Introduction
Renal cell carcinoma (RCC) accounts for
approximately 4% of all adult malignancies. In 2016, ~63,000 new
cases of kidney cancer were estimated to be diagnosed, and more
than 14,000 deaths occurred in the United States (1). Each year, more than 250,000 new
cases of kidney cancer are diagnosed worldwide. In particular,
metastatic RCC has a poor prognosis that seriously affects the
quality of life of patients. The median overall survival of
metastatic RCC is ~12 months, and the 5-year survival rate is
<10% (2). Multiple antagonists
of the vascular endothelial growth factor (VEGF) signalling
pathway, such as sorafenib, sunitinib, axitinib and pazopanib, have
been approved for the treatment of patients with advanced RCC
(3–5). While these anti-VEGF strategies have
shown clinical benefits, many patients are either resistant to such
therapy or, more commonly, acquire resistance to these drugs within
a year of treatment initiation. Therefore, targeting additional
angiogenic signalling pathways may represent a promising
therapeutic approach for patients with advanced RCC.
Vascular endothelial growth inhibitor (VEGI) is a
member of the tumour necrosis factor superfamily, whose members
have been identified as anti-angiogenic cytokines (6,7).
The VEGI gene is located on human chromosome 9q32. The full-length
VEGI gene is ~17 kb, and it consists of four exons and three
introns. Three alternatively spliced isoforms of VEGI, VEGI174,
VEGI192 and VEGI251 have been documented, sharing 151 common
C-terminal amino acids but differing in their N-terminal regions.
The initially reported VEGI protein consists of 174 amino acids,
which can be divided into two parts. AA residues 1–25 at the
N-terminus are the intracellular and transmembrane domains, and AA
residues 26–174 at the C-terminus form an extracellular domain
(8,9). VEGI expression has been observed in
kidney, bladder, prostate, lung, breast and colon tissues (10–13). Studies examining the biological
functions of VEGI revealed that it had potential inhibitory effects
on tumours. For example, our previous studies showed that VEGI
overexpression substantially reduced the motility and adhesion of
prostate and bladder cancer cells and suppressed renal carcinoma
cell growth in vivo (14–16).
In recent years, numerous studies have demonstrated
that activation of epithelial-mesenchymal transition (EMT) is a key
event in the tumour invasion process (17–19). EMT is a biological phenomenon that
frequently occurs in tumour tissues and is associated with local
invasion and distant metastases. During this process, epithelial
cells undergo multiple biochemical changes that enable them to lose
the epithelial-like phenotype and transform into a mesenchymal-like
phenotype. The tumour cells lose cell-cell adhesion, detach from
the primary tumours and disseminate through the vasculature to
other organs. It has been determined that EMT plays a significant
role in tumour progression and metastasis of RCC (20,21).
Currently, there is interest in studying the
relationship between VEGI174 and EMT. The aim of the present study
was to assess whether VEGI174 has an effect on EMT in RCC cells and
which functional domains of VEGI174 play an important role in this
process.
Materials and methods
Cell lines
In this study, the human RCC cell lines A498 and
786-O were provided by Sun Yat-Sen University Laboratory
(Guangzhou, China). Cells were cultured with Dulbecco's modified
Eagle's medium (DMEM) or RPMI-1640 medium supplemented with 10%
fetal bovine serum (FBS) (all from HyClone, Logan, UT, USA),
penicillin and streptomycin (Gibco, Grand Island, NY, USA).
Selection of the effective functional
domains of VEGI174
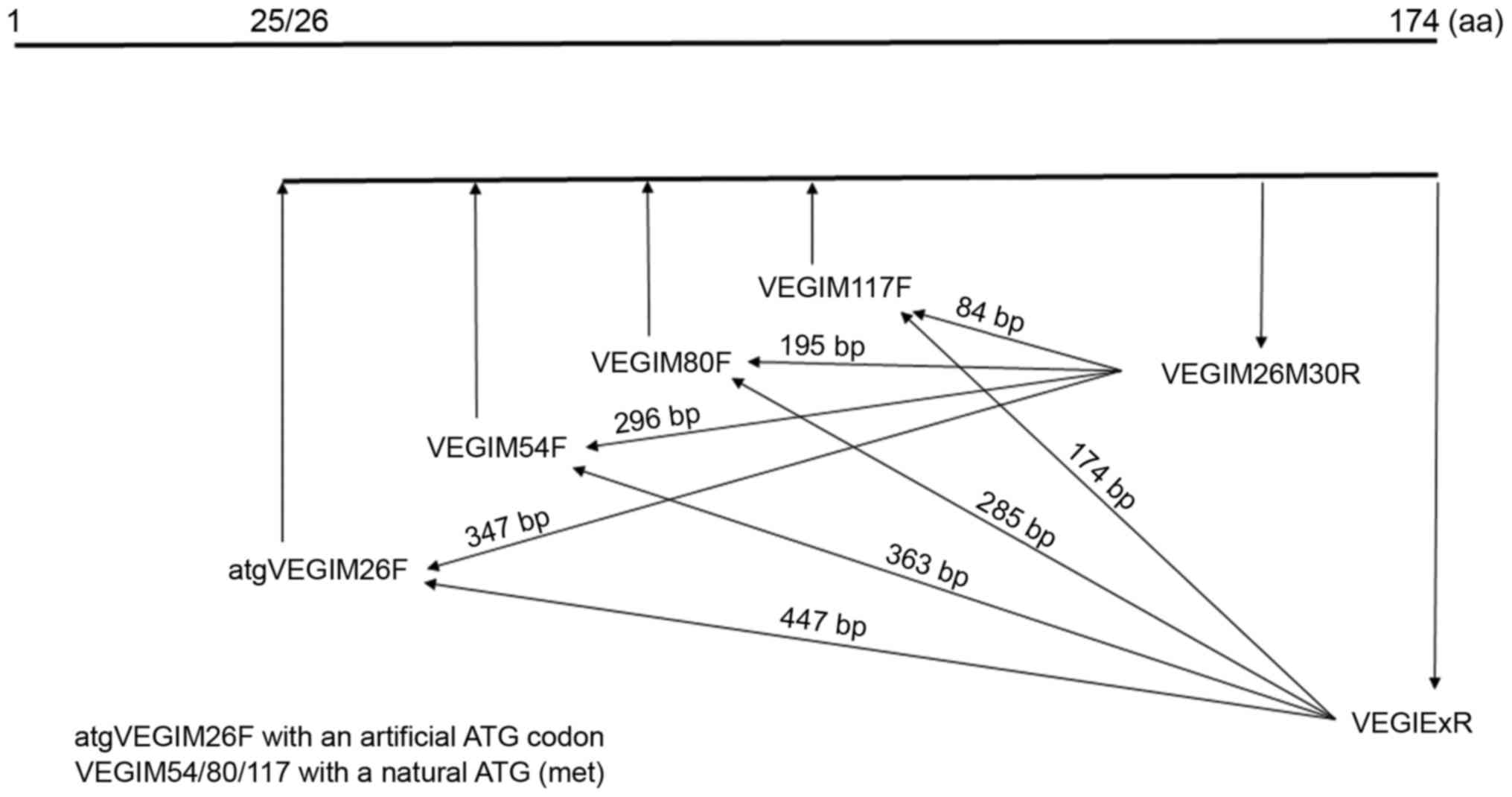
Based on the human VEGI174 sequence (GenBank), we
designed eight different segments of the genes (V1-V8) that encode
VEGI174 extracellular-function domains (Fig. 1). In our preliminary studies,
full-length human VEGI174 and its functional domains (V1–V8) were
