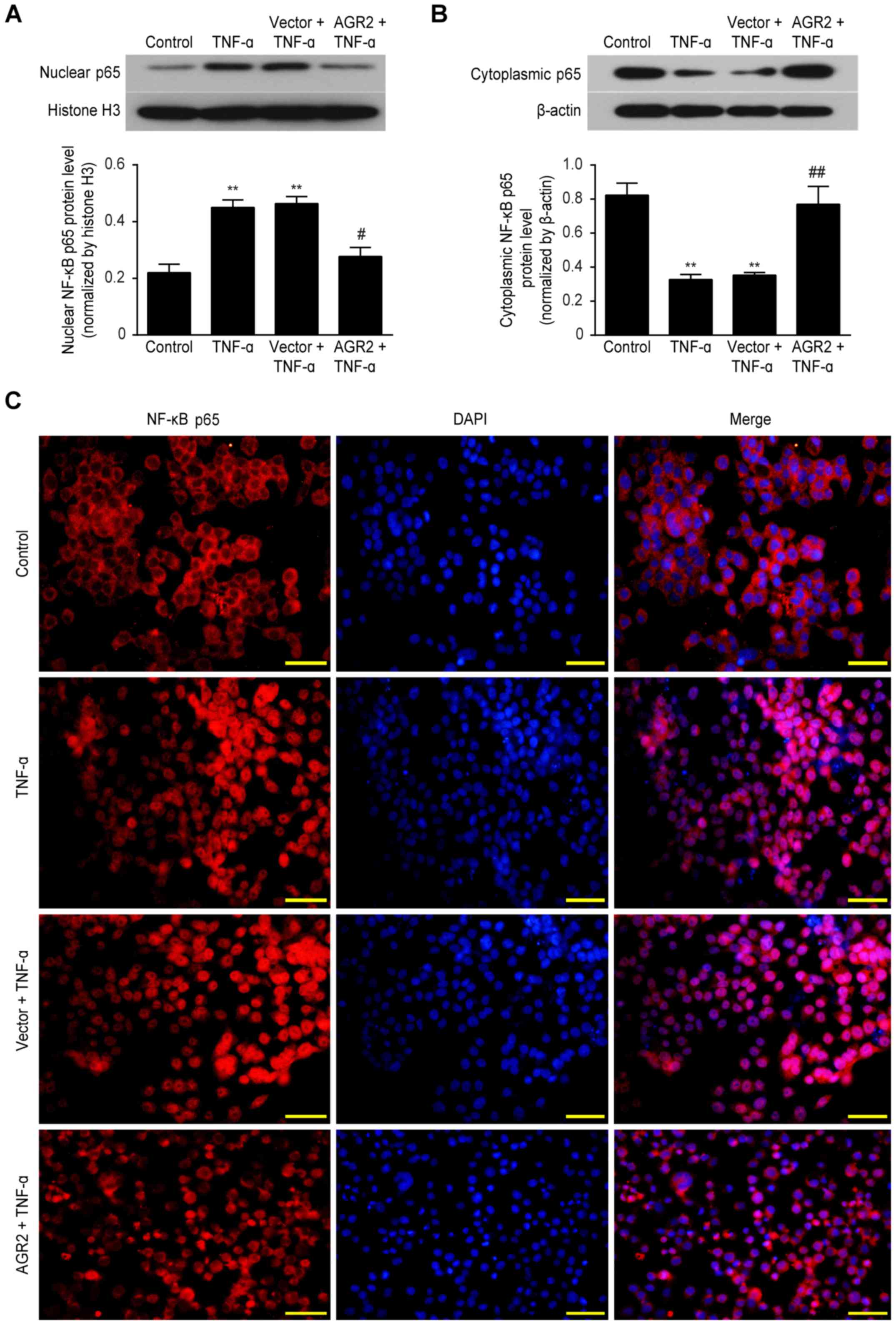
AGR2 reduces TNF-α-induced barrier damage
by inhibiting activation of NF-κB p65
Western blotting showed that TNF-α significantly
increased nuclear NF-κB p65 protein expression and decreased
cytoplasmic NF-κB p65 expression compared with that in the control
group. In contrast, nuclear expression of this protein was
decreased and cytoplasmic expression was increased in the Caco-2
cells transfected with the AGR2 plasmids (Fig. 7A and B). Immunofluorescence
staining further confirmed the above results, showing that in the
control group, the NF-κB p65 protein was mainly localized to the
cytoplasm, where it was inactive. However, in the TNF-α-stimulated
Caco-2 cells, this protein was predominantly nuclear. In the Caco-2
cells transfected with the AGR2 plasmids, nuclear accumulation of
NF-κB p65 in response to TNF-α was significantly reduced (Fig. 7C). The results indicate that AGR2
may reduce intestinal barrier damage by inhibiting the activation
of NF-κB p65.
