Introduction
Oral cancer, i.e., oral squamous cell carcinoma
(OSCC), can affect any part of the oral mucosa and is characterized
by sores in the mouth which bleed easily and do not heal. This
disease is the sixth most common type of cancer, which affects over
263,000 individuals annually worldwide, and has become a
predominant health burden (1,2).
The main cause of mortality due to oral cancer is the failure to
control the pre-cancerous lesions and lymph node metastasis. Both
environmental risk factors, such as betel quid chewing, tobacco use
and alcohol consumption, as well as genetic factors are considered
of particular importance in the etiology of oral cancer (3). In particular, the combination of
these environmental factors and certain genes may increase
susceptibility to oral cancer (3). However, these genes have not yet
been fully identified.
With the advent of the human genome map involved in
different diseases, genetic polymorphisms in certain genes have
been found to play crucial roles in driving carcinogenesis.
Furthermore, although some biomarkers, such as oral habits are
indicators of oral cancer prognostication, genetic variability
among different patients should also be taken into consideration.
Therefore, genetic polymorphisms have gained colossal importance as
novel drug targets or cancer prognostic biomarkers. Furthermore,
previous population-based polymorphism studies which detected the
association of limited candidate genes or genetic polymorphisms
with cancer have not obtained consistent results (4–6).
As carcinogenesis is a complex multistep process, it is usually
caused by somatic mutations in certain genes. Hence, identifying
and assessing polymorphisms in all genes using the method of whole
exome sequencing (WES) may be more effective for cancer prognosis
and treatment.
WES, a type of massively parallel next-generation
sequencing (NGS) technology, is used to effectively identify novel
somatic mutations and establish a new genetic basis of certain
diseases. It consists of capturing exons, and subsequently
high-throughput DNA sequencing. Several small-scale studies have
provided some evidence of the potential of WES in identifying
disease-driving mutations and genes (2,7,8).
For example, using WES within 32 primary oral tumor pairs, Agrawal
et al found that novel mutations in FBXW7 and
NOTCH1 may play roles in carcinogenesis of head and neck
squamous cell carcinoma (2).
In this study, due to the unreliability of previous
clinical markers and the development of NGS, the present study
aimed to obtain a landscape of the somatic mutations underlying
oral cancer using WES.
Materials and methods
Study subjects
Tumor tissues and blood were collected from
treatment-naive Chinese patients with oral cancer, who then
completed the treatment and the follow-up for at least 12 months by
their physician. Tumor tissues were obtained by precise laser
micro-surgical resection, and blood samples were drawn through the
upper arm vein. Ethics approval was obtained from the human Ethics
Committee of Ninth People's Hospital (app. no. 2014011). Written
informed consent was obtained from all patients. All patients were
diagnosed with oral cancer without having any other type of cancer.
Histological sections of lesions were examined by two
oncopathologists, and the diagnosis was based on the World Health
Organization (WHO) guidelines. Clinicopathological staging was
determined according to the Union for International Cancer Control
(UICC) TNM staging system v6. The patient characteristics are
summarized in Table I. All
patients received surgery combined with radiation therapy and/or
chemotherapy. Each patient was followed-up each month after
surgery. The clinical detection of recurrence was confirmed by
histopathology. All patients experienced residual or recurrent
disease.
 | Table IClinical characteristics of the 5
patients with oral cancer. |
Table I
Clinical characteristics of the 5
patients with oral cancer.
| Patient ID | Age (years) | Sex | Risk habita | Tumor stage | Tumor stage
(TNM) | HPV
infectiona |
|---|
| OC1 | 44 | Male | Alcohol
consumption, but no tobacco use | IV | T4a N0 | Absent |
| OC2 | 44 | Male | Alcohol
consumption, but no tobacco use | IV | T4a N0 | Absent |
| OC3 | 46 | Male | Alcohol
consumption, but no tobacco use | IV | T4a N2b | Absent |
| OC4 | 48 | Male | Alcohol
consumption, but no tobacco use | IV | T4a N0 | Absent |
| OC5 | 42 | Male | Alcohol
consumption, but no tobacco use | IV | T4a N2b | Absent |
WES
The bisected tissue samples and blood were stored in
liquid nitrogen. Thereafter, genomic DNA was isolated according to
standard protocols as previously described (9–11).
The genomic DNA libraries were constructed using protocols provided
by Illumina (San Diego, CA, USA). Approximately 50 Mb of sequences
from the whole exons and their flanking regions were enriched from
the fragmented genomic DNA using the Agilent SureSelect DNA Capture
array. The captured DNA libraries were processed using the Illumina
HiSeq 2500 Genome Analyzer with an overall >10X coverage
depth.
Data processing
Sequence data in FASTQ files were quality-checked
using FASTQC v0.11.4 (http://www.bioin-formatics.babraham.ac.uk/projects/fastqc/).
The filtered sequence reads were mapped to the hg19 reference
sequence using Burrows-Wheeler Aligner (BWA) v0.7.12 (http://source-forge.net/projects/bio-bwa/) and SAM
files were generated. Local realignment around indels, duplicate
removal and Base quality score recalibration were performed using
the Genome Analysis Toolkit (GATK) v3.5 (https://www.broadinstitute.org/gatk/). Alignment SAM
files were converted to BAM files using Samtools v1.3. Somatic
variants were called using MuTect v1.1.7 (https://www.broadinstitute.org/gatk/download/auth?package=MuTect)
with default parameters. High confidence variants were identified
with the criteria of a minimum of 10 reads covering a site in the
tumor tissue and 8 reads in the blood, and the maximum alternative
allele frequency in the blood <0.3 of that in the tumor tissue.
