Introduction
Liver injury is generally considered to be a result
of exposure to high levels of environmental toxins and is
associated with metabolic dysfunctions ranging from the transient
elevation of liver enzymes to life-threatening hepatic fibrosis,
liver cirrhosis and even hepatocellular carcinoma (1). Liver inflammation is commonly
associated with hepatocyte necrosis and apoptosis (2). Apoptotic hepatocyte bodies can
activate quiescent hepatic stellate cells (HSCs) and Kupffer cells,
and these activated cell populations in turn promote inflammation
and fibrogenesis (2). Activated
HSCs also increase the production of inflammatory chemokines
(3), the expression of adhesion
molecules (4) and the
presentation of antigens to T lymphocytes and natural killer T
cells (5). These enhanced
inflammation and immune responses can promote hepatocyte necrosis
and apoptosis, and thereby strengthen and perpetuate the stimuli
for fibrogenesis (6). The
sustained suppression of inflammatory activity by eliminating the
etiological agent (7–9) or by dampening the immune response
(10,11) can halt and even reverse fibrotic
progression. The success of current treatments for chronic liver
inflammation in achieving anti-fibrotic effects can be measured by
prolonged survival and possibly by the reduced occurrence of
hepatocellular carcinoma (7,12,13). Therefore, hepatic inflammation is
one of the causes of fibrosis, cirrhosis and hepatocellular
carcinoma.
Serum amyloid P (SAP), a member of the pentraxin
family of proteins, has been shown to inhibit fibrosis in a number
of organ sites in preclinical animal models, in part due to the
inhibition of the differentiation of circulating collagen
I+ cells (14,15). These cells have been demonstrated
to be involved in the pathology associated with bleomycin-induced
lung fibrosis (16). In
previously published articles conducted using the bleomycin model
of lung fibrosis in mice and rats (15), intra-peritoneal injections of
purified rat SAP (rSAP) into rats, or purified mouse SAP (mSAP)
into mice, significantly reduced fibrocyte and macrophage
recruitment to the lungs, as well as myofibroblast activation and
collagen deposition. SAP injections also reduced bleomycin-induced
leukocyte infiltration into rat lungs (15) and transforming growth factor
(TGF)-β1-induced lung inflammation in mice (17). As liver fibrosis shares similar
biological signals with fibrosis in a number of different organ
models, and as hepatic fibrosis commonly follows chronic
inflammation (6), we hypothesized
that human serum amyloid P (hSAP) may exert protective effects on
hepatocytes during hepatic inflammation/fibrosis.
Promedior, Inc. (Malvern, PA, USA) recently
developed PRM-151, a recombinant form of hSAP. Cross-species
comparison assays in vitro demonstrated comparable efficacy
of human serum-derived SAP (hSAP), recombinant hSAP (rhSAP) and
mSAP or rSAP for inhibiting mouse or rat monocytes from fibrocyte
differentiation. Additionally, cross-species comparison assays
in vitro demonstrated comparable efficacy for hSAP and rhSAP
at inhibiting human and cynomolgus monkey fibrocyte differentiation
(18). Taken together, these
results confirmed the conservation of biological function across
species. In this study, we investigated the protective effects of
hSAP against carbon tetrachloride (CCl4)-induced acute
liver injury in mice, including hepatoprotective and
anti-inflammatory effects in acute liver injury, as well as the
potential of hSAP to inhibit the migration and activation of
HSCs.
Materials and methods
hSAP
hSAP is formulated in a P5SP vehicle (10 mM sodium
phosphate, 5% (w/v) sorbitol and 0.01% (w/v) polysorbate 20, pH
7.5) at a concentration of 1.25 mg/ml. The drug was shipped frozen
and was stored at −20°C until initial use. The frozen drug was
gently thawed, and vigorous agitation was avoided.
Animal use and care
Male C57BL/6 8-week-old mice, weighing 20.0±2 g,
were purchased from Vital River Laboratories (Beijing, China). The
mice were maintained in a pathogen-free environment in the animal
facilities at Beijing Friendship Hospital. All protocols were
approved by the Beijing Friendship Hospital Animal Care and Ethics
Committee (13-2006).
The mice were randomly allocated into 7 groups (n=8
in each group). Group I (normal) was administered the same volume
of solvent (olive oil) by intraperitoneal injection. In the second
and third groups (CCl4-exposed groups, 24 and 48 h),
acute liver injury was induced by the administration of a single
intraperitoneal dose of CCl4 (10 μl/g) dissolved
in olive oil (1:7). In the fourth and fifth groups (hSAP-treated +
CCl4-exposed groups, 24 and 48 h), the mice first
received an intravenous dose of hSAP (12.5 mg/kg); they were then
intraperitoneally injected with a single dose of CCl4 2
h later, in the same manner as the CCl4-exposed group.
In the sixth and seventh groups (vehicle-treated +
CCl4-exposed group, 24 and 48 h), the mice received the
same procedure, but received the vehicle at the same volume as
hSAP. At 24 h (24 h group) and 48 h (48 h group) after the
CCl4 injection, the mice were sacrificed under
anesthesia. Some liver tissues were fixed in 4% paraformaldehyde
for subsequent histological examination and some liver tissues were
stored at −80°C for further experiments.
Histological examination and
terminal-deoxynucleotidyl transferase mediated nick-end labeling
(TUNEL) assay
Liver samples were fixed in 4% paraformaldehyde,
paraffin embedded and sectioned. Hematoxylin and eosin (H&E)
staining was performed by Wuhan Goodbio Technology Co., Ltd.
(Wuhan, China). Necrotic hepatocytes show distinctive cytoplasmic
eosinophilia, abnormal sizes and contours and nuclear pyknosis and
karyorrhexis. Five high-power fields at ×200 magnification were
randomly selected, and the degree of cellular death due to necrosis
was analyzed semiquantitatively using image analysis with the
National Institutes of Health image program (ImageJ) following the
user's guide (http://imagej.net/docs/guide). Immunohistochemistry
was performed on paraffin-embedded mouse liver sections using the
specific antibody for CD45 (GB11066; Wuhan Goodbio Technology Co.,
Ltd.), desmin (D93F5; Cell Signaling Technology, Danvers, MA, USA)
and α-smooth muscle actin (α-SMA; ab5694; Abcam, Cambridge, MA,
USA) to detect inflammatory cells and HSCs in livers. The negative
control was the replacement of primary antibody with non-immune
serum. For the detection of cell apoptosis, TUNEL assay was
performed following the manufacturer's instructions (11684795910;
Roche, Indianapolis, IN, USA). In each tissue specimen, 5
high-power fields at ×200 magnification were randomly selected, and
the percentage of positive cells was quantified using ImageJ
software.
Primary HSC isolation and cell
culture
Primary HSCs were isolated from 3 wild-type C57BL/6
8-week-old mice by a 2-step collagenase-pronase perfusion of mouse
livers followed by 8.2% Nycodenz (Accurate Chemical and Scientific
Corp., Westbury, NY, USA). Two-layer discontinuous density gradient
centrifugation was performed as previously described (19). Isolated HSCs were cultured in
Dulbecco's modified Eagle's medium (DMEM; Gibco, Grand Island, NY,
USA) containing 10% fetal bovine serum. For HSC activation
experiments in vitro, the cells were cultured overnight in
serum-free medium prior to the experiments. The HSCs were then
cultured with hSAP (30 μg/ml) or vehicle for 4 h before
stimulation with TGF-β1 (10 ng/ml for 24 h; PeproTech, Rocky Hill,
NJ, USA) (20). Following 24 h of
incubation, all cells were harvested for further experiments.