Discussion
The TJ complex is critical for maintaining mucosal
barrier integrity after injury (30); however, the exact mechanism for
primary repair of TJs has not been fully elucidated. The present
study describes a novel mechanism by which AGR2 regulates TJ
proteins and the cytoskeleton in response to TNF-α-induced
intestinal barrier dysfunction. We report that TNF-α decreased AGR2
expression in the Caco-2 monolayers, while AGR2 overexpression
inhibited the TNF-α-mediated intestinal barrier injury, reduced the
permeability of Caco-2 monolayers, increased the expression of TJ
proteins and stabilized the cytoskeletal structure by inhibiting
NF-κB p65 mediated MLCK/p-MLC pathway activation.
Numerous previous studies, including those by our
group, have confirmed that inflammatory cytokine-mediated
intestinal epithelial barrier dysfunction contributes to multiple
enteropathies, including IBD (31–34); such cytokines disrupt the
integrity of the TJ complex and increase intestinal barrier
permeability. A study by Chen et al showed that stimulating
Caco-2 monolayers with 10 ng/ml TNF-α did not affect TJ protein
expression levels but that TNF-α played a role in altering the
distribution of TJ proteins (35). Other studies revealed that 10
ng/ml TNF-α led to both decrease expression and abnormal
distribution of TJ proteins (36,37). In our study, stimulating Caco-2
monolayers with 100 ng/ml TNF-α significantly increased cell
membrane permeability, accompanied by abnormal distribution and
decreases in the expression of ZO-1, claudin-1 and occludin, as
well as rearrangement of cytoskeletal F-actin; these findings are
consistent with a study by Zhang et al (38). Our data further strengthens the
evidence that TNF-α can disrupt the intestinal epithelial barrier
by reducing TJ protein levels and reorganizing cytoskeletal
structure.
More recently, it has been demonstrated that
activation of the MLCK/p-MLC pathway plays a key role in
inflammatory cytokine-mediated intestinal mucosal barrier
dysfunction (39,40). To investigate the molecular
mechanism by which TNF-α injures the intestinal barrier, we
determined MLCK, MLC and p-MLC protein expression and found that
TNF-α-induced barrier injury was accompanied by increases in both
MLCK expression and MLC phosphorylation, which was also consistent
with the findings of Cunningham et al (41).
Additionally, activation of NF-κB p65 also plays a
crucial role in TNF-α-induced intestinal barrier injury. Previous
research has shown that NF-κB binds to the promoter region of the
MLCK gene and increases MLCK transcription and MLC
phosphorylation, leading to the altered permeability and TJ
disruption known to be associated with proinflammatory cytokines
(42). Here, in our study, TNF-α
significantly increased nuclear protein expression and distribution
of NF-κB p65, indicating that NF-κB p65 is related to the
intestinal barrier injury caused by TNF-α, consistent with the
findings of Al-Sadi et al (43).
AGR2 is a recently discovered gene with a protective
effect of mucosal barrier in IBD; however, the mechanism by which
it regulates intestinal epithelial barrier function remains
unclear. In the present study, we investigated the role of AGR2 in
TNF-α-induced intestinal barrier injury and its related mechanisms.
The results demonstrated that overexpression of AGR2 inhibited the
TNF-α-induced decrease in TEER and increase in FD-40 flux,
increased the expression and improved the distribution of TJ
proteins, and stabilized the cytoskeletal structure, thereby
ameliorating the TNF-α-induced cell membrane hyperpermeability. The
impact of AGR2 on TNF-α-induced activation of the MLCK/p-MLC
signaling pathway was then detected; AGR2 inhibited the changes in
MLCK, MLC and p-MLC expression in response to TNF-α stimulation,
preliminarily confirming that AGR2 improved intestinal barrier
dysfunction through regulation of the MLCK/p-MLC pathway.