Somatic mutations which are defined as called mutations in the
absence of corresponding reads in the blood DNA samples were
annotated using the Annovar main package (http://annovar.openbioinformatics.org/en/latest/)
to infer the locations of the variants within the genes. Somatic
single nucleotide variants (SNVs) were then combined and searched
in the dbSNP and COSMIC databases. The reference human genome hg19
and dbSNP132 were used to call and annotate the SNVs. The novel
variant identified by WES was verified by Sanger sequencing. The
primers used for PCR and sequencing were 5′-AAGTTGTCGTAGAGGCAGGC-3′
as the forward primer and 5′-GACCCCTGGCGGCAATG-3′ as the reverse
primer. PCR was used for Sanger sequencing validation of the new
variant. The primers used for PCR and sequencing were forward,
5′-AAGTTGTCGTAGAGGCAGGC-3′ and reverse, 5′-GACCCCTGGCGGCAATG-3′.
PCR was performed on an ABI9700 PCR instrument (Applied Biosystems,
Inc., Foster City, CA, USA). in a 30 µl reaction volume
including 19 HotStarTaq buffer, 2.8 mM Mg2+, 0.1 U of
HotStarTaq polymerase (Qiagen, Inc., Valencia, CA, USA), 2 ng of
blood genomic DNA, 0.5 pmol of each primer, 0.5 mmol of dNTPs and
0.5 mmol of ddNTPs. Thermocycling was carried out at 94°C for 15
min, followed by 45 cycles at 94°C for 20 sec, 56°C for 30 sec, and
72°C for 1 min, with a final incubation at 72°C for 3 min.
Evaluation of missense variants
First of all, the effects of the SNVs were evaluated
using Mutation Taster (http://www.mutationtaster.org/) (12), which automatically yielded types
of predictions, i.e., 'disease_causing_automatic',
'disease_causing', 'polymorphism' and 'polymorphism_automatic', and
the probability P-value of the prediction. A P-value close to 1
indicates a high possibility of the prediction results. A
transcript ID and a sequence covering each single-nucleotide
polymorphism (SNP) were required as input.
Sorting Intolerant From Tolerant (SIFT; http://sift.jcvi.org/) uses a query sequence and
multiple alignment information to predict roles of variants
(13). The alleles with
calculated probabilities <0.05 are predicted to be deleterious;
probabilities ≥0.05 are tolerated (14). The protein FASTA AA sequence was
used as the query sequence and novel mutated SNPs were
analyzed.
Polymorphism Phenotyping v2.0 (PolyPhen-2;
http://genetics.bwh.harvard.edu/pph2/) predicts the
influence of variants through automatically selecting 8
sequence-based and 3 structure-based predictive features by an
iterative greedy algorithm. If the classifier probability is
<0.15, the allele is predicted to be benign, and if the
probability is ≥0.15, the allele is predicted to be probably or
possibly damaging. Protein FASTA AA sequence, amino acid position
and amino acid variants are required (15).
PANTHER (http://www.pantherdb.org/tools/csnpScore-Form.jsp)
predicts the functional consequences of substitutions on the
protein by calculating the substitution position-specific
evolutionary conservation (subPSEC) score derived from the Hidden
Markov Model and alignment score (16). The SNPs with subPSEC ≤-3 were
considered damaging.
SNPs&GO (http://snps.biofold.org/snps-and-go/) is based on SVM
and functional information codified by gene ontology (GO) terms to
predict the impact of protein variations. The present analysis was
protein sequence-based. The prediction (either disease or neutral)
and RI (probability of disease-related class) are given (17).
SNAP2.0 (https://rostlab.org/services/snap/) predicts the
effect of mutations by identifying all variants based on neutral
network method that utilizes protein information. It requires
protein sequence as input and yields 80% accuracy (18).
Modeling protein structures
I-Mutant v2.0 (http://folding.biofold.org/i-mutant/i-mutant2.0.html)
is a neural network-based tool for the routine analysis of protein
stability and alterations caused by the single-site mutations,
which also provides the scores for the Gibbs free energy change
(DDG) calculated with the FOLD-X (19). The FOLD-X analysis tool provides
the comparison between wild-type and mutant models in the form of
van der Waals clashes, which greatly influence the energy
decomposition. The protein FASTA AA sequence was used as the query
sequence.
The residues mutation was performed using the
SwissPDB Viewer viewer and energy minimization for 3D structures
was performed by the NOMAD-Ref server (http://lorentz.immstr.pasteur.fr/gromacs/minimization_submission.php)(20).
This server uses Gromacs as the default force field for energy
minimization based on the methods of steepest descent, conjugate
gradient, and L-BFGS. We used the conjugate gradient method for
optimizing the 3D structures. The deviation between the two
structures is evaluated by their root-mean-square deviation (RMSD)
values.
To examine the stability of the native and mutant
modeled structures, the identification of the stabilizing residues
is useful. We used the server SRide (http://sride.enzim.hu/) (21) for identifying the stabilizing
residues in native protein and mutant models. Stabilizing residues
were computed using parameters, such as long-range order,
surrounding hydrophobicity, conservation score and stabilization
center.