Dual-fluorescent immunohistochemistry and
Oil Red O staining for the identification of isolated primary
HSCs
Isolated primary HSCs seeded on sterile slides were
fixed with 4% paraformaldehyde and permeated with 0.1% Triton
X-100. Non-specific binding was blocked with 1% bovine serum
albumin for 30 min. The cells were then incubated with primary
antibodies against desmin (D93F5; Cell Signaling Technology) at 4°C
overnight, followed by secondary antibodies for 40 min. Nuclei were
counterstained with 4′,6-diamidino-2-phenylindole (DAPI)
(Invitrogen, Carlsbad, CA, USA) for 5 min. To detect lipid droplets
in freshly isolated HSCs, the cells were fixed in ice-cold 4%
paraformaldehyde for 20 min at room temperature prior to incubation
for 10 min in a saturated solution of Oil Red O (G1016; Wuhan
Goodbio Technology Co., Ltd.) in isopropanol. The slides were
counterstained with Mayer's haematoxylin. Images were captured
using a fluorescence microscope (Nikon, Tokyo, Japan).
Hepatocyte cell line and apoptosis
assay
To avoid the adverse effects of hepatocyte injury
produced during perfusion on the detection of cell death, we used
the mouse hepatocyte cell line, NCTC 1469 (Cell Resource Center,
Chinese Academy of Medical Sciences and Peking Union Medical
College) to detect the potential protective effects of hSAP on
hepatocyte death induced by CCl4. NCTC 1469 cells were
cultured with hSAP (30 μg/ml) or the vehicle for 4 h prior
to stimulation with CCl4 (2.5 mM for 4 h), as previously
described (21). We used DMSO to
dissolve CCl4 and DMSO is the vehicle control. Apoptosis
was assessed using a PE Annexin V apoptosis detection kit (559763;
BD Pharmingen, San Jose, CA, USA) following the manufacturer's
instructions. The cells were analyzed with a FACSCalibur flow
cytometer (Becton-Dickinson, San Jose, CA, USA), and the cells
considered viable were PE-negative and 7-AAD-negative. Cells in
early apoptosis were PE Annexin V-positive and 7-AAD-negative, and
cells in late apoptosis or dead were both PE Annexin V-positive and
7-AAD-positive.
RNA extraction, reverse transcription and
quantitative PCR
Total RNA was extracted from the cell pellets or
tissues using TRIzol reagent (Invitrogen) according to the
manufacturer's instructions. The reverse transcription reaction was
performed using a high capacity cDNA reverse transcription kit
(4375575; Applied Biosystems, Foster City, CA, USA). The cDNA was
subjected to PCR in the presence of SYBR-Green dye, with the ABI
power SYBR-Green PCR Master Mix kit (4367659; Applied Biosystems).
Quantitative PCR was performed on a 7500 real-time PCR instrument
(Applied Biosystems). Mouse primers were designed using Primer 3
and were synthesized by SBS Genetech Co., Ltd. (Beijing, China)
(Table I). The relative mRNA
levels of genes were calculated using the 2−∆∆Ct
formula, and mouse β-actin was used as a housekeeping gene. All
experiments were performed independently 3 times, and the average
was used for comparison.
 | Table IPrimers used for quantitative
PCR. |
Table I
Primers used for quantitative
PCR.
| Gene | Forward primer
(5′→3′) | Reverse primer
(5′→3′) |
|---|
| IL-1β | GGT CAA AGG TTT GGA
AGC AG | TGT GAA ATG CCA CCT
TTT GA |
| IL-6 |
ACCAGAGGAAATTTTCAATAGGC |
TGATGCACTTGCAGAAAACA |
| TNF-α |
AGGGTCTGGGCCATAGAACT |
CCACCACGCTCTTCTGTCTAC |
| MCP-1 |
ATTGGGATCATCTTGCTGGT |
CCTGCTGTTCACAGTTGCC |
| MIP-2 |
TCCAGGTCAGTTAGCCTTGC |
CGGTCAAAAAGTTTGCCTTG |
| CD11b |
GTTTGTTGAAGGCATTTCCC |
ATTCGGTGATCCCTTGGATT |
| Bcl-2 | CTT TCT GCT TTT TAT
TTC ATG AGG | CAG AAG ATC ATG CCG
TCC TT |
| Bax | GAT CAG CTC GGG CAC
TTT AG | TTG CTG ATG GCA ACT
TCA AC |
| α-SMA |
GTTCAGTGGTGCCTCTGTCA |
ACTGGGACGACATGGAAAAG |
| TIMP-1 |
AGGTGGTCTCGTTGATTTCT |
GTAAGGCCTGTAGCTGTGCC |
| β-actin |
ATGGAGGGGAATACAGCCC |
TTCTTTGCAGCTCCTTCGTT |
Protein extraction and western blot
analysis
The preparation of protein extracts from frozen
livers or isolated cells, electrophoresis, and subsequent blotting
were performed as previously described (22,23). We incubated the blots with primary
antibodies to B cell lymphoma/leukemia (Bcl)-2 (1:1,000; 50E3),
Bcl-2-associated X protein (Bax, 1:1,000; 2772), cleaved caspase-3
(1:1,000; 5A1E) (all from Cell Signaling Technology), α-SMA
(1:2,000; 5694), tissue inhibitor of metalloproteinases (TIMP)-1
(1:1,000; 38978) (both from Abcam) and β-actin (1:5,000; A1978;
Sigma, St. Louis, MO, USA) at 4°C overnight. This was followed by
the addition of the appropriate horseradish peroxidase-conjugated
secondary antibody (1:10,000; ZB2301 or ZB2306; ZSGB Bio, Beijing,
China) and incubation for 60 min and specific antibody-antigen
complexes were detected with the ECL western blot detection kit
(Pierce, Rockford, IL, USA). All experiments were performed
independently at least 3 times, and protein expression was
quantified by densitometric analysis of immunoblots using Quantity
One software (Thermo Fisher Scientific, Waltham, MA, USA).
Statistical analysis
Data are expressed as the means ± standard deviation
(SD). Two-group comparisons were conducted using the Student's
t-test, and comparisons of the means of 3 or more groups were
performed by ANOVA. A value of P<0.05 was considered to indicate
a statistically significant difference.