Subsequently, we investigated whether activity of the transcription
factor NF-κB p65 was also influenced by AGR2. The results revealed
that AGR2 inhibited nulear protein expression of NF-κB p65, and
immunofluorescence staining revealed that AGR2 inhibited NF-κB p65
translocation into the nucleus. Thus, these data suggest that
inhibition of NF-κB p65-mediated MLCK/p-MLC pathway activation may
be the molecular mechanism by which AGR2 ameliorates intestinal
barrier dysfunction caused by TNF-α.
One limitation of the present study is that we
tested the effect of AGR2 on NF-κB p65 and MLCK/p-MLC pathway
expression in TNF-α-induced intestinal epithelial barrier
dysfunction, but we did not specific block NF-κB p65 or MLCK gene
expression. Further study is needed to investigate other potential
mechanisms by which AGR2 ameliorates TNF-α-induced intestinal
epithelial barrier injury.
In conclusion, to the best of our knowledge, this is
the first study showing that the AGR2 gene inhibits
TNF-α-induced intestinal epithelial hyperpermeability by enhancing
TJ function and cytoskeletal stability. These effects may be
exerted through the inhibition of NF-κB and MLCK/p-MLC pathway
activation. In addition, this study may provide new evidence for
treating IBD by upregulating AGR2 expression, and we hope that this
study potentially helps improve the prognosis of IBD patients.
Abbreviations:
|
AGR2
|
anterior gradient protein 2
homologue
|
|
IBD
|
inflammatory bowel disease
|
|
TNF-α
|
tumor necrosis factor-α
|
|
MLCK
|
myosin light chain kinase
|
|
p-MLC
|
phosporylated myosin light chain
|
|
TEER
|
transepithelial electrical
resistance
|
|
TJ
|
tight junction
|
Acknowledgments
The authors are grateful to the members of the
Department of Pediatric Gastroenterology, China Medical University.
We would also like to thank Ms. Dongyan Liu for assisting with the
manuscript revision.
References
|
1
|
Salim SY and Söderholm JD: Importance of
disrupted intestinal barrier in inflammatory bowel diseases.
Inflamm Bowel Dis. 17:362–381. 2011. View Article : Google Scholar
|
|
2
|
Petersson J, Schreiber O, Hansson GC,
Gendler SJ, Velcich A, Lundberg JO, Roos S, Holm L and Phillipson
M: Importance and regulation of the colonic mucus barrier in a
mouse model of colitis. Am J Physiol Gastrointest Liver Physiol.
300:G327–G333. 2011. View Article : Google Scholar
|
|
3
|
Fernández-Blanco JA, Estévez J,
Shea-Donohue T, Martínez V and Vergara P: Changes in epithelial
barrier function in response to parasitic infection: implications
for IBD pathogenesis. J Crohns Colitis. 9:463–476. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Edelblum KL and Turner JR: The tight
junction in inflammatory disease: communication breakdown. Curr
Opin Pharmacol. 9:715–720. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Alipour M, Zaidi D, Valcheva R, Jovel J,
Martínez I, Sergi C, Walter J, Mason AL, Wong GK, Dieleman LA, et
al: Mucosal barrier depletion and loss of bacterial diversity are
primary abnormalities in paediatric ulcerative colitis. J Crohns
Colitis. 10:462–471. 2016. View Article : Google Scholar :
|
|
6
|
Vetrano S, Rescigno M, Cera MR, Correale
C, Rumio C, Doni A, Fantini M, Sturm A, Borroni E, Repici A, et al:
Unique role of junctional adhesion molecule-A in maintaining
mucosal homeostasis in inflammatory bowel disease.
Gastroenterology. 135:173–184. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Neurath MF and Travis SP: Mucosal healing
in inflammatory bowel diseases: a systematic review. Gut.
61:1619–1635. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Suzuki T, Yoshinaga N and Tanabe S:
Interleukin-6 (IL-6) regulates claudin-2 expression and tight
junction permeability in intestinal epithelium. J Biol Chem.
286:31263–31271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Amasheh M, Grotjohann I, Amasheh S, Fromm
A, Söderholm JD, Zeitz M, Fromm M and Schulzke JD: Regulation of
mucosal structure and barrier function in rat colon exposed to
tumor necrosis factor alpha and interferon gamma in vitro: a novel
model for studying the pathomechanisms of inflammatory bowel
disease cytokines. Scand J Gastroenterol. 44:1226–1235. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu H, Wang P, Cao M, Li M and Wang F:
Protective role of oligomycin against intestinal epithelial barrier
dysfunction caused by IFN-γ and TNF-α. Cell Physiol Biochem.