Results
Subject characteristics
In total, tissue and blood samples from 5 patients.
All 5 patients were male with a mean age of 44.8±2.28, ranging from
42 to 48 years (Table I). The
patients with oral cancer were all HPV-negative and were all
alcohol consumers, but were not tobacco users. All patients
presented with advanced disease (stage IV)s.
WES and validation
Coding exons of approximately 20,000 protein coding
genes were sequenced from DNA isolated from the blood and primary
tumor of each patient. The mean depths of sequencing were
136.90±11.96 for blood DNA and 139.87±16.29 for tumor DNA. The
details for each patient are provided in Table II. The coding regions of genomes
of the 5 patients contained 253 somatic variants, of which 51
(20.2%) were predicted to be synonymous, 138 (54.5%) were missense
and 5 (2%) nonsense. The average mutation rate/Mb was estimated to
be 2.04±0.73 (Table III).
| Table IISequencing summary for 5 sample
exomes. |
Table II
Sequencing summary for 5 sample
exomes.
| Sample (no.) | Tumor tissue
(5) | Blood (5) |
|---|
| Reads passed
filtration |
100021213±7006327 |
99561798±3048213 |
| Reads mapped
(%) | 99.86
(99.72–99.91) | 99.80
(99.64–99.89) |
| Capture efficiency
(%)b | 75.12
(69.9–78.2) | 74.56
(69–77.8) |
| Mean deptha | 139.87±16.29 | 136.90±11.96 |
| ≥1 Coverage
(%)c | 96.78
(96.6–96.9) | 96.8
(96.6–97.0) |
| ≥10 Coverage
(%)c | 94.7
(94.3–95.0) | 94.66
(94.5–94.8) |
| ≥20 Coverage
(%)c | 92.26
(91.1–93.0) | 92.2
(91.7–92.7) |
| ≥40 Coverage
(%)c | 85.86
(82.9–87.9) | 85.7
(84.3–87.2) |
| Table IIISummary of somatic mutation types and
prevalence in 5 patients with oral cancer. |
Table III
Summary of somatic mutation types and
prevalence in 5 patients with oral cancer.