separately cloned into mammalian expression plasmid vectors and
then transfected into HUVECs. Our results confirmed that
overexpression of VEGI174 or domains V1 through V8 was able to
inhibit the motility and adhesion of HUVECs to varying degrees. In
particular, the inhibitory effects produced by overexpression of V7
and V8 were more significant than those produced by overexpression
of domains V1 to V6. Therefore, we selected the full-length VEGI174
and domains V7 and V8 as the target genes in this study.
Construction of VEGI174-, V7- and
V8-expressing transgenes
The coding sequences for full-length VEGI174 and for
its functional domains V7 and V8 were cloned into mammalian
expression plasmid vectors (pEF/His TOPO TA; Invitrogen, Inc.,
Paisley, UK). The recombinant plasmid vectors were transformed into
chemically competent TOP10 E. coli (Invitrogen). After
verification and amplification, plasmids containing VEGI174, V7 or
V8 transgenes or empty control plasmids were then transfected into
A498 and 786-O cells using electroporation (Easyjet; EquiBio Ltd.,
Kent, UK). The transfectants were selected with blasticidin and
then applied in experiments (plasmid expression groups:
A498VEGIexp, A498V7exp, A498V8exp,
786-OVEGIexp, 786-OV7exp,
786-OV8exp; empty control plasmid groups:
A498pEF/His, 786-OpEF/His cells). Detailed
experimental methods can be found in our previous studies (14–16). Full primer sequences used in this
study are provided in Table
I.
 | Table IPrimers of the relevant genes used in
the present study. |
Table I
Primers of the relevant genes used in
the present study.
| Primer | Forward | Reverse |
|---|
| VEGI
(expression) |
5′-ATGAGACGCTTTTTAAGCAA-3′ |
5′-CTATAGTAAGGCTCCAAAG-3′ |
| V7
(expression) |
5′-ATGACCTCTGAGTGCAGTGA-3′ |
5′-ATTAGCTTGTGGGGTTCTTGCAAG-3′ |
| V8
(expression) |
5′-ATGGGGACCAAGTCTGTA-3′ |
5′-ATTAGCTTGTCCCCTTCTTGCAAG-3′ |
| E-cadherin |
5′-TGCCCAGAAAATGAAAAAGG-3′ |
5′-GTGTATGTGGCAATGCGTTC-3′ |
| Vimentin |
5′-GAGAACTTTGCCGTTGAAGC-3′ |
5′-GCTTCCTGTAGGTGGCAATC-3′ |
| β-catenin |
5′-TGGATGGGCTGCCTCCAGGTGAC-3′ |
5′-ACCAGCCCACCCCTCGAGCCC-3′ |
| Slug |
5′-CATGCCTGTCATACCACAAC-3′ |
5′-GGTGTCAGATGGAGGAGGG-3′ |
| GAPDH
(reference) |
5′-AGAAGGCTGGGGCTCATTTG-3′ |
5′-AGGGGCCATCCACAGTCTTC-3′ |
Cell growth assay
A total of 5,000 cells were seeded into each well of
a 96-well plate and cultured in a humidified incubator at 37°C and
5% CO2. During cell culture, cyto-activity was tested
with a Cell Counting kit-8 (Dojindo, Kumamoto, Japan) at 24, 48 and
72 h.
Quantitative polymerase chain reaction
(qPCR)
After the transfectants were cultured for 36 h, RNA
was extracted using total RNA isolation reagent (ABgene, Epsom,
UK). The first cDNA strand was synthesised with HiScript II reverse
transcriptase (Vanzyme, Nanjing, China). qPCR was performed using
an iCycler iQ5 system (Bio-Rad, Hemel Hemstead, UK) to determine
the expression level of EMT-related markers (E-cadherin, vimentin,
β-catenin and Slug) in the RCC cell lines. Glyceraldehyde
3-phosphate dehydrogenase (GAPDH) served as the housekeeping gene.
Full primer sequences are provided in Table I.
Western blot analysis of the expression
of EMT markers
Protein quantification was determined using a
bicinchoninic acid colorimetric assay (Pierce BCA Protein assay
kit; Thermo Fisher Scientific, Waltham, MA, USA). Proteins
undergoing sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE) were transferred to PVDF membranes
(Invitrogen) by electroblotting and probed with the primary
antibodies E-cadherin (1:1,000, 5296s; Cell Signaling Technology,
Danvers, MA, USA), vimentin (1:1,000, ab187380), β-catenin
(1:1,000, ab32572), Slug (1:1,000, ab51772) (all from Abcam,
Cambridge, MA, USA) and secondary anti-rabbit IgG, HRP-linked
antibody (1:2,000, #7074; Cell Signaling Technology). Monoclonal
mouse anti-human α-tubulin (1:1,000, #2125; Cell Signaling
Technology) was used as an internal reference. The electrophoretic
bands were analysed by an enhanced chemiluminescence system
(Bio-Rad) and ImageJ software (NIH, Bethesda, MD, USA).