29:799–808. 2012. View Article : Google Scholar
|
|
11
|
Michielan A and D'Incà R: Intestinal
permeability in inflammatory bowel disease: pathogenesis, clinical
evaluation, and therapy of leaky gut. Mediators Inflamm.
2015:6281572015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Suzuki T: Regulation of intestinal
epithelial permeability by tight junctions. Cell Mol Life Sci.
70:631–659. 2013. View Article : Google Scholar
|
|
13
|
Shen L, Weber CR, Raleigh DR, Yu D and
Turner JR: Tight junction pore and leak pathways: a dynamic duo.
Annu Rev Physiol. 73:283–309. 2011. View Article : Google Scholar
|
|
14
|
Chen Y, Li D, Dai Z, Piao X, Wu Z, Wang B,
Zhu Y and Zeng Z: L-methionine supplementation maintains the
integrity and barrier function of the small-intestinal mucosa in
post-weaning piglets. Amino Acids. 46:1131–1142. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Turner JR: Intestinal mucosal barrier
function in health and disease. Nat Rev Immunol. 9:799–809. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Marchiando AM, Shen L, Graham WV, Edelblum
KL, Duckworth CA, Guan Y, Montrose MH, Turner JR and Watson AJ: The
epithelial barrier is maintained by in vivo tight junction
expansion during pathologic intestinal epithelial shedding.
Gastroenterology. 140:1208–1218. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Assasi N, Blackhouse G, Xie F, Marshall
JK, Irvine EJ, Gaebel K, Robertson D, Campbell K, Hopkins R and
Goeree R: Patient outcomes after anti TNF-alpha drugs for Crohn's
disease. Expert Rev Pharmacoecon Outcomes Res. 10:163–175. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang QY, Sun AM, Song J, Chen Y, Wang JD
and Li CG: Cytokine tumor necrosis factor alpha induces intestinal
epithelial barrier dysfunction. Cytokine. 58:226–230. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huys L, Van Hauwermeiren F, Dejager L,
Dejonckheere E, Lienenklaus S, Weiss S, Leclercq G and Libert C:
Type I interferon drives tumor necrosis factor-induced lethal
shock. J Exp Med. 206:1873–1882. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Takahashi N, Vanlaere I, de Rycke R,
Cauwels A, Joosten LA, Lubberts E, van den Berg WB and Libert C:
IL-17 produced by Paneth cells drives TNF-induced shock. J Exp Med.
205:1755–1761. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Suzuki M, Nagaishi T, Yamazaki M, Onizawa
M, Watabe T, Sakamaki Y, Ichinose S, Totsuka M, Oshima S, Okamoto
R, et al: Myosin light chain kinase expression induced via tumor
necrosis factor receptor 2 signaling in the epithelial cells
regulates the development of colitis-associated carcinogenesis.
PLoS One. 9:e883692014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Feng Y and Teitelbaum DH: Tumour necrosis
factor-induced loss of intestinal barrier function requires TNFR1
and TNFR2 signalling in a mouse model of total parenteral
nutrition. J Physiol. 591:3709–3723. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Du J, Chen Y, Shi Y, Liu T, Cao Y, Tang Y,
Ge X, Nie H, Zheng C and Li YC: 1,25-Dihydroxyvitamin D protects
intestinal epithelial barrier by regulating the myosin light chain
kinase signaling pathway. Inflamm Bowel Dis. 21:2495–2506. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu X, Xu J, Mei Q, Han L and Huang J:
Myosin light chain kinase inhibitor inhibits dextran sulfate
sodium-induced colitis in mice. Dig Dis Sci. 58:107–114. 2013.
View Article : Google Scholar
|
|
25
|
Park SW, Zhen G, Verhaeghe C, Nakagami Y,
Nguyenvu LT, Barczak AJ, Killeen N and Erle DJ: The protein
disulfide isomerase AGR2 is essential for production of intestinal
mucus. Proc Natl Acad Sci USA. 106:6950–6955. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zheng W, Rosenstiel P, Huse K, Sina C,
Valentonyte R, Mah N, Zeitlmann L, Grosse J, Ruf N, Nürnberg P, et
al: Evaluation of AGR2 and AGR3 as candidate genes for inflammatory
bowel disease. Genes Immun. 7:11–18. 2006. View Article : Google Scholar
|
|
27
|
Lai YR, Lu YF, Lien HW, Huang CJ and Hwang
SP: Foxa2 and Hif1ab regulate maturation of intestinal goblet cells
by modulating agr2 expression in zebrafish embryos. Biochem J.