| Sample | SNV in coding
regions
| Total | Mutation/Mb
DNA | NS/S |
|---|
| Synonymous | Missense | Stop gained | Others |
|---|
| OC1 | 6 | 13 | 1 | 3 | 23 | 0.97 | 2.33 |
| OC2 | 10 | 21 | 0 | 13 | 44 | 1.74 | 2.10 |
| OC3 | 10 | 32 | 1 | 14 | 57 | 2.21 | 3.30 |
| OC4 | 13 | 41 | 1 | 18 | 73 | 2.91 | 3.23 |
| OC5 | 12 | 31 | 2 | 11 | 56 | 2.35 | 2.75 |
| Total | 51 | 138 | 5 | 59 | 253 | | |
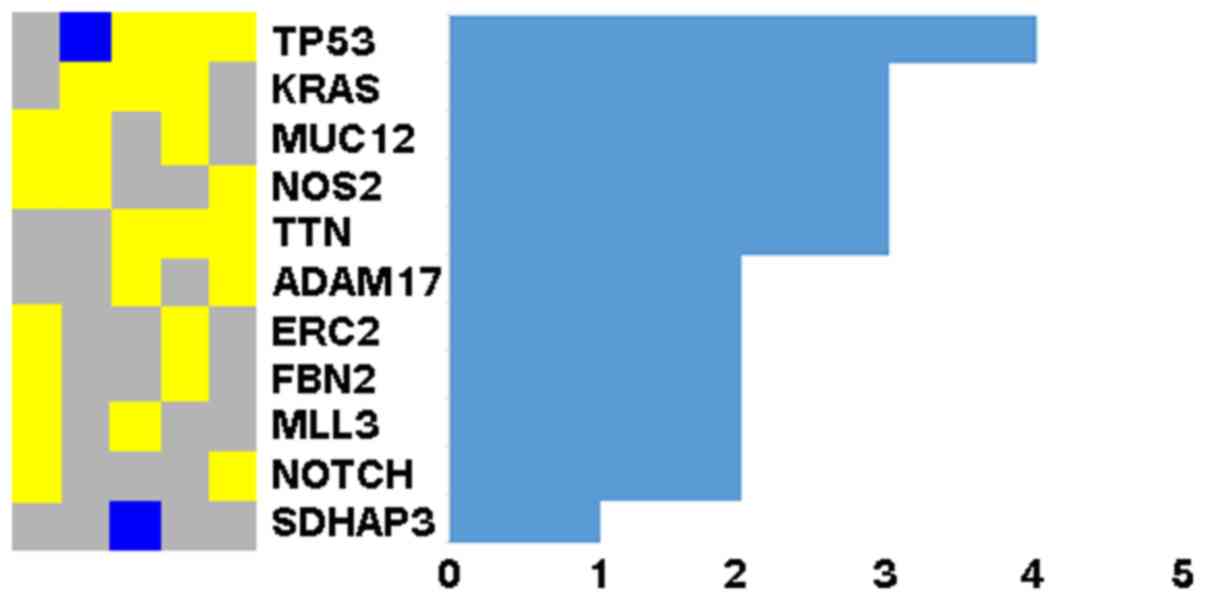
The TP53 gene was mutated in at least 4
samples and 10 genes in 2 or more samples (Fig. 1). Since NOTCH signaling has been
suggested to play important roles in head and neck squamous cell
carcinoma (2), a newly identified
variant (chr19:15288426A>C) in the NOTCH3 gene, a
homology of NOTCH1, and a known variant
(chr6:32168732C>T) in the NOTCH4 gene, another homology
of NOTCH1 attracted our attention. The variant
(chr6:32168732C>T) had a frequency of 8.35E-06 in the database
of ExAC (http://exac.broadin-stitute.org) and was a silent
mutation, resulting in no amino acid substitution. We then
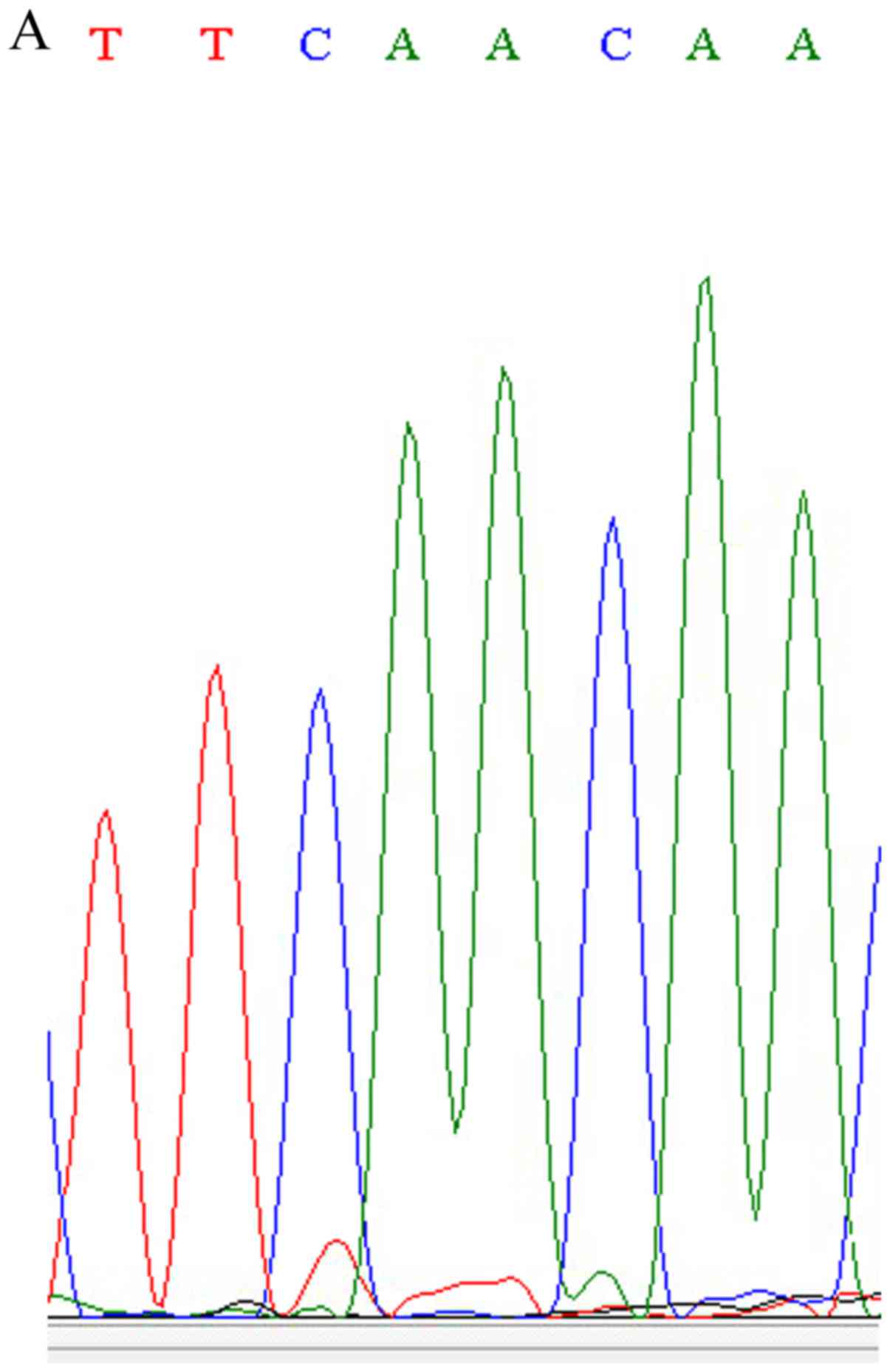
confirmed the newly identified variant (chr19:15288426A>C) in
the NOTCH3 gene using Sanger sequencing (Fig. 2). This nonsense SNP (nsSNP) has
the same location with the known nsSNP of rs761506399, but has a
different allele mutation. The nsSNP, rs761506399, is an A>G
mutation and results in 1438 Asn changing into Ser, while the new
variant is an A>C mutation and results in 1438 Asn changing into
Thr.
Evaluation of the variant
We first evaluated the functional effect of this
nsSNP by Mutation Taster, which was predicted to be 'disease
causing' with a probability of 0.9997 and suggested to possibly
affect protein features.