Statistical analysis
Statistical analyses were performed with SPSS 19.0
software (IBM Corp., Armonk, NY, USA). Statistical significance was
determined by ANOVA and LSD multiple comparison t-test. A value of
P<0.05 was considered to indicate a statistically significant
difference.
Results
Manipulation of VEGI174, V7 and V8
overexpression has no impact on cell growth
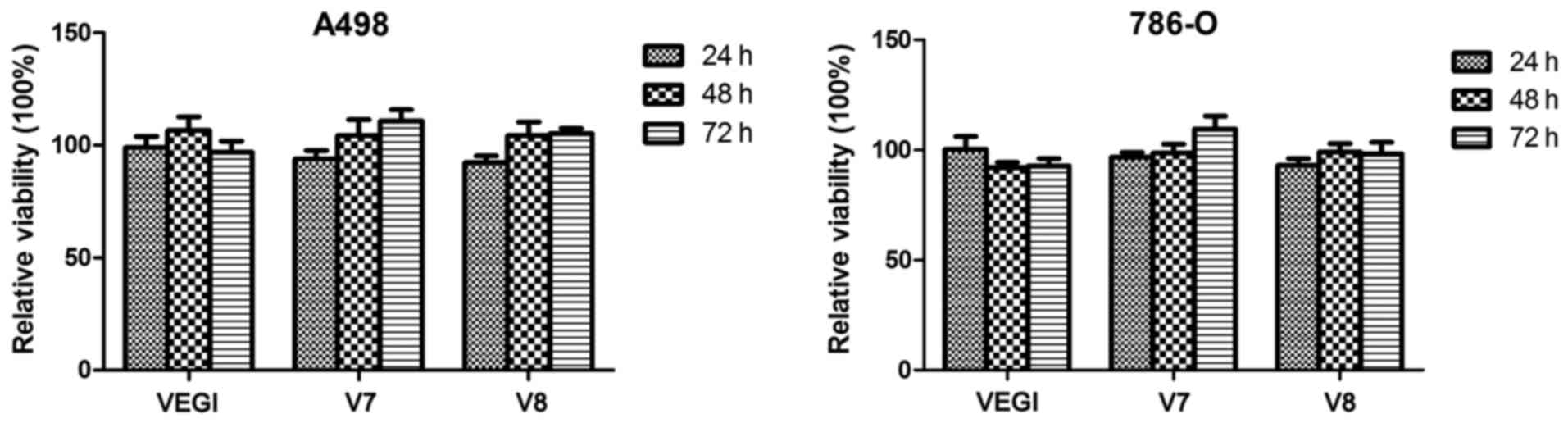
We examined the effect of the overexpression of
VEGI174, V7 and V8 on the viability of A498 and 786-O cell lines.
The cell activity of the empty plasmid control group was set as the
baseline. The relative cell viability between
A498VEGIexp, A498V7exp, A498V8exp,
786-OVEGIexp, 786-OV7exp and
786-OV8exp cells and empty plasmid control cells was not
significantly different (P>0.05) (Fig. 2).
The influence of VEGI174, V7 and V8
overexpression on EMT markers at the genetic level
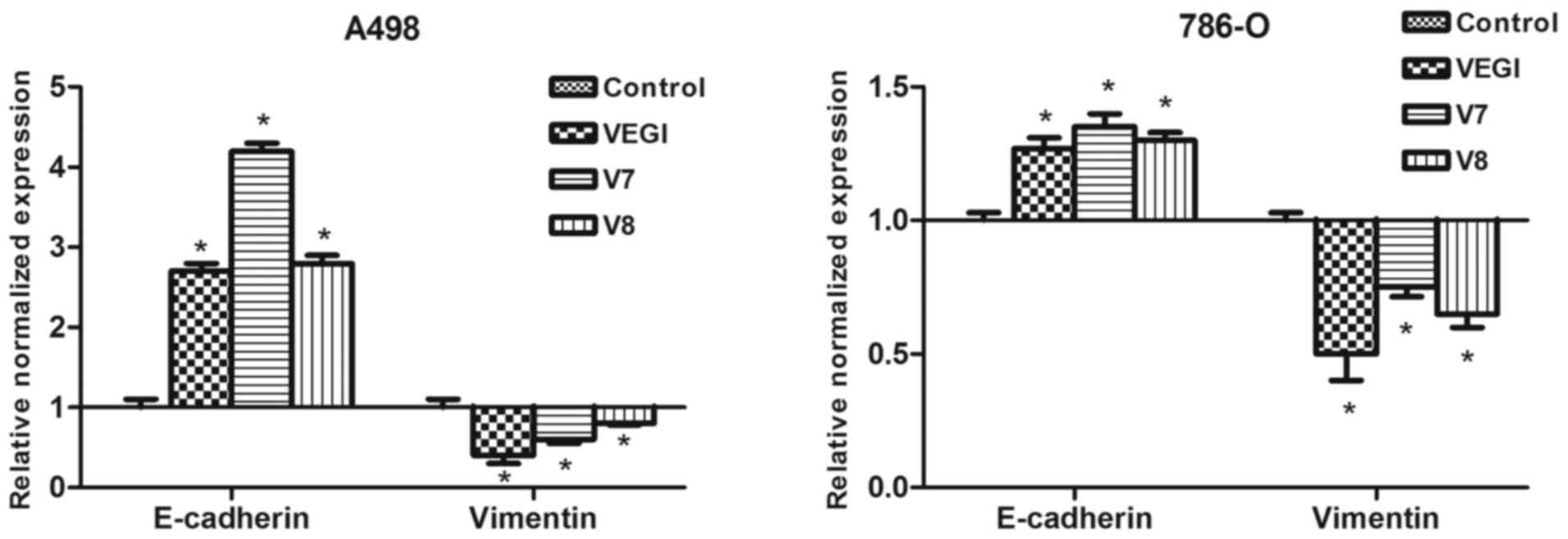
A498 and 786-O cells were transfected with VEGI174,
V7 or V8 recombinant plasmids to create sublines expressing
enhanced or suppressed levels of EMT-related markers. According to
the qPCR results, mRNA expression of E-cadherin was markedly
increased, while that of vimentin was decreased in the
A498VEGIexp, A498V7exp, A498V8exp,
786-OVEGIexp, 786-OV7exp and
786-OV8exp cells compared with these levels in the empty
plasmid controls (P<0.05) (Fig.