473:2205–2218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao F, Edwards R, Dizon D, Afrasiabi K,
Mastroianni JR, Geyfman M, Ouellette AJ, Andersen B and Lipkin SM:
Disruption of Paneth and goblet cell homeostasis and increased
endoplasmic reticulum stress in Agr2−/− mice. Dev Biol.
338:270–279. 2010. View Article : Google Scholar
|
|
29
|
Su L, Shen L, Clayburgh DR, Nalle SC,
Sullivan EA, Meddings JB, Abraham C and Turner JR: Targeted
epithelial tight junction dysfunction causes immune activation and
contributes to development of experimental colitis.
Gastroenterology. 136:551–563. 2009. View Article : Google Scholar :
|
|
30
|
Liu Z, Li N and Neu J: Tight junctions,
leaky intestines, and pediatric diseases. Acta Paediatr.
94:386–393. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lin N, Xu LF and Sun M: The protective
effect of trefoil factor 3 on the intestinal tight junction barrier
is mediated by toll-like receptor 2 via a I3K/Akt dependent
mechanism. Biochem Biophys Res Commun. 440:143–149. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xu LF, Teng X, Guo J and Sun M: Protective
effect of intestinal trefoil factor on injury of intestinal
epithelial tight junction induced by platelet activating factor.
Inflammation. 35:308–315. 2012. View Article : Google Scholar
|
|
33
|
Blander JM: Death in the intestinal
epithelium-basic biology and implications for inflammatory bowel
disease. FEBS J. 283:2720–2730. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xiao YT, Yan WH, Cao Y, Yan JK and Cai W:
Neutralization of IL-6 and TNF-α ameliorates intestinal
permeability in DSS-induced colitis. Cytokine. 83:189–192. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen S, Zhu J, Chen G, Zuo S, Zhang J,
Chen Z, Wang X, Li J, Liu Y and Wang P: 1,25-Dihydroxyvitamin D3
preserves intestinal epithelial barrier function from TNF-alpha
induced injury via suppression of NF-kB 65 mediated MLCK-P-MLC
signaling pathway. Biochem Biophys Res Commun. 460:873–878. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
He F, Peng J, Deng XL, Yang LF, Camara AD,
Omran A, Wang GL, Wu LW, Zhang CL and Yin F: Mechanisms of tumor
necrosis factor-alpha-induced leaks in intestine epithelial
barrier. Cytokine. 59:264–272. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cao M, Wang P, Sun C, He W and Wang F:
Amelioration of IFN-γ and TNF-α-induced intestinal epithelial
barrier dysfunction by berberine via suppression of MLCK-MLC
phosphorylation signaling pathway. PLoS One. 8:e619442013.
View Article : Google Scholar
|
|
38
|
Zhang J, Lu Y, Wei J, Li L and Han L:
Protective effect of carboxytmethylpachymaran on TNF-alpha-induced
damage in Caco-2 cell monolayers. Int J Biol Macromol. 93(Pt A):
506–511. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kuhlmann CR, Tamaki R, Gamerdinger M,
Lessmann V, Behl C, Kempski OS and Luhmann HJ: Inhibition of the
myosin light chain kinase prevents hypoxia-induced blood-brain
barrier disruption. J Neurochem. 102:501–507. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Boivin MA, Ye D, Kennedy JC, Al-Sadi R,
Shepela C and Ma TY: Mechanism of glucocorticoid regulation of the
intestinal tight junction barrier. Am J Physiol Gastrointest Liver
Physiol. 292:G590–G598. 2007. View Article : Google Scholar
|
|
41
|
Cunningham KE and Turner JR: Myosin light
chain kinase: pulling the strings of epithelial tight junction
function. Ann NY Acad Sci. 1258:34–42. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ye D and Ma TY: Cellular and molecular
mechanisms that mediate basal and tumour necrosis
factor-alpha-induced regulation of myosin light chain kinase gene
activity. J Cell Mol Med. 12:1331–1346. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Al-Sadi R, Guo S, Ye D, Rawat M and Ma TY:
TNF-alpha modulation of intestinal tight junction permeability is
mediated by NIK/IKK-alpha axis activation of the canonical NF-κB
pathway. Am J Pathol. 186:1151–1165. 2016. View Article : Google Scholar : PubMed/NCBI
|