Subsequently, SIFT, which predicts tolerated and
deleterious substitutions at each position of the query sequence
with multiple alignment score (13), indicated that this nsSNP was
tolerated (score, 0.24) (Table
IV). PolyPhen-2, which predicts the functional importance of an
allele replacement using a training set and naive Bayes classifier,
indicated that this nsSNP was 'possibly damaging' with a score of
0.986. We also examined the probability of the variant causing a
deleterious functional change by PANTHER, which is based on the
Hidden Markov model and alignment score in a very low value of
subPSEC, −2.0575. Another method utilized was SNAP2.0, which is a
neutral network method combined with a training set. It designated
that this nsSNP affects the function of the NOTCH3 protein.
Finally, the mutants were analyzed using SNPs&GO, which is an
SVM-based classifier and obtains quite accurate (79%) results.
SNPs&GO predicted that this nsSNP was neutral (RI=5) (Table IV).
| Table IVList of results of SIFT, PolyPhen,
Panther, SNPs&GO, I-Mutant and SNAP2. |
Table IV
List of results of SIFT, PolyPhen,
Panther, SNPs&GO, I-Mutant and SNAP2.
| SNP location | AA change | SIFT
(score)a | PolyPhen (PSIC
score) | PANTHER
(subPSEC)b | SNPs&GO
(RI) | I-Mutant (DDG) | SNAP2 (expected
accuracy) |
|---|
| chr19:1528842
6T>G | N1438T | Tolerated
(0.24) | Probably damaging
(0.986) | −2.0575 | Neutral (5) | Stability decreases
(−1.67) | Effect (85%) |
3D structure analysis
I-Mutant v2.0, which is a neural network based tool
for the routine analysis of protein stability and alterations
caused by the single-site mutations, indicated that it can decrease
protein stability with a DDG value of −1.67 (Table IV).
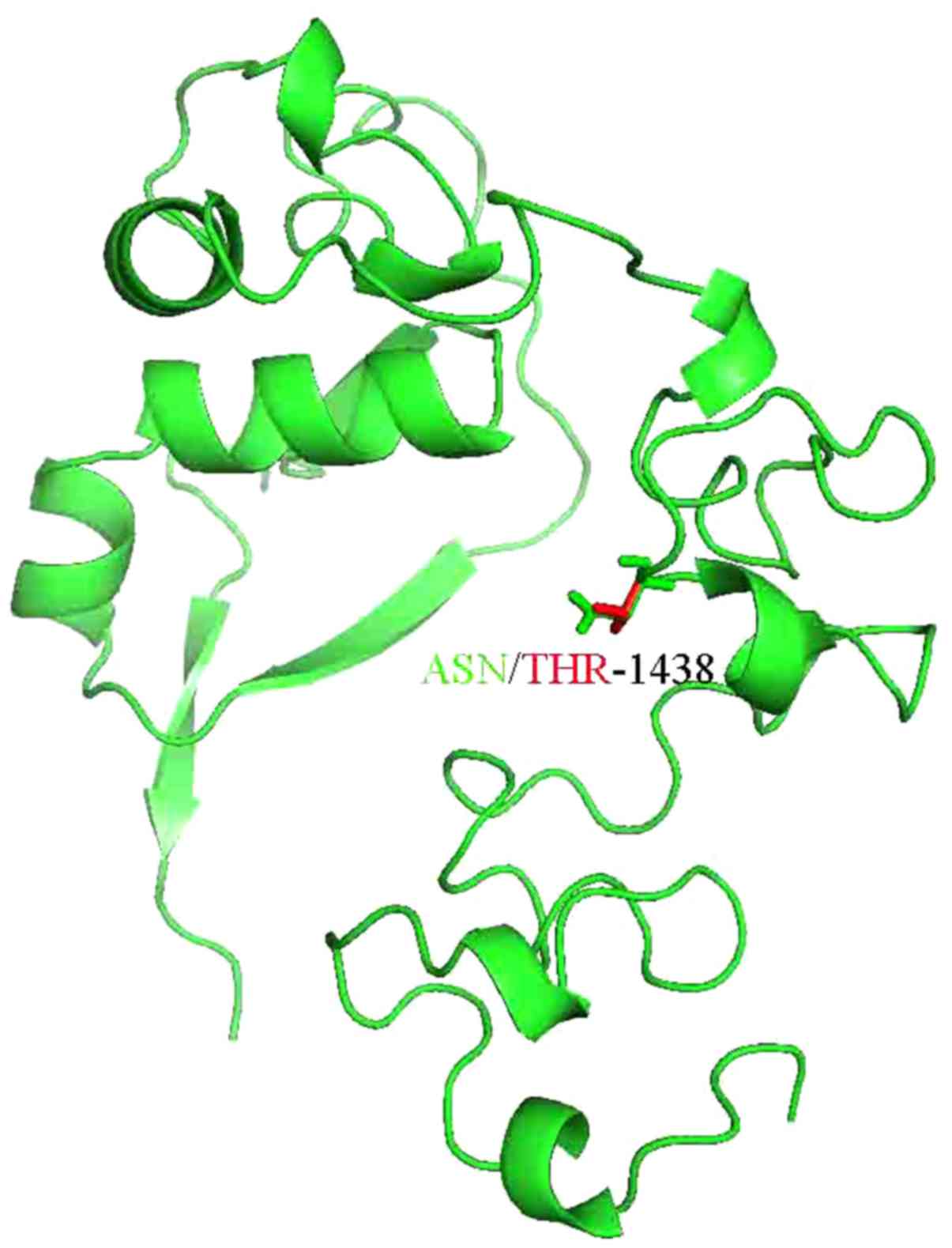
Thus, the 3D structure of NOTCH3 or the domain of
NOTCH3 was analyzed. The 3D structure of part of NOTCH3 was
resolved as 4ZLP.pdb and the novel mutation was located within the
sequence of 4ZLP.pdb. Subsequently, the mutation residues of NOTCH3
were anlayzed using the SwissPDB Viewer and energy minimization for
3D structures was performed using the NOMAD-Ref server (20) (Fig.