3). Furthermore, mRNA expression of E-cadherin was found to be
higher in the A498V7exp cells than that in the
A498VEGIexp and A498V8exp cells (P<0.01).
This result indicated that the bioactivity of the V7 functional
domain was higher in regard to regulating the expression of
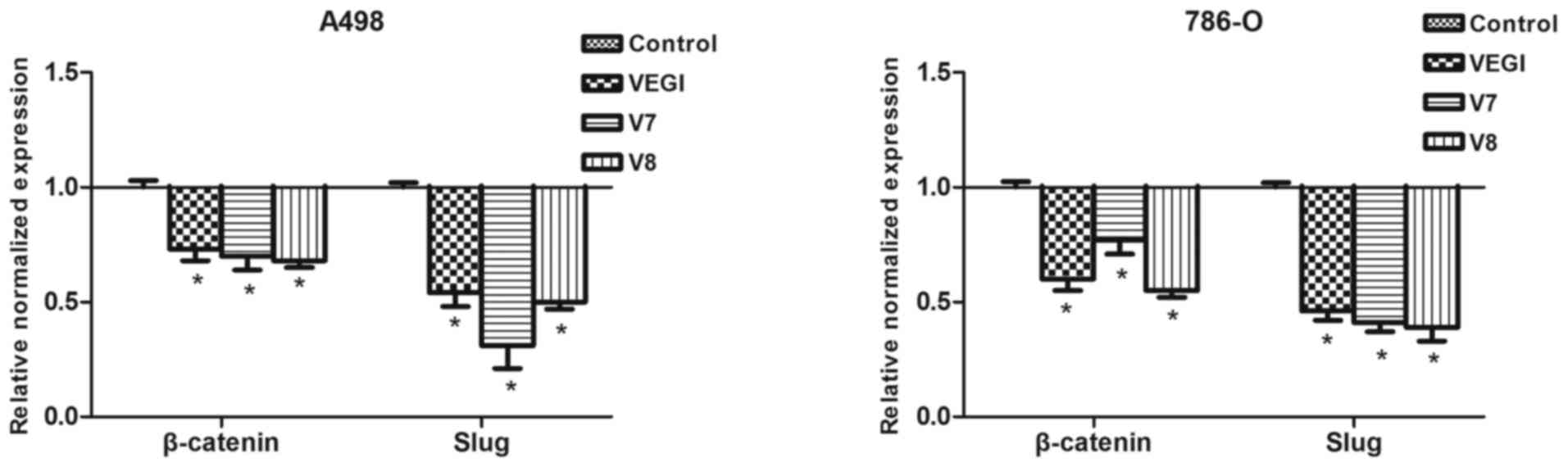
E-cadherin. β-catenin and Slug mRNA expression was downregulated in
the A498VEGIexp, A498V7exp,
A498V8exp, 786-OVEGIexp,
786-OV7exp and 786-OV8exp cells compared with
the corresponding controls (P<0.05) (Fig. 4). However, there were no
significant differences in β-catenin and Slug mRNA expression among
the A498 or 786-O cells that overexpressed VEGI174, V7 or V8
(P>0.05).
Effect of VEGI174, V7 and V8