Results
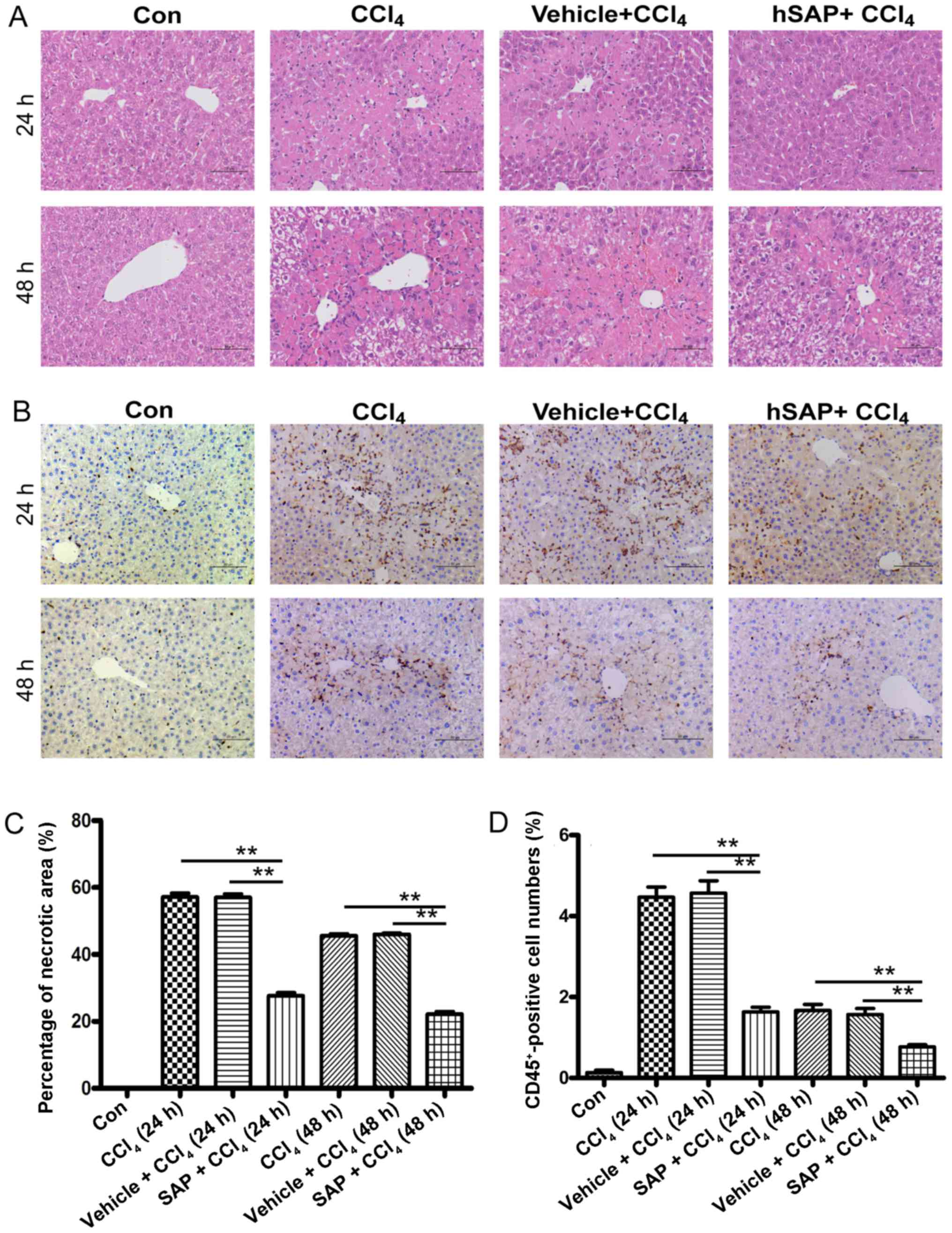
Effects of hSAP pre-treatment on the
histopathological changes in the livers of mice
H&E staining of the liver tissue sections
revealed severe and diffuse centrilobular necrosis in the mice at
24 h following CCl4 administration. By contrast, only
spotty necrosis of the hepatocytes was found in the livers of
CCl4-challenged mice pre-treated with hSAP. In addition,
less inflammatory cell infiltration was observed in the hSAP +
CCl4 group compared with the CCl4 group and
the vehicle + CCl4 group. Diffuse centrilobular necrosis
and inflammatory reactions were decreased in each group 48 h after
the CCl4 administration; however, hSAP administration
continued to decrease the necrotic area and inflammatory reaction
at this time point (Fig. 1A and
C).
hSAP pre-treatment inhibits
CCl4-induced inflammatory cell infiltration and
pro-inflammatory factors, and chemokine expression
CD45 cell surface antigen is a transmembrane protein
expressed by all nucleated cells of hematopoietic origin, apart
from erythrocytes and platelets (24). In this study, we used this marker
to lable inflammatory cells in the injured liver.
Immunohistochemical staining revealed that the number of
CD45-positive cells accumulating in the liver sections was
decreased by hSAP treatment compared with that of the
CCl4 and vehicle + CCl4 groups (P<0.01;
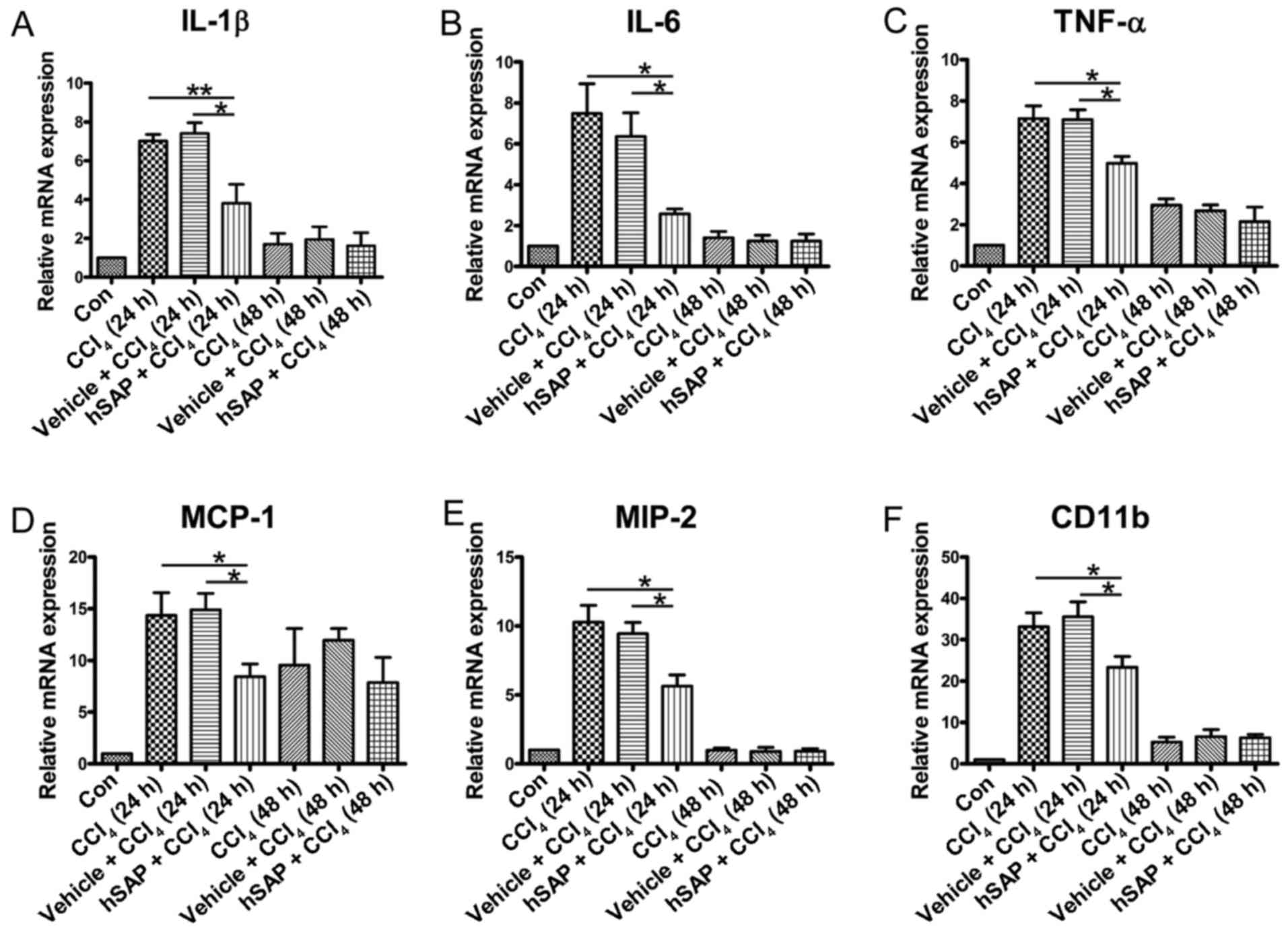
Fig. 1B and D). Exposure to
CCl4 significantly increased the hepatic interleukin
(IL)-1β, IL-6 and tumor necrosis factor (TNF)-α mRNA expression
levels compared with those of the normal group, suggesting the
induction of a severe inflammatory response. However, the
pre-administration of hSAP suppressed the mRNA expression of
hepatic IL-1β, IL-6 and TNF-α (Fig.