3). We used the conjugate gradient method for optimizing the 3D
structures. The total energy for the native structure was −1,229.84
kcal/mol, and that for the mutant structure (N1438T) was −1,051.39
kcal/mol. The difference between the wild-type structure and the
N1438T structure was evaluated by the RMSD value of 1.63.
The computed total solvent accessible surface area
of the wild-type protein was 10,287 Å2 and thesurface
area excluding the solvent was 8,285 Å2, while the total
solvent accessible surface area of theN1438T protein was 10,268
Å2, and the surface area excluding the solvent was 8,268
Å2. The mutant amino acid reduces the solvent accessible
surface area by 19 Å2 for N1438T, and solvent excluded
surface area by 17 Å2 in spite of having more number of
atoms as compared to the wild-type. The reduction in surface area
may have little effect on the solubility of the protein, and thus
on its function. The mutant amino acid is also an AA without charge
just like the wide type AA. The hydrophobicity slightly increases
for domain with mutant AA (data not shown).
The SRide server (21) was used to identify the stabilizing
residues of the native structure and mutant modeled structures with
the conservation score threshold of 6, LRO threshold of 0.012, and
surrounding hydrophobicity threshold of 20. Four stabilizing
residues were identified in the native structure, which were CYS67,
ASP70, GLY110 and PRO173. The single-mutant model (N1438T) has 3
identified stabilizing residues, which were CYS67, ASP70 and
GLY110. This analysis revealed that one stabilizing residue in the
N1438T-mutant model less than the native protein structure may
decrease the stability of the N1438T mutant protein.
Discussion
Oral cancer has become a serious health concern. It
is characterized by a high incidence, low survival rate and severe
functional impairment and cosmetic deformity accompanying
treatment. Both genetic and environmental factors are widely
recognized to result in individual susceptibility to this disease
(3). In this study, we identified
a novel NOTCH3 mutation in patients with oral cancer using
WES.
Since WES has been proven to be an effective method
for a more comprehensive dissection to genomic variation in gene
coding regions, it is often used reveal subtle genetic variations
in cancer genomes, to understand the process of tumorigenesis, and
fulfill personalized therapies. However, there are some technical
limitations to WES. One of these is the identified mutations
without underlying a mendelian disorder; the other is the mutant
alleles located in the coding regions that are not well covered by
WES; the last is the obscured specific copy to which the variant
maps due to the presence of pseudogenes or repetitive regions
(22). Thus, we only studied
somatic variants in the present study and confirmed them with
Sanger sequencing.
NOTCH, initially identified in Drosophila, is
a large transmembrane protein containing epidermal growth
factor-like (EGFL) repeats (23,24). There are 4 NOTCH receptors in
mammals, i.e., NOTCH 1, 2, 3 and 4, which can bind with NOTCH
ligands. They are also transmembrane proteins containing multiple
EGFL repeats, including Jagged1, Jagged2, Delta1, Delta3 and Delta4
(25). NOTCH signaling is
activated by receptor-ligand interaction during cell-cell contacts,
resulting in the proteolytic release of the NOTCH intracellular
(NIC) domain into the nucleus and the interaction of NIC with
DNA-binding protein to activate transcription of genes involved in
a number of cellular properties (25). NOTCH activity affects the
implementation of differentiation, proliferation and apoptotic
programs to control a broad spectrum of developmental processes,
such as neurogenesis, hematopoiesis, vasculogenesis, keratinocyte
growth or differentiation (26).
Since NOTCH activation can maintain cancer stem cells in some
tissues, whereas it can terminate their differentiation in others,
NOTCH signaling may play dual roles in cancer, depending on the
cellular and tissue context (27), such as an oncogene in non-small
cell lung cancer, ovarian carcinoma and osteosarcoma (28–30), whereas it can act as a suppressor
gene in chronic myelomonocytic leukemia (CMML) and skin cancer
(31,32). As regards the role of NOTCH
signaling in oral cancer, both Köse et al (33) and Agrawal et al (2) found that NOTCH1 was involved
in normal oral mucosa or head and neck squamous cell carcinoma. It
has been suggested that NOTCH signaling is functionally activated
in OSCC (34). Previous
expression array studies have suggested the significant
upregulation of NOTCH4 and Jagged1 in OSCC compared to normal oral
tissue (35,36). Furthermore, higher expression
rates of NOTCH1 and NOTCH3 have been observed in tongue carcinoma
compared to adjacent nonneoplastic tissues (37). These data indicated that the NOTCH
signaling pathway may play important roles in the development of
oral cancer.
In this study, we reported one variant in the
NOTCH3 and NOTCH4 gene, respectively. The variant in
the NOTCH4 gene was known with the frequency of 8.35E-06 in
database of ExAC. While the variant (chr19:15288426A>C) in the
NOTCH3 gene was novel. Multiple in silico analyses
was then performed to identify the roles of the variant on the
NOTCH3.