overexpression on EMT markers at the protein level
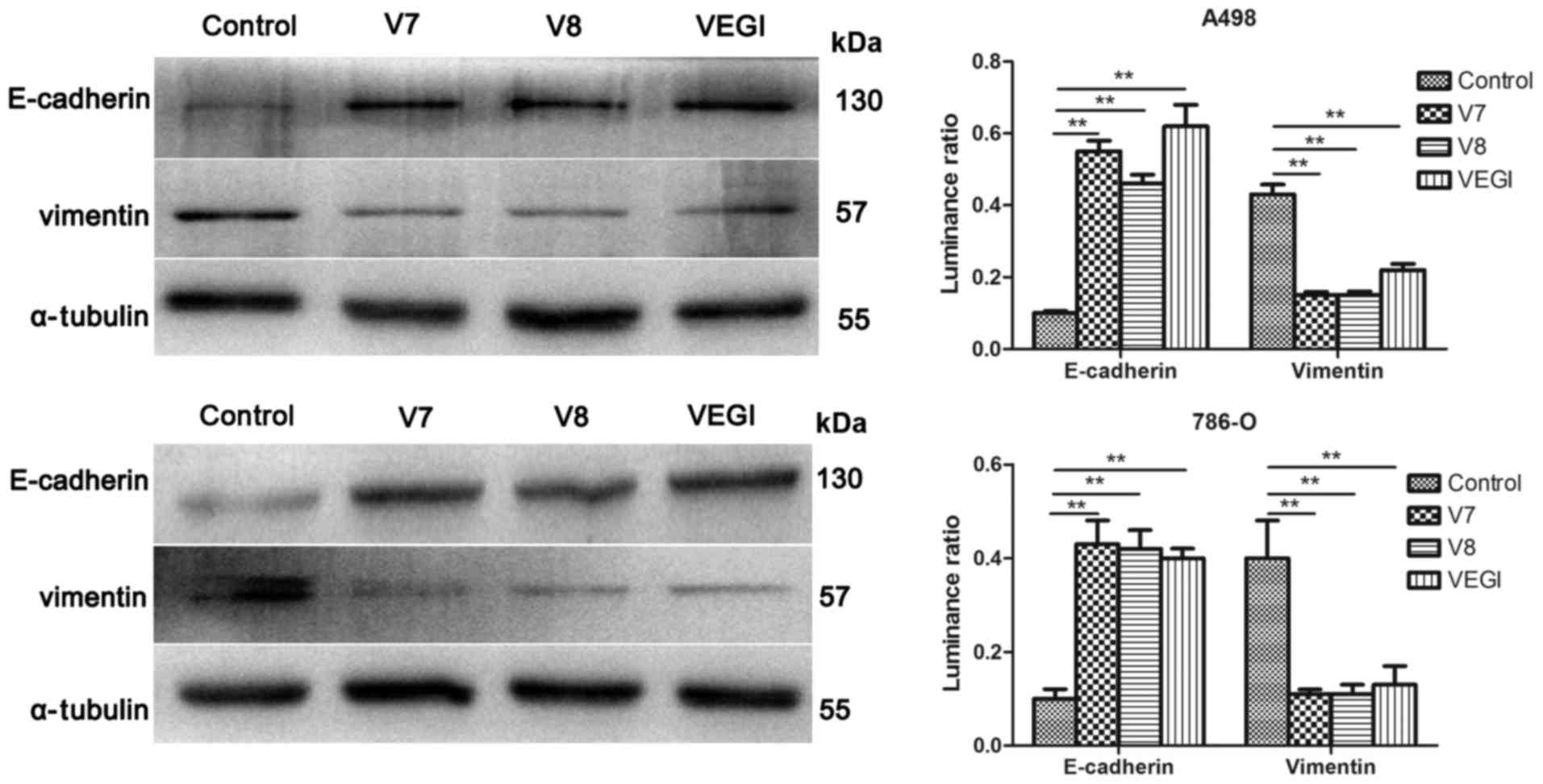
Compared with the empty plasmid controls, a
significant increase in E-cadherin and a decrease in vimentin were
noted in the A498VEGIexp, A498V7exp,
A498V8exp, 786-OVEGIexp,
786-OV7exp and 786-OV8exp cells as determined
by western blot analysis (P<0.01) (Fig. 5). The protein expression of
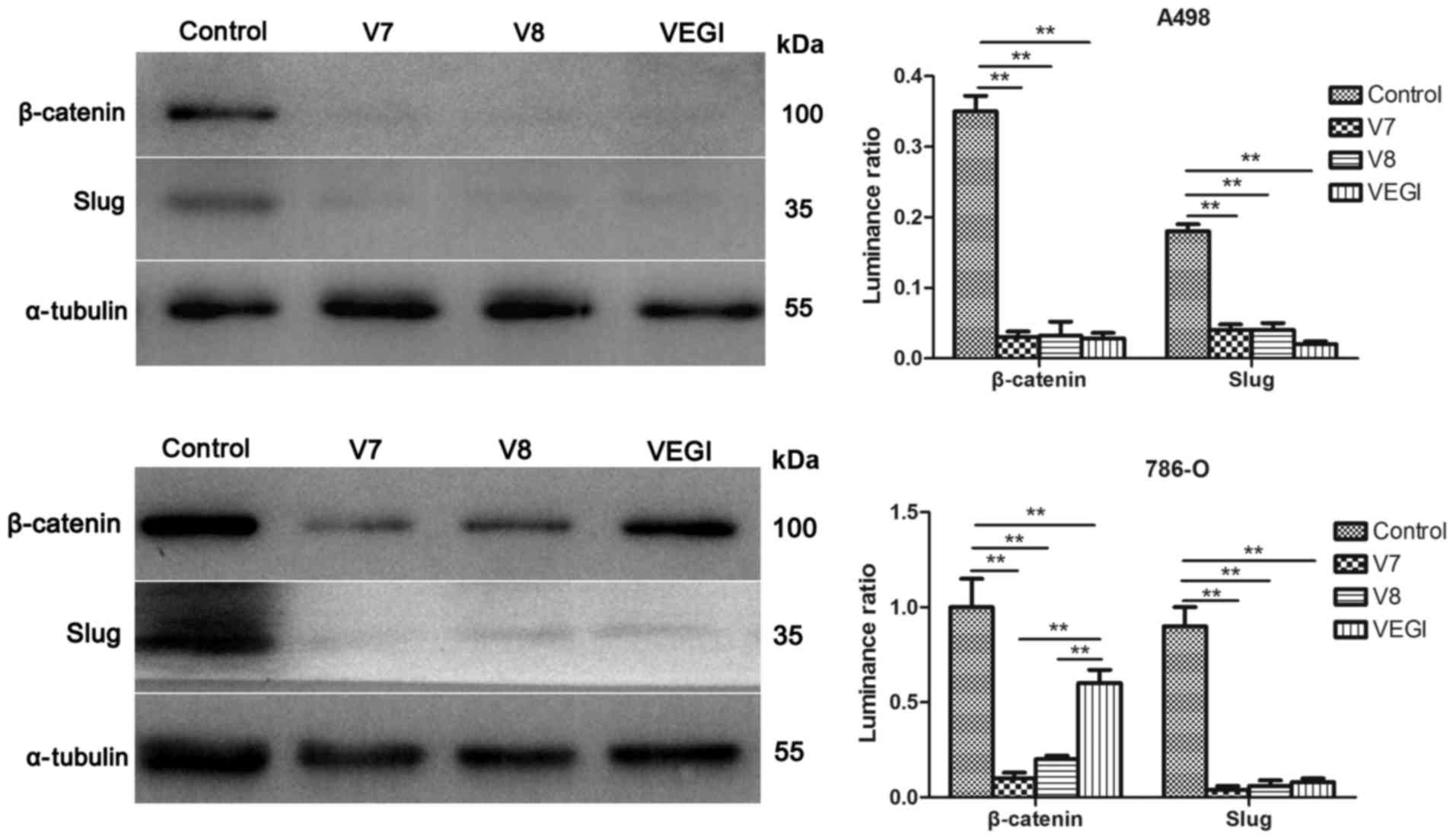
β-catenin and Slug declined in the A498VEGIexp,
A498V7exp, A498V8exp,
786-OVEGIexp, 786-OV7exp and
786-OV8exp cells compared with these levels in the
corresponding controls (P<0.01) (Fig. 6). However, differences in the
effect of VEGI174, V7 and V8 overexpression on EMT markers in A498
or 786-O cells were not significant (P>0.05).