2A–C). In addition, the levels of chemokines that regulate
inflammation, such as monocyte chemotactic protein (MCP)-1 and
macrophage inflammatory protein (MIP)-2, were also detected. High
expression levels of MCP-1 and MIP-2 were observed in the
CCl4 and vehicle + CCl4 groups, whereas hSAP
administration down-regulated the expression of these chemokines
(Fig. 2D and E). To confirm the
inflammatory infiltration, we detected CD11b (a biomarker for
neutrophils) expression in the liver. CD11b expression in the liver
sections was decreased by hSAP treatment compared with the injury
group (Fig. 2F). The levels of
all pro-inflammatory factors and chemokine expression in the 24-h
group were higher than those in the 48-h group, indicating that
acute liver injury induced by CCl4 reached a peak value
at 24 h after CCl4 administration.
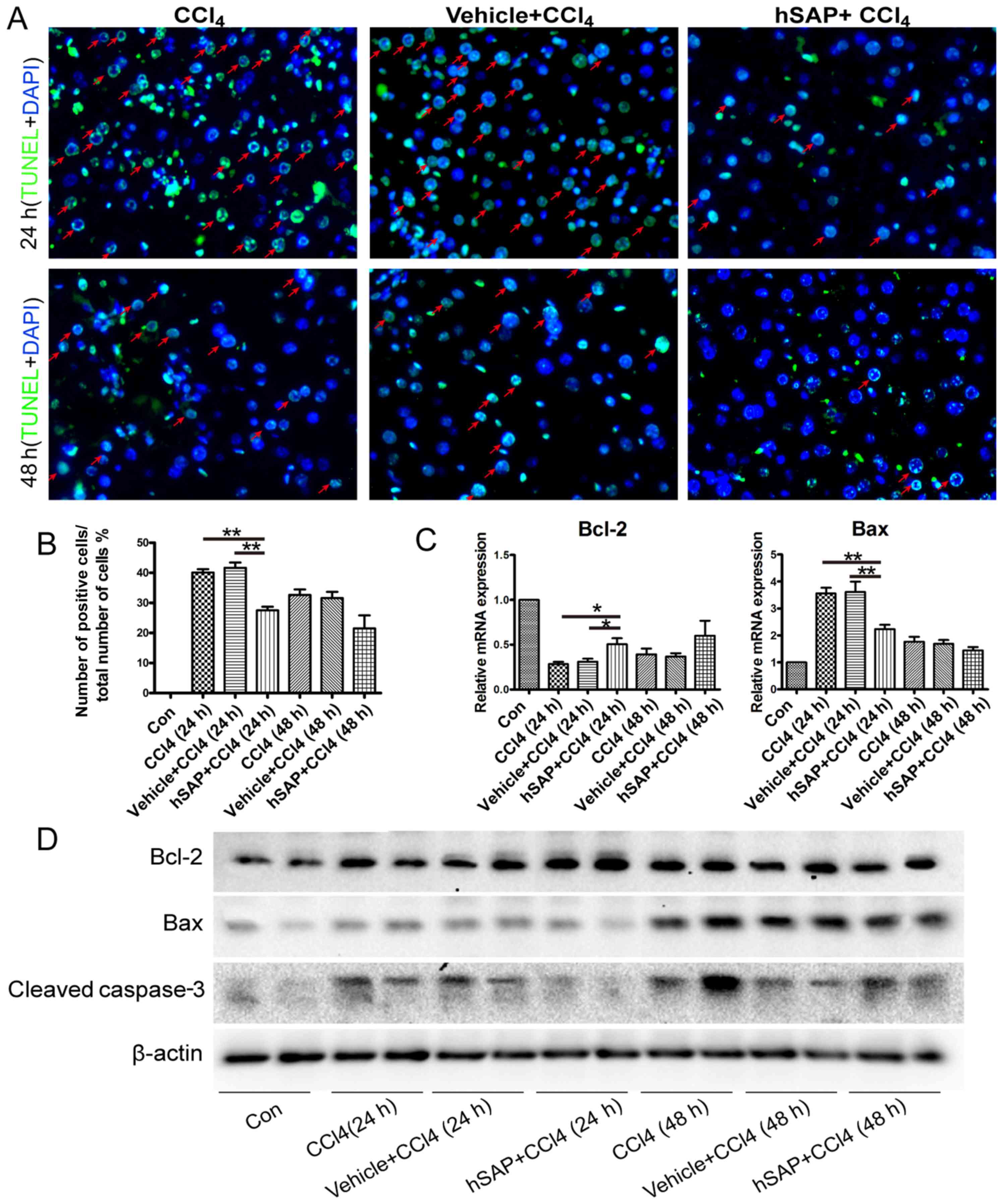
hSAP pre-treatment decreases
CCl4-induced apoptosis in vivo
Previous studies have reported severe hepatocyte
apoptosis in CCl4-induced acute liver injury (25,26). In this study, to clarify whether
the effect of hSAP on acute liver injury was primarily due to its
inhibitory effect on hepatocyte apoptosis, we performed TUNEL
staining to assess the protective ability of hSAP against
CCl4-induced hepatocyte apoptosis. The analysis of
cleaved caspase-3 expression in the liver also revealed that the
pre-administration of hSAP inhibited the increased apoptosis
induced by exposure to CCl4 (Fig. 3A, B and D). To determine the
mechanisms underlying the anti-apoptotic effects of hSAP, the
expression of the apoptosis-related genes, Bcl-2 and Bax, in
hepatocytes was detected using quantitative PCR and western blot
analysis. The pre-administration of hSAP significantly upregulated
the expression levels of Bcl-2 and significantly downregulated the
expression levels of Bax compared with those in the model group
(Fig. 3C and D). The expression
of Bcl-2, Bax and cleaved caspase-3 exhibited a significant
difference in the hSAP pre-treatment group at 24 h following the
CCl4 administration compared with the model group
(P<0.05 or P<0.01).