SIFT is an advantageous analysis tool that be used
to distinguish damaging SNPs with only ~20% false-positive error
and ~90% true-positive prediction (13). PolyPhen-2 is a structural
modification analysis tool and can achieve true positive prediction
rates of 92 and 73% on a training dataset and test dataset at a
false-positive rate of 20% (15).
Both SIFT and PolyPhen suggested that this variant had mildly
damaging effects on the NOTCH3 gene, which was supported by
the results of the analysis using PANTHER, SNAP and SNPs&GO.
However, this SNP had a probability of 0.9997 to be 'disease
causing', as shown by Mutation Taster analysis. Thus, 3D structure
simulation analysis was performed. Our 3D structure models
suggested this variant hadlittle effects on the solubility and
hydrophobicity of the protein and thus its function, but can
decrease the stability of the rotein by increasing the total energy
following minimization and decreasing the stabilizing residues of
the protein. Furthermore, I-Mutant supported that this variant can
decrease protein stability with a DDG value of −1.67.
In conclusion, the present study identified a novel
variant (chr19:15288426A>C) in the NOTCH3 gene using WES
and confirmed it using Sanger sequencing. With multiple in
silico analyses and 3D structure simulation, it is suggested
that this variant has mildly damaging effects on the function of
NOTCH3 gene, but can decrease protein stability. Thus, this
variant may be helpful in cancer prognosis and therapeutic
decision-making.
Acknowledgments
This study was supported by grants from the Project
of the National Natural Science Foundation of China (grant nos.
31140007 and 81472516), the Natural Science Foundation of Shanghai
(no. 14ZR1424200) and the Shanghai Leading Academic Discipline
Project (no. S30206).
References
|
1
|
Haddad RI and Shin DM: Recent advances in
head and neck cancer. N Engl J Med. 359:1143–1154. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Agrawal N, Frederick MJ, Pickering CR,
Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, et
al: Exome sequencing of head and neck squamous cell carcinoma
reveals inactivating mutations in NOTCH1. Science. 333:1154–1157.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Su CW, Huang YW, Chen MK, Su SC, Yang SF
and Lin CW: Polymorphisms and plasma levels of tissue inhibitor of
metalloproteinase-3: Impact on genetic susceptibility and clinical
outcome of oral cancer. Medicine (Baltimore). 94:e20922015.
View Article : Google Scholar
|
|
4
|
Liu H, Jia J, Mao X and Lin Z: Association
of CYP1A1 and GSTM1 polymorphisms with oral cancer susceptibility:
A Meta-analysis. Medicine (Baltimore). 94:e8952015. View Article : Google Scholar
|
|
5
|
Liu J, Song J, Wang MY, He L, Cai L and
Chou KC: Association of EGF rs4444903 and XPD rs13181 polymorphisms
with cutaneous melanoma in Caucasians. Med Chem. 11:551–559. 2015.
View Article : Google Scholar
|
|
6
|
Cai L, Huang W and Chou KC: Prostate
cancer with variants in CYP17 and UGT2B17 genes: A meta-analysis.
Protein Pept Lett. 19:62–69. 2012. View Article : Google Scholar
|
|
7
|
Li C, Gao Z, Li F, Li X, Sun Y, Wang M, Li
D, Wang R, Li F, Fang R, et al: Whole exome sequencing identifies
frequent somatic mutations in cell-cell adhesion genes in Chinese
patients with lung squamous cell carcinoma. Sci Rep. 5:142372015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bell D, Berchuck A, Birrer M, Chien J,
Cramer DW, Dao F, Dhir R, DiSaia P, Gabra H, Glenn P, et al Cancer
Genome Atlas Research Network: Integrated genomic analyses of
ovarian carcinoma. Nature. 474:609–615. 2011. View Article : Google Scholar
|
|
9
|
Jiang SY, Li LL, Yue J, Chen WZ, Yang C,
Wan CL, He L, Cai L and Deng SL: The effects of SP110's associated
genes on fresh cavitary pulmonary tuberculosis in Han Chinese
population. Clin Exp Med. 16:219–225. 2016. View Article : Google Scholar
|
|
10
|
Hao CQ, Zhou Y, Wang JP, Peng MJ, Xie YM,
Kang WZ, Sun L, Wang PZ, Wan CL, He L, et al: Role of Nogo-A in the
regulation of hepatocellular carcinoma SMMC-7721 cell apoptosis.
Mol Med Rep. 9:1743–1748. 2014.PubMed/NCBI
|
|
11
|
Cai L, Deng SL, Liang L, Pan H, Zhou J,
Wang MY, Yue J, Wan CL, He G and He L: Identification of genetic
associations of SP110/MYBBP1A/RELA with pulmonary tuberculosis in
the Chinese Han population. Hum Genet. 132:265–273. 2013.
View Article : Google Scholar
|
|
12
|
Schwarz JM, Rödelsperger C, Schuelke M and
Seelow D: MutationTaster evaluates disease-causing potential of
sequence alterations. Nat Methods. 7:575–576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ng PC and Henikoff S: SIFT: Predicting
amino acid changes that affect protein function. Nucleic Acids Res.
