1. Introduction
Cell death is an essential process in the
physiology, pathology, growth, development, senility and death of
an organism. There are two major modes of cell death: Apoptosis and
necrosis, whose molecular mechanisms have been extensively studied
(1). In the last few years, other
forms of cell death have been identified and reported, including
autophagy and necroptosis (2).
Necroptosis is another form of cell death which was recently
discovered; it not only has some similar phenotypes with necrosis,
but can also be regulated by a series of exact mechanisms. It can
be activated by stimulation with ligands of death receptors (DRs),
such as TNF-α (3). However, the
few studies on necroptosis in cerebrovascular diseases, such as
stroke, are insufficient to form a comprehensive understanding of
the topic. The present review mainly expounds the features,
molecular mechanism [focusing on the tumor necrosis factor α-tumor
necrosis factor receptor 1 (TNFα-TNFR1) signaling pathway] and
identification of necroptosis, and the latest study of this type of
cell death in stroke.
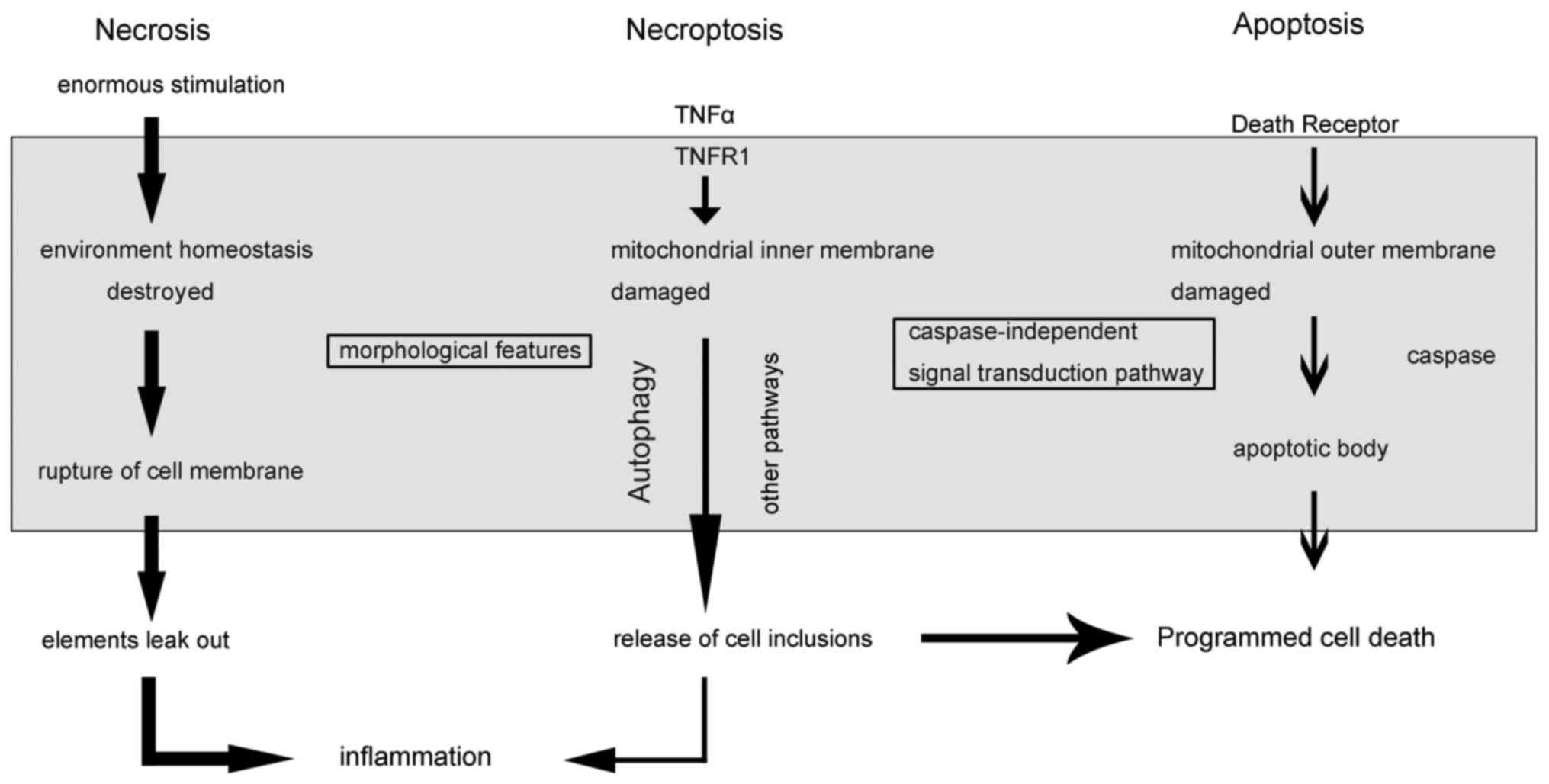
2. Cell death
Cell death is an irreversible biological phenomenon
and a termination of life. The death of nerve cells has different
forms and features. Cell death was previously divided into necrosis
and apoptosis. Necrosis can be recognized early and defined through
pathomorphic characteristics, which occur when cells or the body is
faced with enormous external pressure, and is a passive process.