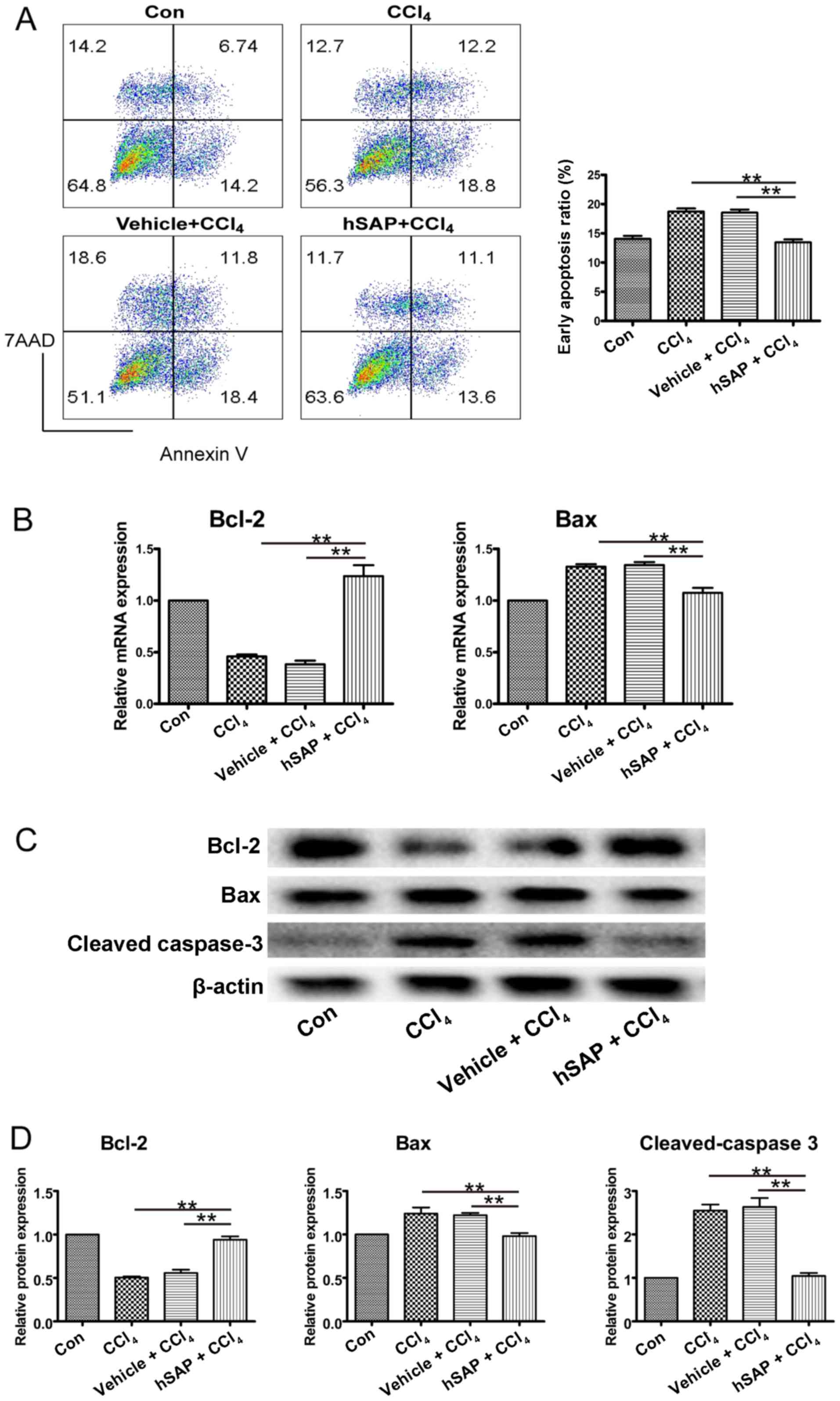
hSAP pre-treatment decreases the
CCl4-induced apoptosis of hepatocytes in vitro
To confirm the hepatocyte protective effects of
hSAP, NCTC 1469 cells were cultured with hSAP (30 μg/ml) or
the vehicle for 4 h prior to stimulation with CCl4 (2.5
mM). Four hours later, the cells were collected, and cell death was
detected using an apoptosis detection kit. The number of cells in
early apoptosis stained with Annexin V was significantly reduced by
28–30% in the hSAP treatment group compared with the
CCl4 group (13.4±0.9 vs. 18.5±1.5%) (Fig. 4A). The expression of Bcl-2 was
increased, whereas the expression levels of Bax and cleaved
caspase-3 were significantly inhibited in the hSAP pre-treatment
group compared with the CCl4 group (P<0.01),
indicating that hSAP has a direct anti-apoptotic function on
hepatocytes exposed to CCl4 (Fig. 4B–D).
hSAP inhibits the migration and
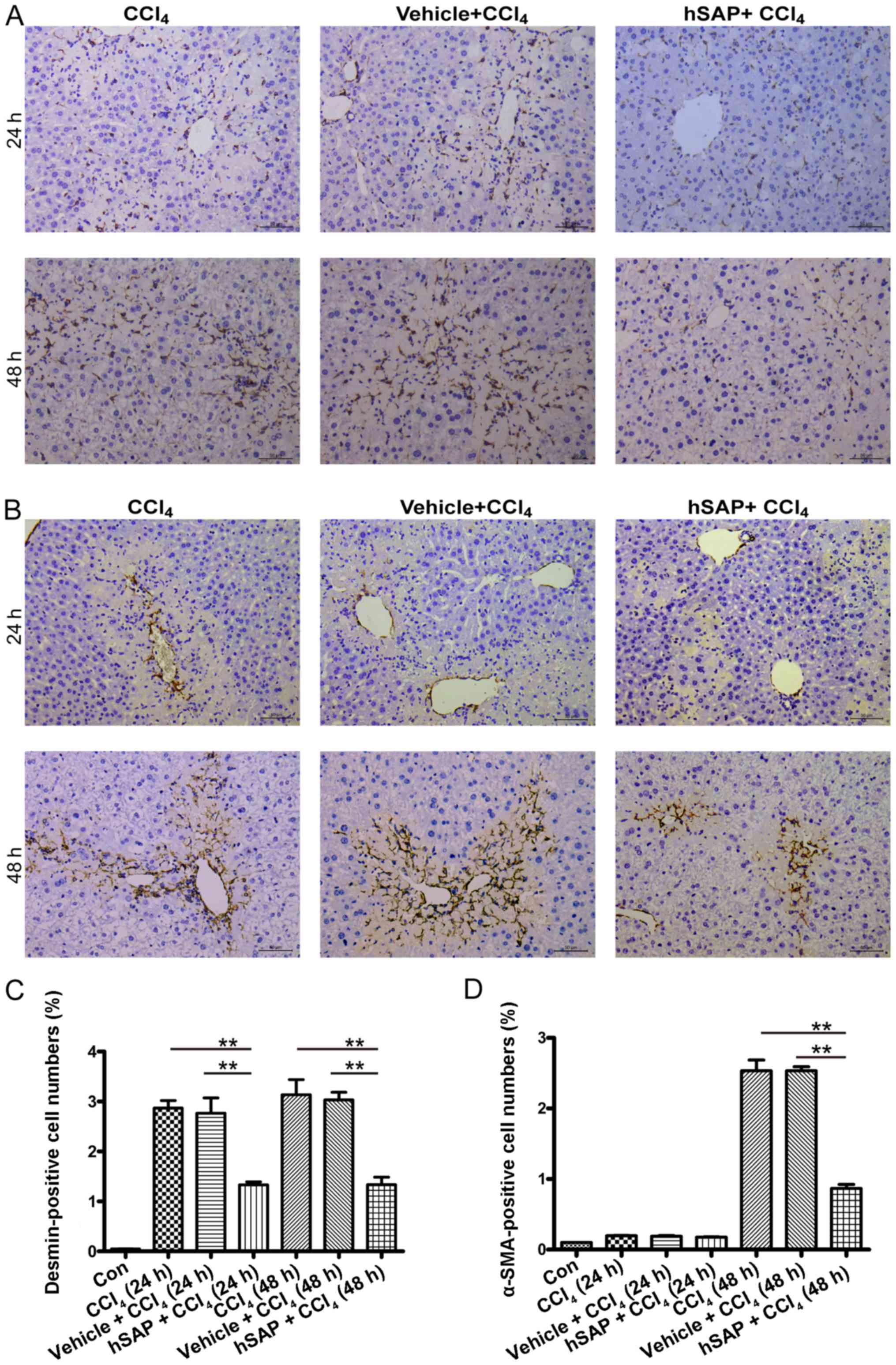
activation of HSCs in vivo
As activated HSCs secrete high levels of MCP-1 and
MCP-1 in turn can promote the migration and positioning of HSCs
(27,28), we further detected the migration
of HSCs in injured livers. Although aggregated HSCs were found
around the necrotic area at 24 and 48 h following the
CCl4 administration, fewer HSCs migrated to these areas
following hSAP administration, as confirmed by desmin staining
(P<0.05; Fig. 5A and C).
Subsequently, we used α-SMA, a biomarker of activated HSCs, to
detect the activation of HSCs in the injured liver. Although there
were a limited number of activated HSCs in each group at 24 h
following the CCl4 administration, immunostaining of the
tissue sections for α-SMA expression revealed intense staining
patterns around the damaged hepatocytes in the mice from the
CCl4 and the vehicle + CCl4 groups 48 h
following the CCl4 administration. The administration of
hSAP resulted in approximately 65% decreased positive staining in
the sinusoids, demonstrating fewer activated HSCs following hSAP
treatment (Fig. 5B and D).