Discussion
VEGI, an endogenous inhibitor of endothelial cell
proliferation, was first identified in human umbilical vein
endothelial cells. VEGI is widely expressed in endothelial cells.
Many studies have investigated the roles of VEGI in human cancers.
This inhibitor is a promising candidate for cancer therapy. Parr
et al (13) reported that
VEGI is aberrantly expressed in breast cancer and displays
prognostic relevance. Patients with breast tumours expressing
reduced levels of VEGI had a poorer prognosis than those patients
expressing high levels of VEGI. Chew et al (8) reported that overexpression of a
secretable VEGI fusion protein abrogated xenograft tumour
progression by reducing the tumour growth rate and microvessel
density. VEGI also inhibited the growth of other human tumour cell
lines, for instance, breast carcinoma (MCF-7), epithelial (HeLa)
and myeloid (U-937 and ML-1a) tumour cells (22,23). Our previous studies showed that
overexpression of VEGI inhibited cell motility in vitro and
vascular endothelial tube formation and tumour growth in
vivo (14–16).
The EMT process is linked to its role in tumour
invasion and metastasis. EMT has been established in multiple
cancer types, such as digestive tract, pancreas, liver, prostate
and breast cancers (24–30). Zhang et al (31) demonstrated that hypoxia was able
to induce EMT and enhance the invasion and migration ability of HCC
cells. Chu et al (32)
showed that EMT was closely related to in vitro cell
migration and invasion in breast cancer. Therefore, further studies
are needed to investigate the relationships between EMT development
and cancer. These studies will offer insights into the oncogenic
EMT pathways of tumour metastasis and provide potential therapeutic
targets. Currently, the relationship between VEGI and EMT is not
clear. In this study, we studied the effects of VEGI174 and its
functional domains (V7 and V8) on EMT in RCC cells in
vitro.
Our results confirmed that overexpression of
VEGI174, V7 or V8 inhibited EMT in A498 and 786-O cell lines. The
mRNA level of E-cadherin was significantly upregulated, while
vimentin was downregulated in the A498VEGIexp,
A498V7exp, A498V8exp,
786-OVEGIexp, 786-OV7exp and
786-OV8exp cells compared with that noted in the empty
plasmid controls (P<0.05). Elevated protein expression of
E-cadherin and decreased expression of vimentin were also observed
in these cells compared with the corresponding controls
(P<0.01). Thus, changes in the level of protein expression were
consistent with the changes in mRNA expression. E-cadherin belongs
to a family of calcium-dependent transmembrane glycoproteins. This
glycoprotein is an intercellular adhesion molecule that enhances
connections between cells and maintains the stability of the
cytoskeleton. Many signalling pathways promote EMT by suppressing
the expression of E-cadherin (33,34). Importantly, vimentin, the
mesenchymal intermediate filament and a hallmark of EMT, is
overexpressed in malignant epithelial cancers and correlates with a
poor prognosis (35). Thus,
vimentin provides a further link between EMT and malignancy.
Abundant evidence indicates that vimentin regulates mesenchymal
cell shape and mammary epithelial cell migration and that it plays
a role in regulating signal transduction, which is necessary for
EMT induction (36–38).
Moreover, we also detected the expression of
EMT-related regulatory factors (β-catenin and Slug). β-catenin is a
key mediator in the Wnt/β-catenin signalling pathway. β-catenin
translocates into the cell nucleus and promotes downstream pathways
by binding to target genes (39,40). Studies have shown that
Wnt/β-catenin signalling is associated with EMT (41–43). Slug, a member of the Snail family,
is known to play diverse roles in the cell. Slug a transcriptional
repressor, and its deregulation has been observed in a variety of
cancers. Slug-mediated regulation of EMT is often associated with
its ability to transcriptionally repress the expression of
E-cadherin (44–47), and it has been shown to promote
cancer cell invasion, migration and metastasis. In this study,
compared with empty plasmid controls, β-catenin and Slug mRNA and
protein expression levels were downregulated in the A498 and 786-O
cells that overexpressed VEGI174, V7 or V8. These two proteins may
be key regulatory factors in the mechanisms involved in suppression
of the EMT process by VEGI174, V7 or V8 overexpression.
In conclusion, the present study showed that
overexpression of VEGI174, V7 or V8 was able to inhibit the EMT
process in the A498 and 786-O cell lines. This finding indicates
that VEGI may be a potential tumour suppressor and target for RCC
therapy. Furthermore, V7 and V8 are two effective functional
domains of VEGI174, which can be considered for synthesis as
peptides and further studied for the treatment of RCC.
Acknowledgments
This study was supported by the 2014 Beijing Natural
Science Foundation (grant no. 7142059).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu L, Zhang W, Qi X, Li H, Yu J, Wei S,
Hao X and Ren X: Randomized study of autologous cytokine-induced
killer cell immunotherapy in metastatic renal carcinoma. Clin
Cancer Res. 18:1751–1759. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bianconi M, Faloppi L, Loretelli C, Zizzi
A, Giampieri R, Bittoni A, Andrikou K, Del Prete M, Burattini L,
Montironi R, et al: Angiogenesis genotyping in the selection of
first-line treatment with either sunitinib or pazopanib for
advanced renal cell carcinoma. Oncotarget. 7:37599–37607.