Meanwhile, apoptosis is regulated cell death, also known as
programmed cell death (PCD) (4).
A recent study showed, however, that necrosis can also be regulated
in a programmed manner (3).
Cells undergo a type of passive death when organisms
face up to enormous external pressure, and this is defined as
necrosis. Necrosis was previously defined by its pathomorphic
characteristics. Several causes, including physiological trauma,
can lead to a change of internal environmental osmotic pressure,
destroying the balance of an organism's internal environmental
homeostasis and leading to necrosis (1). Furthermore, following necrosis, a
series of reactions occur, including loss of cell membrane
integrity, cell membrane swelling (due to absorption of moisture)
and mitochondrial dysfunction (5). Due to the rupture of the cell
membrane, a large number of intracellular elements leak out, which
can cause and aggravate inflammation of the surrounding tissues
(6).
Apoptosis is a natural phenomenon of cell death
regulated by genes that occurs when organisms are affected by
environmental stimulation (7).
Damaged cells or cells without functions can be induced by protease
(caspase) and then start the self-elimination procedure. As
apoptosis occurs, a cell undergoes shrinkage, an increase in
density, chromatin condensation, cracking of the nucleus and the
formation of an apoptotic body (via cell membrane invagination).
Due to the formation of the apoptotic body, the inflammatory
response is avoided after apoptosis (8). In the majority of cases, apoptosis
may be triggered in two ways: i) Intrinsic stimulation through the
mitochondrial signaling pathway; or ii) extrinsic stimulation
through cell surface DRs, including TNFα, TNF-related
apoptosis-inducing ligand (TRAIL) receptors and Fas (CD95/APO1). In
either case, activation of cysteine aspartyl proteases (caspases)
is necessary, and is known as the caspase-dependent manner
(9). However, one previous study
showed that apoptosis can occur even after caspases are inhibited.
This result suggests that PCD can also occur in a
caspase-independent manner (10).
Macroautophagy (referred to herein as autophagy)
involves the engulfment of cytoplasmic material and intracellular
organelles via a self-cannibalization mechanism, leading to the
formation of autophagosomes. This is followed by fusion with
lysosomes to form an autolysosome, where the encapsulated material
is degraded by specific acidic hydrolases (11). In this way, cell would survive in
a nutrient- or growth factor-deficient environment for a longer
period; and there are some autophagy vacuolations formed in the
cytoplasm, but not chromatin condenses (12). Autophagy is observed in
physiological and pathological processes, however, the pros and
cons of its role have not been fully elucidated.
Necroptosis is a form of cell death similar to
necrosis in terms of morphological features (such as early
destruction of membrane integrity, and cell and intracellular
organelle swelling), but it can be regulated in a
caspase-independent manner, that is to say that necroptosis can be
triggered by a combination of death ligands and DRs under the
inhibition of caspase (13).
Additionally, necroptosis can be blocked by a small specific
molecular compound known as necroptosis-specific inhibitor-1
(Nec-1), but not by any of the specific inhibitors of apoptosis or
autophagy. At the same time, Nec-1 is unable to inhibit apoptosis
and autophagy, and does not have any other significant effects on
cells (14). As necroptosis
occurs, the occurrence of autophagy can often be observed, however,
necroptosis is not blocked after the application of inhibitors of
autophagy, suggesting that autophagy is downstream of the
necroptosis pathways (3). The
most evident difference between necroptosis and apoptosis is the
local inflammation caused by the release of the cell contents. The
inflammation in necroptosis mainly presents as a large amount of
inflammatory cell invasion and activation (15). Different forms and features of
cell death, including certain connections and distinctions between
these different forms are revealed in Fig. 1.