31:3812–3814. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ng PC and Henikoff S: Predicting the
effects of amino acid substitutions on protein function. Annu Rev
Genomics Hum Genet. 7:61–80. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Thomas PD, Campbell MJ, Kejariwal A, Mi H,
Karlak B, Daverman R, Diemer K, Muruganujan A and Narechania A:
PANTHER: A library of protein families and subfamilies indexed by
function. Genome Res. 13:2129–2141. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Capriotti E, Calabrese R, Fariselli P,
Martelli PL, Altman RB and Casadio R: WS-SNPs&GO: A web server
for predicting the deleterious effect of human protein variants
using functional annotation. BMC Genomics. 14(Suppl 3): S62013.
View Article : Google Scholar :
|
|
18
|
Bromberg Y and Rost B: SNAP: Predict
effect of non-synonymous polymorphisms on function. Nucleic Acids
Res. 35:3823–3835. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Capriotti E, Fariselli P and Casadio R:
I-Mutant2.0: Predicting stability changes upon mutation from the
protein sequence or structure. Nucleic Acids Res. 33:W306–W310.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lindahl E, Azuara C, Koehl P and Delarue
M: NOMAD-Ref: Visualization, deformation and refinement of
macromolecular structures based on all-atom normal mode analysis.
Nucleic Acids Res. 34:W52–W56. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Magyar C, Gromiha MM, Pujadas G, Tusnády
GE and Simon I: SRide: A server for identifying stabilizing
residues in proteins. Nucleic Acids Res. 33:W303–W35. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang Y, Muzny DM, Reid JG, Bainbridge MN,
Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al:
Clinical whole-exome sequencing for the diagnosis of mendelian
disorders. N Engl J Med. 369:1502–1511. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Leong KG and Karsan A: Recent insights
into the role of Notch signaling in tumorigenesis. Blood.
107:2223–2233. 2006. View Article : Google Scholar
|
|
24
|
Mohr OL: Character changes caused by
mutation of an entire region of a chromosome in Drosophila.
Genetics. 4:275–282. 1919.PubMed/NCBI
|
|
25
|
Callahan R and Egan SE: Notch signaling in
mammary development and oncogenesis. J Mammary Gland Biol
Neoplasia. 9:145–163. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mitsiadis TA, Lardelli M, Lendahl U and
Thesleff I: Expression of Notch 1, 2 and 3 is regulated by
epithelial-mesenchymal interactions and retinoic acid in the
developing mouse tooth and associated with determination of
ameloblast cell fate. J Cell Biol. 130:407–418. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Roy M, Pear WS and Aster JC: The
multifaceted role of Notch in cancer. Curr Opin Genet Dev.
17:52–59. 2007. View Article : Google Scholar
|
|
28
|
Jin MM, Ye YZ, Qian ZD and Zhang YB: Notch
signaling molecules as prognostic biomarkers for non-small cell
lung cancer. Oncol Lett. 10:3252–3260. 2015.
|
|
29
|
Park JT, Li M, Nakayama K, Mao TL,
Davidson B, Zhang Z, Kurman RJ, Eberhart CG, Shih IeM and Wang TL:
Notch3 gene amplification in ovarian cancer. Cancer Res.
66:6312–6318. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Engin F, Bertin T, Ma O, Jiang MM, Wang L,
Sutton RE, Donehower LA and Lee B: Notch signaling contributes to
the pathogenesis of human osteosarcomas. Hum Mol Genet.
18:1464–1470. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Klinakis A, Lobry C, Abdel-Wahab O, Oh P,
Haeno H, Buonamici S, van De Walle I, Cathelin S, Trimarchi T,
Araldi E, et al: A novel tumour-suppressor function for the Notch
pathway in myeloid leukaemia. Nature. 473:230–233. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nicolas M, Wolfer A, Raj K, Kummer JA,
Mill P, van Noort M, Hui CC, Clevers H, Dotto GP and Radtke F:
Notch1 functions as a tumor suppressor in mouse skin. Nat Genet.
33:416–421. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Köse O, Lalli A, Kutulola AO, Odell EW and
Waseem A: Changes in the expression of stem cell markers in oral
lichen planus and hyperkeratotic lesions. J Oral Sci. 49:133–139.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hijioka H, Setoguchi T, Miyawaki A, Gao H,
Ishida T, Komiya S and Nakamura N: Upregulation of Notch pathway
molecules in oral squamous cell carcinoma. Int J Oncol. 36:817–822.
2010.PubMed/NCBI
|
|
35
|
Ha PK, Benoit NE, Yochem R, Sciubba J,
Zahurak M, Sidransky D, Pevsner J, Westra WH and Califano J: A
transcriptional progression model for head and neck cancer. Clin
Cancer Res. 9:3058–3064. 2003.PubMed/NCBI
|
|
36
|
Leethanakul C, Patel V, Gillespie J,
Pallente M, Ensley JF, Koontongkaew S, Liotta LA, Emmert-Buck M and
Gutkind JS: Distinct pattern of expression of differentiation and
growth-related genes in squamous cell carcinomas of the head and
neck revealed by the use of laser capture microdissection and cDNA
arrays. Oncogene. 19:3220–3224. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang TH, Liu HC, Zhu LJ, Chu M, Liang YJ,
Liang LZ and Liao GQ: Activation of Notch signaling in human tongue
carcinoma. J Oral Pathol Med. 40:37–45. 2011. View Article : Google Scholar
|