2016.PubMed/NCBI
|
|
4
|
Buchler T, Bortlicek Z, Poprach A, Pavlik
T, Veskrnova V, Honzirkova M, Zemanova M, Fiala O, Kubackova K,
Slaby O, et al: Outcomes for patients with metastatic renal cell
carcinoma achieving a complete response on targeted therapy: A
Registry-based Analysis. Eur Urol. 70:469–475. 2015. View Article : Google Scholar
|
|
5
|
Fishman MN, Tomshine J, Fulp WJ and
Foreman PK: A systematic review of the efficacy and safety
experience reported for sorafenib in advanced renal cell carcinoma
(RCC) in the post-approval setting. PLoS One. 10:e01208772015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tan KB, Harrop J, Reddy M, Young P,
Terrett J, Emery J, Moore G and Truneh A: Characterization of a
novel TNF-like ligand and recently described TNF ligand and TNF
receptor superfamily genes and their constitutive and inducible
expression in hemato-poietic and non-hematopoietic cells. Gene.
204:35–46. 1997. View Article : Google Scholar
|
|
7
|
Zhai Y, Yu J, Iruela-Arispe L, Huang WQ,
Wang Z, Hayes AJ, Lu J, Jiang G, Rojas L, Lippman ME, et al:
Inhibition of angiogenesis and breast cancer xenograft tumor growth
by VEGI, a novel cytokine of the TNF superfamily. Int J Cancer.
82:131–136. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chew LJ, Pan H, Yu J, Tian S, Huang WQ,
Zhang JY, Pang S and Li LY: A novel secreted splice variant of
vascular endothelial cell growth inhibitor. FASEB J. 16:742–744.
2002.PubMed/NCBI
|
|
9
|
Zhang N, Sanders AJ, Ye L and Jiang WG:
Vascular endothelial growth inhibitor in human cancer (Review). Int
J Mol Med. 24:3–8. 2009.PubMed/NCBI
|
|
10
|
Zhai Y, Ni J, Jiang GW, Lu J, Xing L,
Lincoln C, Carter KC, Janat F, Kozak D, Xu S, et al: VEGI, a novel
cytokine of the tumor necrosis factor family, is an angiogenesis
inhibitor that suppresses the growth of colon carcinomas in vivo.
FASEB J. 13:181–189. 1999.PubMed/NCBI
|
|
11
|
Liang PH, Tian F, Lu Y, Duan B, Stolz DB
and Li LY: Vascular endothelial growth inhibitor (VEGI; TNFSF15)
inhibits bone marrow-derived endothelial progenitor cell
incorporation into Lewis lung carcinoma tumors. Angiogenesis.
14:61–68. 2011. View Article : Google Scholar :
|
|
12
|
Yamanegi K, Kawabe M, Futani H, Nishiura
H, Yamada N, Kato-Kogoe N, Kishimoto H, Yoshiya S and Nakasho K:
Sodium valproate, a histone deacetylase inhibitor, modulates the
vascular endothelial growth inhibitor-mediated cell death in human
osteosarcoma and vascular endothelial cells. Int J Oncol.
46:1994–2002. 2015.PubMed/NCBI
|
|
13
|
Parr C, Gan CH, Watkins G and Jiang WG:
Reduced vascular endothelial growth inhibitor (VEGI) expression is
associated with poor prognosis in breast cancer patients.
Angiogenesis. 9:73–81. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang N, Sanders AJ, Ye L, Kynaston HG and
Jiang WG: Vascular endothelial growth inhibitor, expression in
human prostate cancer tissue and the impact on adhesion and
migration of prostate cancer cells in vitro. Int J Oncol.
35:1473–1480. 2009.PubMed/NCBI
|
|
15
|
Zhang N, Sanders AJ, Ye L, Kynaston HG and
Jiang WG: Expression of vascular endothelial growth inhibitor
(VEGI) in human urothelial cancer of the bladder and its effects on
the adhesion and migration of bladder cancer cells in vitro.
Anticancer Res. 30:87–95. 2010.PubMed/NCBI
|
|
16
|
Zhang N, Wu P, Shayiremu D, Wu L, Shan H,
Ye L, Zhao X, Cai J, Jiang WG, Gong K, et al: Suppression of renal
cell carcinoma growth in vivo by forced expression of vascular
endothelial growth inhibitor. Int J Oncol. 42:1664–1673.
2013.PubMed/NCBI
|
|
17
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wong IY, Javaid S, Wong EA, Perk S, Haber
DA, Toner M and Irimia D: Collective and individual migration
following the epithelial-mesenchymal transition. Nat Mater.
13:1063–1071. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Saad S, Stanners SR, Yong R, Tang O and
Pollock CA: Notch mediated epithelial to mesenchymal transformation
is associated with increased expression of the Snail transcription
factor. Int J Biochem Cell Biol. 42:1115–1122. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Piva F, Giulietti M, Santoni M, Occhipinti
G, Scarpelli M, Lopez-Beltran A, Cheng L, Principato G and
Montironi R: Epithelial to mesenchymal transition in renal cell
carcinoma: Implications for cancer therapy. Mol Diagn Ther.
20:111–117. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xiao Q, Hsu CY, Chen H, Ma X, Xu J and Lee
JM: Characterization of cis-regulatory elements of the vascular
endothelial growth inhibitor gene promoter. Biochem J. 388:913–920.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Haridas V, Shrivastava A, Su J, Yu GL, Ni
J, Liu D, Chen SF, Ni Y, Ruben SM, Gentz R, et al: VEGI, a new
member of the TNF family activates nuclear factor-kappa B and c-Jun
N-terminal kinase and modulates cell growth. Oncogene.