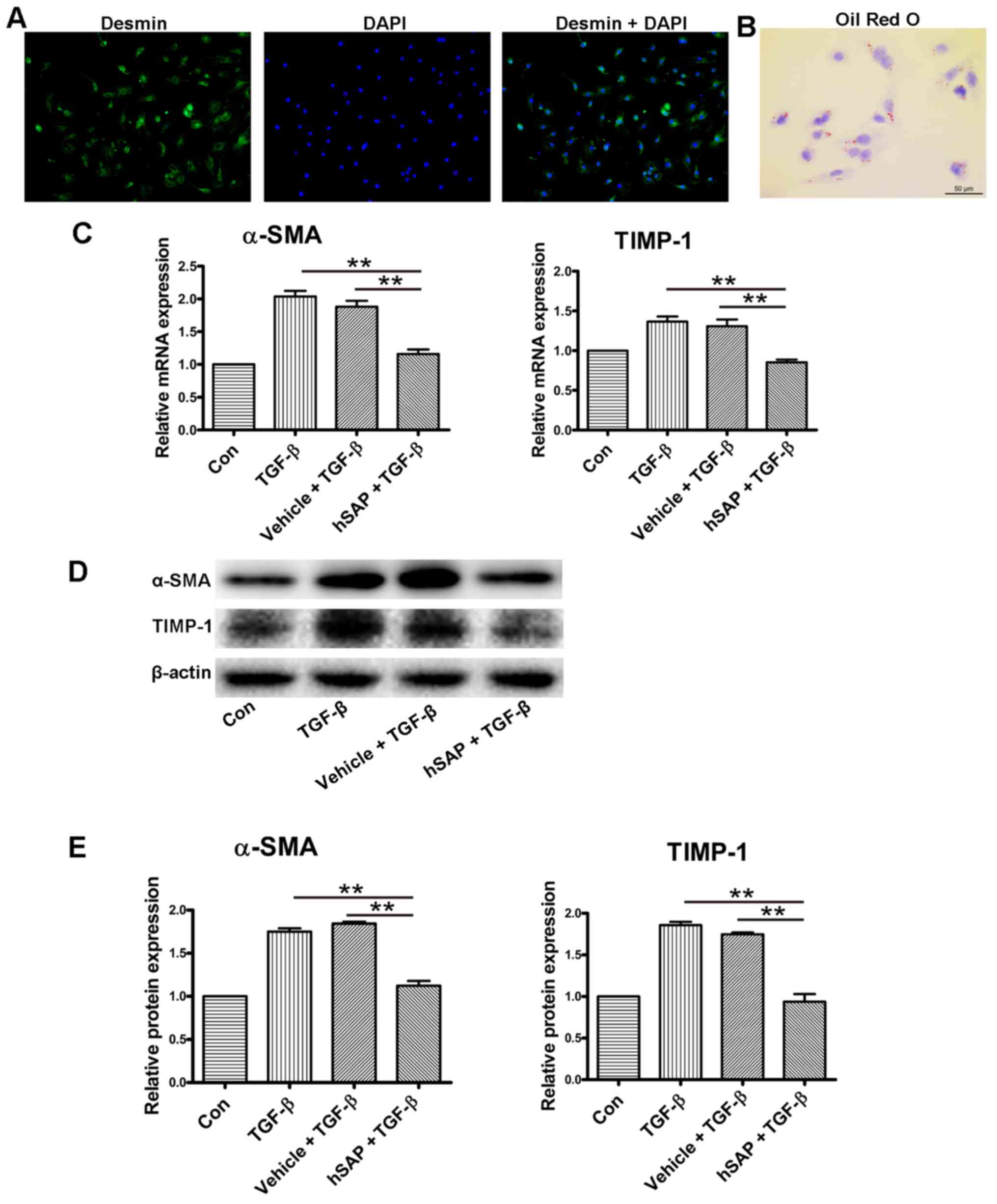
hSAP inhibits the TGF-β1-induced
activation of HSCs in vitro
CCl4 is a hepatotoxin, which causes the
apoptosis of damaged hepatocytes. Apoptotic hepatocytes release
factors that activate Kupffer cells, the major source of TGF-β1.
Quiescent HSCs are induced by TGF-β1 to transdifferentiate into
myofibroblasts that secrete extracellular matrix (2). Based on the observed weaker
activation of HSCs in the injured liver of hSAP-treated mice, we
wished to elucidate whether hSAP inhibits the TGF-β1-induced
activation of HSCs directly in vitro. Primary HSCs were
isolated from wild-type C57BL/6 mice, and the purity was >98%,
as assessed by desmin immunofluorescence staining and Oil Red O
staining (Fig. 6A and B).
Following 24 h of incubation with TGF-β1, the isolated HSCs
exhibited a significantly lower activation in the 30 μg/ml
hSAP treatment group. The mRNA and protein levels of α-SMA and
TIMP-1 in the hSAP treatment group were significantly reduced
compared with those in the TGF-β1 group (Fig. 6C–E).
Discussion
Carbon tetrachloride is a widely used hepatotoxin to
establish animal models for evaluating the hepatoprotective
activities of drugs (29). In the
present study, mice injected with CCl4 exhibited
characteristics of acute liver injury, including distorted hepatic
parenchyma, inflammatory cell infiltration and hepatocyte necrosis
(visualized by H&E staining). However, pre-treatment with hSAP
significantly decreased the CCl4-induced
histopathological changes in the livers. These findings indicate
that hSAP can exert protective effects against
CCl4-induced liver damage.
The inflammatory response is involved in the process
of CCl4-induced acute liver injury (30). The traditional inflammatory
response is characterized as a complex reaction to an injuring
agent that involves the loss of vascular wall integrity, the
effusion of inflammatory cells, the activation of leukocytes and
their extravasations, and the release of pro-inflammatory
cytokines, such as TNF-α, IL-6 and IL-1β (31,32). In this study, the CD45 antibody
was used to stain inflammatory cells in liver sections, and we
found that hSAP treatment significantly reduced inflammatory
infiltration compared with the injury group. Exposure to
CCl4 significantly upregulated the expression of
pro-inflammatory cytokines (TNF-α, IL-6 and IL-1β), chemokines
(MCP-1 and MIP-2) and CD11b (marker of neutrophil activation) in
injured mouse livers. However, the pre-administration of hSAP
markedly inhibited the upregulation of these pro-inflammatory
cytokines, chemokines and leukocyte infiltration. These results
suggested that hSAP can meliorate liver injury caused by
CCl4 by inhibiting the inflammatory response.
Previous studies have demonstrated that hepatocyte
apoptosis can be triggered by CCl4 (25,26). In this study, the rate of
apoptosis evaluated by TUNEL staining was significantly increased
in the CCl4 group, which was decreased by the
pre-administration of hSAP. To investigate whether hSAP treatment
modulates the molecular mechanisms involved in apoptosis, we
detected the expression of apoptosis-related molecules,
particularly Bcl-2 (anti-apoptotic protein), Bax (pro-apoptotic
protein) and caspase-3. The present study demonstrated that the
pre-administration of hSAP suppressed the upregulation of Bax
expression, inhibited the activation of caspase-3, and restored
Bcl-2 expression which was decreased by CCl4. The result
that the mRNA expression of Bax reached a peak value at 24 h
following CCl4 administration, whereas Bax and
cleaved-caspase-3 protein bands in the western blots were far
denser at 48 h, could partially be explained by the timeline
difference between the regulation of mRNA and protein expression.
Our in vitro experiments also confirmed that hSAP
significantly reduced the CCl4-induced early apoptotic
rate in hepatocytes by regulating the expression of the
apoptosis-related proteins, Bax and Bcl-2.
Several studies have proposed that the phagocytosis
of apoptotic bodies by HSCs links cell death to HSC activation,
showing increased HSC activation and survival after the
phagocytosis of apoptotic bodies in vitro (33–35). DNA from apoptotic hepatocytes can
provide a stop signal to mobile HSCs when they reach an area of
apoptotic hepatocytes and induce a stationary phenotype-associated
upregulation of collagen production (36). Apoptotic body engulfment in HSCs
also stimulates TGF-β1 expression and induces collagen I,
indicating a fibrogenic response (33). In this study, the
immunohistochemical staining of desmin (a biomarker for HSCs) and
α-SMA (a biomarker for activated HSCs) showed that the
pre-administration of hSAP inhibited the migration and activation
of HSCs to the injured liver site and may be mediated by less
apoptotic hepatocyte DNA in hSAP-pre-treated livers.
The short pentraxin, SAP, was recently described to
reduce fibrosis in a number of different organ models, including
pulmonary, renal, cardiac and oral submucous fibrosis, partially
through the inhibition of fibrocyte and macrophage accumulation and
activation in the injured organ (15,17,37–40). It has been demonstrated that the
anti-fibrotic effects of SAP in TGF-β1-induced lung fibrosis are
mediated through the modulation of monocyte responses (17), and that SAP also inhibits fibrosis
through Fcγ receptor (FcγR)-dependent monocyte-macrophage
regulation (37). HSCs are the
main cell type responsible for liver fibrosis, and TGF-β1 is the
most potent cytokine that can promote the activation of HSCs
(2). In this study, to determine
whether hSAP inhibits the activation of HSCs directly, we isolated
primary HSCs from normal mouse liver and incubated these primary
HSCs with or without hSAP and with TGF-β1 stimulation. Although
TGF-β1 activated HSCs by upregulating the expression of α-SMA and
TIMP-1, the mRNA and protein levels of these profibrogenic genes in
the hSAP treatment group were significantly reduced by 43–45%
compared with the control group. It has been shown that hSAP serves
as a ligand for activating FcγRs and downregulates the activation
of monocytes and macrophages (37). Primary rat HSCs express FcγRs
(41), suggesting that hSAP may
also bind directly to HSCs via FcRs. Rat hepatocytes may express
MHC class I-related Fc receptor for IgG (42). For our in vitro experiment,
we hypothesized that SAP could act on hepatocytes and HSCs by
binding between SAP and FcγRs. Additional studies are required to
fully investigate the mechanism through which hSAP inhibits the
activation of HSCs.
In the present study, we demonstrated that hSAP has
a strong anti-inflammatory and hepatoprotective effect in
CCl4-induced acute liver injury in mice, most likely
through the combined effects of inhibiting leukocyte infiltration,
inflammatory cytokine expression and hepatocyte apoptosis. hSAP may
also inhibit HSCs activation directly or indirectly due to fewer
apoptotic hepatocytes. Based on the intimate association between
inflammation and fibrosis, the important role of HSCs in
fibrogenesis and the inhibitory effects of hSAP on HSC activation,
hSAP may also affect the development of liver fibrosis. Further
studies are required to evaluate the role of SAP in liver fibrosis
in vitro and in vivo.
Abbreviations:
|
α-SMA
|
α-smooth muscle actin
|
|
Bax
|
Bcl-2-associated X protein
|
|
Bcl-2
|
B cell lymphoma/leukemia-2
|
|
CCl4
|
carbon tetrachloride
|
|
HSCs
|
hepatic stellate cells
|
|
IL
|
interleukin
|
|
MCP
|
monocyte chemotactic protein
|
|
MIP
|
macrophage inflammatory protein
|
|
SAP
|
serum amyloid P
|
|
hSAP
|
human serum-derived SAP
|
|
TGF-β1
|
transforming growth factor-β1
|
|
TIMP-1
|
tissue inhibitor of
metalloproteinases-1
|
|
TNF-α
|
tumor necrosis factor-α
|
|
TUNEL
|
terminal-deoxynucleotidyl transferase
mediated nick end labeling
|
Acknowledgments
We would like to thank Promedior, Inc. for providing
hSAP. This study was supported by grants from the National Natural
Science Foundation of China (no. 81570542), the Natural Science
Foundation of Beijing Municipality (no. 7142043), and the Beijing
Health System Talents Plan (no. 2013-3-057).
References
|
1
|
Reuber MD and Glover EL: Cirrhosis and
carcinoma of the liver in male rats given subcutaneous carbon
tetrachloride. J Natl Cancer Inst. 44:419–427. 1970.PubMed/NCBI
|
|
2
|
Lee UE and Friedman SL: Mechanisms of
hepatic fibrogenesis. Best Pract Res Clin Gastroenterol.
25:195–206. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schwabe RF, Bataller R and Brenner DA:
Human hepatic stellate cells express CCR5 and RANTES to induce
proliferation and migration. Am J Physiol Gastrointest Liver
Physiol. 285:G949–G958. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hellerbrand SC, Wang SC, Tsukamoto H,
Brenner DA and Rippe RA: Expression of intracellular adhesion
molecule 1 by activated hepatic stellate cells. Hepatology.
24:670–676. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Winau F, Hegasy G, Weiskirchen R, Weber S,
Cassan C, Sieling PA, Modlin RL, Liblau RS, Gressner AM and
Kaufmann SH: Ito cells are liver-resident antigen-presenting cells
for activating T cell responses. Immunity. 26:117–129. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Czaja AJ: Hepatic inflammation and
progressive liver fibrosis in chronic liver disease. World J
Gastroenterol. 20:2515–2532. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mallet V, Gilgenkrantz H, Serpaggi J,
Verkarre V, Vallet-Pichard A, Fontaine H and Pol S: Brief
communication: The relationship of regression of cirrhosis to
outcome in chronic hepatitis C. Ann Intern Med. 149:399–403. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kweon YO, Goodman ZD, Dienstag JL, Schiff
ER, Brown NA, Burchardt E, Schoonhoven R, Brenner DA and Fried MW:
Decreasing fibrogenesis: An immunohistochemical study of paired
liver biopsies following lamivudine therapy for chronic hepatitis
B. J Hepatol. 35:749–755. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lau DT, Kleiner DE, Park Y, Di Bisceglie
AM and Hoofnagle JH: Resolution of chronic delta hepatitis after 12
years of interferon alfa therapy. Gastroenterology. 117:1229–1233.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Czaja AJ and Carpenter HA: Decreased
fibrosis during corticosteroid therapy of autoimmune hepatitis. J
Hepatol. 40:646–652. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mohamadnejad M, Malekzadeh R,
Nasseri-Moghaddam S, Hagh-Azali S, Rakhshani N, Tavangar SM,
Sedaghat M and Alimohamadi SM: Impact of immunosuppressive
treatment on liver fibrosis in autoimmune hepatitis. Dig Dis Sci.
50:547–551. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lok AS, Everhart JE, Wright EC, Di
Bisceglie AM, Kim HY, Sterling RK, Everson GT, Lindsay KL, Lee WM,
Bonkovsky HL, et al HALT-C Trial Group: Maintenance peginterferon
therapy and other factors associated with hepatocellular carcinoma
in patients with advanced hepatitis C. Gastroenterology.
140:840–849. 2011. View Article : Google Scholar :
|
|
13
|
Roberts SK, Therneau TM and Czaja AJ:
Prognosis of histological cirrhosis in type 1 autoimmune hepatitis.
Gastroenterology. 110:848–857. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Haudek SB, Xia Y, Huebener P, Lee JM,
Carlson S, Crawford JR, Pilling D, Gomer RH, Trial J, Frangogiannis
NG, et al: Bone marrow-derived fibroblast precursors mediate
ischemic cardio-myopathy in mice. Proc Natl Acad Sci USA.