18:6496–6504. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ho QT and Kuo CJ: Vascular endothelial
growth factor: Biology and therapeutic applications. Int J Biochem
Cell Biol. 39:1349–1357. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Byles V, Zhu L, Lovaas JD, Chmilewski LK,
Wang J, Faller DV and Dai Y: SIRT1 induces EMT by cooperating with
EMT transcription factors and enhances prostate cancer cell
migration and metastasis. Oncogene. 31:4619–4629. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen J, Imanaka N, Chen J and Griffin JD:
Hypoxia potentiates Notch signaling in breast cancer leading to
decreased E-cadherin expression and increased cell migration and
invasion. Br J Cancer. 102:351–360. 2010. View Article : Google Scholar :
|
|
27
|
Sato F, Kubota Y, Natsuizaka M, Maehara O,
Hatanaka Y, Marukawa K, Terashita K, Suda G, Ohnishi S, Shimizu Y,
et al: EGFR inhibitors prevent induction of cancer stem-like cells
in esophageal squamous cell carcinoma by suppressing
epithelial-mesenchymal transition. Cancer Biol Ther. 16:933–940.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jiao L, Li DD, Yang CL, Peng RQ, Guo YQ,
Zhang XS and Zhu XF: Reactive oxygen species mediate
oxaliplatin-induced epithelial-mesenchymal transition and invasive
potential in colon cancer. Tumour Biol. 37:8413–8423. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liang B, Zheng W, Fang L, Wu L, Zhou F,
Yin X, Yu X and Zou Z: Overexpressed targeting protein for Xklp2
(TPX2) serves as a promising prognostic marker and therapeutic
target for gastric cancer. Cancer Biol Ther. 17:824–832. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Krantz SB, Shields MA, Dangi-Garimella S,
Munshi HG and Bentrem DJ: Contribution of epithelial-to-mesenchymal
transition and cancer stem cells to pancreatic cancer progression.
J Surg Res. 173:105–112. 2012. View Article : Google Scholar :
|
|
31
|
Zhang L, Huang G, Li X, Zhang Y, Jiang Y,
Shen J, Liu J, Wang Q, Zhu J, Feng X, et al: Hypoxia induces
epithelial-mesenchymal transition via activation of SNAI1 by
hypoxia-inducible factor-1α in hepatocellular carcinoma. BMC
Cancer. 13:1082013. View Article : Google Scholar
|
|
32
|
Chu CY, Jin YT, Zhang W, Yu J, Yang HP,
Wang HY, Zhang ZJ, Liu XP and Zou Q: CA IX is upregulated in
CoCl2-induced hypoxia and associated with cell invasive
potential and a poor prognosis of breast cancer. Int J Oncol.
48:271–280. 2016.
|
|
33
|
Lombaerts M, van Wezel T, Philippo K,
Dierssen JW, Zimmerman RM, Oosting J, van Eijk R, Eilers PH, van de
Water B, Cornelisse CJ, et al: E-cadherin transcriptional
downregulation by promoter methylation but not mutation is related
to epithelial-to-mesenchymal transition in breast cancer cell
lines. Br J Cancer. 94:661–671. 2006.PubMed/NCBI
|
|
34
|
Natalwala A, Spychal R and Tselepis C:
Epithelial-mesenchymal transition mediated tumourigenesis in the
gastrointestinal tract. World J Gastroenterol. 14:3792–3797. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Satelli A and Li S: Vimentin in cancer and
its potential as a molecular target for cancer therapy. Cell Mol
Life Sci. 68:3033–3046. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gilles C, Polette M, Zahm JM, Tournier JM,
Volders L, Foidart JM and Birembaut P: Vimentin contributes to
human mammary epithelial cell migration. J Cell Sci. 112:4615–4625.
1999.PubMed/NCBI
|
|
37
|
Mendez MG, Kojima S and Goldman RD:
Vimentin induces changes in cell shape, motility, and adhesion
during the epithelial to mesenchymal transition. FASEB J.
24:1838–1851. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vuoriluoto K, Haugen H, Kiviluoto S,
Mpindi JP, Nevo J, Gjerdrum C, Tiron C, Lorens JB and Ivaska J:
Vimentin regulates EMT induction by Slug and oncogenic H-Ras and
migration by governing Axl expression in breast cancer. Oncogene.
30:1436–1448. 2011. View Article : Google Scholar
|
|
39
|
Polakis P: Wnt signaling and cancer. Genes
Dev. 14:1837–1851. 2000.PubMed/NCBI
|
|
40
|
Giles RH, van Es JH and Clevers H: Caught
up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta.
1653:1–24. 2003.PubMed/NCBI
|
|
41
|
Kwon YJ, Baek HS, Ye DJ, Shin S, Kim D and
Chun YJ: CYP1B1 enhances cell proliferation and metastasis through
induction of EMT and activation of Wnt/β-catenin signaling via Sp1
upregulation. PLoS One. 11:e01515982016. View Article : Google Scholar
|
|
42
|
Lee SC, Kim OH, Lee SK and Kim SJ: IWR-1
inhibits epithelial-mesenchymal transition of colorectal cancer
cells through suppressing Wnt/β-catenin signaling as well as
survivin expression. Oncotarget. 6:27146–27159. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ai R, Sun Y, Guo Z, Wei W, Zhou L, Liu F,
Hendricks DT, Xu Y and Zhao X: NDRG1 overexpression promotes the
progression of esophageal squamous cell carcinoma through
modulating Wnt signaling pathway. Cancer Biol Ther. 17:943–954.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bolós V, Peinado H, Pérez-Moreno MA, Fraga
MF, Esteller M and Cano A: The transcription factor Slug represses
E-cadherin expression and induces epithelial to mesenchymal
transitions: A comparison with Snail and E47 repressors. J Cell
Sci. 116:499–511. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Storci G, Sansone P, Trere D, Tavolari S,
Taffurelli M, Ceccarelli C, Guarnieri T, Paterini P, Pariali M,
Montanaro L, et al: The basal-like breast carcinoma phenotype is
regulated by SLUG gene expression. J Pathol. 214:25–37. 2008.
View Article : Google Scholar
|
|
46
|
Hajra KM, Chen DY and Fearon ER: The SLUG
zinc-finger protein represses E-cadherin in breast cancer. Cancer
Res. 62:1613–1618. 2002.PubMed/NCBI
|
|
47
|
Martin TA, Goyal A, Watkins G and Jiang
WG: Expression of the transcription factors snail, slug, and twist
and their clinical significance in human breast cancer. Ann Surg
Oncol. 12:488–496. 2005. View Article : Google Scholar : PubMed/NCBI
|