103:18284–18289. 2006. View Article : Google Scholar
|
|
15
|
Pilling D, Roife D, Wang M, Ronkainen SD,
Crawford JR, Travis EL and Gomer RH: Reduction of bleomycin-induced
pulmonary fibrosis by serum amyloid P. J Immunol. 179:4035–4044.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Phillips RJ, Burdick MD, Hong K, Lutz MA,
Murray LA, Xue YY, Belperio JA, Keane MP and Strieter RM:
Circulating fibrocytes traffic to the lungs in response to CXCL12
and mediate fibrosis. J Clin Invest. 114:438–446. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Murray LA, Chen Q, Kramer MS, Hesson DP,
Argentieri RL, Peng X, Gulati M, Homer RJ, Russell T, van Rooijen
N, et al: TGF-beta driven lung fibrosis is macrophage dependent and
blocked by Serum amyloid P. Int J Biochem Cell Biol. 43:154–162.
2011. View Article : Google Scholar
|
|
18
|
Duffield JS and Lupher ML Jr: PRM-151
(recombinant human serum amyloid P/pentraxin 2) for the treatment
of fibrosis. Drug News Perspect. 23:305–315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kisseleva T, Cong M, Paik Y, Scholten D,
Jiang C, Benner C, Iwaisako K, Moore-Morris T, Scott B, Tsukamoto
H, et al: Myofibroblasts revert to an inactive phenotype during
regression of liver fibrosis. Proc Natl Acad Sci USA.
109:9448–9453. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang L, Roh YS, Song J, Zhang B, Liu C,
Loomba R and Seki E: Transforming growth factor beta signaling in
hepatocytes participates in steatohepatitis through regulation of
cell death and lipid metabolism in mice. Hepatology. 59:483–495.
2014. View Article : Google Scholar
|
|
21
|
Iwaisako K, Haimerl M, Paik YH, Taura K,
Kodama Y, Sirlin C, Yu E, Yu RT, Downes M, Evans RM, et al:
Protection from liver fibrosis by a peroxisome
proliferator-activated receptor δ agonist. Proc Natl Acad Sci USA.
109:E1369–E1376. 2012. View Article : Google Scholar
|
|
22
|
Cong M, Liu T, Wang P, Fan X, Yang A, Bai
Y, Peng Z, Wu P, Tong X, Chen J, et al: Antifibrotic effects of a
recombinant adeno-associated virus carrying small interfering RNA
targeting TIMP-1 in rat liver fibrosis. Am J Pathol. 182:1607–1616.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cong M, Liu T, Wang P, Xu Y, Tang S, Wang
B, Jia J, Liu Y, Hermonat PL and You H: Suppression of tissue
inhibitor of metalloproteinase-1 by recombinant adeno-associated
viruses carrying siRNAs in hepatic stellate cells. Int J Mol Med.
24:685–692. 2009.PubMed/NCBI
|
|
24
|
Gredelj-Simec N, Jelić-Puskarić B, Ostojić
A, Siftar Z, Fiala D, Kardum-Skelin I, Vrhovac R and Jaksić B:
Diagnostic and prognostic significance of CD45 cell surface antigen
expression in hematologic malignancies with main focus on acute
leukemias. Acta Med Croatica. 65(Suppl 1): 45–52. 2011.In
Croatian.
|
|
25
|
Karakus E, Karadeniz A, Simsek N, Can I,
Kara A, Yildirim S, Kalkan Y and Kisa F: Protective effect of Panax
ginseng against serum biochemical changes and apoptosis in liver of
rats treated with carbon tetrachloride (CCl4). J Hazard
Mater. 195:208–213. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang BY, Zhang XY, Guan SW and Hua ZC:
Protective effect of procyanidin B2 against
CCl4-induced acute liver injury in mice. Molecules.
20:12250–12265. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ramm GA: Chemokine (C-C motif) receptors
in fibrogenesis and hepatic regeneration following acute and
chronic liver disease. Hepatology. 50:1664–1668. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Marra F, Romanelli RG, Giannini C, Failli
P, Pastacaldi S, Arrighi MC, Pinzani M, Laffi G, Montalto P and
Gentilini P: Monocyte chemotactic protein-1 as a chemoattractant
for human hepatic stellate cells. Hepatology. 29:140–148. 1999.
View Article : Google Scholar
|
|
29
|
Weber LW, Boll M and Stampfl A:
Hepatotoxicity and mechanism of action of haloalkanes: Carbon
tetrachloride as a toxicological model. Crit Rev Toxicol.
33:105–136. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang F, Wang X, Qiu X, Wang J, Fang H,
Wang Z, Sun Y and Xia Z: The protective effect of Esculentoside A
on experimental acute liver injury in mice. PLoS One.
9:e1131072014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Spicer J, Brodt P and Ferri L: Role of
inflammation in the early stages of liver metastasis. Liver
Metastasis: Biology and Clinical Management. Brodt P: Springer; pp.
155–185. 2011, View Article : Google Scholar
|
|
32
|
Kumar V, Abbas A, Fausto N and Aster J:
Cellular Responses to Stress and Toxic Insults: Adaptation, Injury
and Death. Robbins and Cotran Pathologic Basis of Disease. 8th
edition. Saunders Elsevier; Philidelphia: pp. 18–19. 2009
|
|
33
|
Canbay A, Taimr P, Torok N, Higuchi H,
Friedman S and Gores GJ: Apoptotic body engulfment by a human
stellate cell line is profibrogenic. Lab Invest. 83:655–663. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhan SS, Jiang JX, Wu J, Halsted C,
Friedman SL, Zern MA and Torok NJ: Phagocytosis of apoptotic bodies
by hepatic stellate cells induces NADPH oxidase and is associated
with liver fibrosis in vivo. Hepatology. 43:435–443. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiang JX, Mikami K, Venugopal S, Li Y and
Török NJ: Apoptotic body engulfment by hepatic stellate cells
promotes their survival by the JAK/STAT and Akt/NF-kappaB-dependent
pathways. J Hepatol. 51:139–148. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Watanabe A, Hashmi A, Gomes DA, Town T,
Badou A, Flavell RA and Mehal WZ: Apoptotic hepatocyte DNA inhibits
hepatic stellate cell chemotaxis via toll-like receptor 9.
Hepatology. 46:1509–1518. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Castaño AP, Lin SL, Surowy T, Nowlin BT,
Turlapati SA, Patel T, Singh A, Li S, Lupher ML Jr and Duffield JS:
Serum amyloid P inhibits fibrosis through Fc gamma R-dependent
monocyte-macrophage regulation in vivo. Sci Transl Med.
1:5ra132009. View Article : Google Scholar
|
|
38
|
Murray LA, Rosada R, Moreira AP, Joshi A,
Kramer MS, Hesson DP, Argentieri RL, Mathai S, Gulati M, Herzog EL,
et al: Serum amyloid P therapeutically attenuates murine
bleomycin-induced pulmonary fibrosis via its effects on
macrophages. PLoS One. 5:e96832010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Murray LA, Kramer MS, Hesson DP, Watkins
BA, Fey EG, Argentieri RL, Shaheen F, Knight DA and Sonis ST: Serum
amyloid P ameliorates radiation-induced oral mucositis and
fibrosis. Fibrogenesis Tissue Repair. 3:112010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Haudek SB, Trial J, Xia Y, Gupta D,
Pilling D and Entman ML: Fc receptor engagement mediates
differentiation of cardiac fibroblast precursor cells. Proc Natl
Acad Sci USA. 105:10179–10184. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shen H, Zhang M, Kaita K, Minuk GY, Rempel
J and Gong Y: Expression of Fc fragment receptors of immunoglobulin
G (Fc gammaRs) in rat hepatic stellate cells. Dig Dis Sci.
50:181–187. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Blumberg RS, Koss T, Story CM, Barisani D,
Polischuk J, Lipin A, Pablo L, Green R and Simister NE: A major
histocompatibility complex class I-related Fc receptor for IgG on
rat hepatocytes. J Clin Invest. 95:2397–2402. 1995. View Article : Google Scholar : PubMed/NCBI
|