Distinguishing features of
necroptosis
Although it has similar morphological
characteristics, necroptosis is different from traditional
necrosis, which is regulated by a series of signal transduction
pathways and is a process of positive consumption of energy.
Necroptosis ensures that cells are not completely passive when
faced with external harm, in order to reduce damage to the cells
via regulating a series of signals (1,2).
Necroptosis has typical morphological features of necrosis
(organelles swelling, collapse, loss of cell membrane integrity and
deficiency of nuclear chromatin), and the process can be identified
through a light/electron microscope with propidium iodide (PI)
staining (3). Due to the release
of cell inclusions, necroptosis causes local inflammation, and
infiltration and activation of inflammatory cells (15). Necroptosis also has the following
characteristics: Cell death with necrotic characterization is more
beneficial to maintain embryonic development and homeostasis during
the adult period; necrotic cell death can be induced by a
combination of ligands with their specific membrane receptors;
necroptosis can be regulated via genetics, pharmacology and other
factors; and inactivation of caspase can translate apoptosis into
complete necrosis or a specific cell death with mixed
characteristics of apoptosis and necrosis (3).
Identification of necroptosis
The basic identification of necroptosis can be
divided into morphological and biochemical methods. Morphological
methods include: Electron microscopy judgment and PI staining
(16). Biochemical methods
include: Use of necroptosis-specific inhibitor-1 (Nec-1), receptor
interacting protein 1 (RIP1)-knockout and receptor interacting
protein 3 (RIP3)-knockout. In addition, the formation of compounds
(RIP1-RIP3), known as necrosomes, and the phosphorylation of RIP1
and RIP3 can serve as complementary judgment points (17,18).
3. Mechanisms of necroptosis and the
TNFα-TNFR1-related signaling pathway
Following TNF signaling or other harmful stimulation
that acts on corresponding cell death receptors, successive changes
in mitochondria and the different forms of cell death occur
(19). The mitochondrial outer
membrane permeability is changed which leads to the release of the
proteins in the mitochondrial intermembrane space, including
cytochrome c. The release of cytochrome c could
activate apoptosis via mitochondrial pathways. Additionally, the
release of apoptosis-inducing factor and endonuclease G is the
start of caspase-independent apoptosis. Furthermore, changes in
mitochondrial inner membrane permeability induce a rapid decline in
mitochondrial membrane potential, destroying respiratory chain
electron transport (4). As a
result, adenosine triphosphate synthesis and cell membranes are
damaged, with necrosis-like cell death occurring (20). In short, damage of the
mitochondrial outer membrane leads to apoptosis and damage of the
mitochondrial inner membrane leads to necroptosis. Therefore,
compared with apoptosis, necroptosis does not involve the key
regulators of apoptosis factors (caspase) and does not involve the
release of mitochondrial cytochrome c.
According to previous studies, there are several
mechanisms of necroptosis, including the TNFα-TNFR1-related
signaling pathway, TRAIL and factors associated with the apoptosis
ligand signaling pathway, the RIP3-mitochondrial-reactive oxygen
species (ROS) metabolic pathway and the zVAD-mediated
PKC-mitogen-activated protein kinase (MAPK)-AP-1-related signaling
pathway (21-23). Among these mechanisms, the
signaling cascade downstream of TNFα leading to necroptosis has
been deeply studied (24-28). In brief, TNFR1 exists in the cell
membrane and its subunits can spontaneously trimerize at the plasma
membrane (24). As ligands bind,
the trimer conformation of these receptors would be changed,
allowing their cytosolic tails to recruit multiple proteins, and
then a complex (complex I) would develop near the cell membrane,
which can include TNFα receptor-associated death domain (TRADD),
RIP1 kinase, TNFR-associated factor 2 (TRAF2), TRAF5, cellular
inhibitor of apoptosis 1 (cIAP1) and cIAP2. Ubiquitylation of RIP1
by cIAP1 and cIAP2 stabilized complex I, and then combined with
transforming growth factor β-activated kinase 1-binding protein 2,
IκB kinase (IKK) and nuclear factor-κB essential modulator (NEMO)
regulatory subunit (25).
Afterwards, IKKα and IKKβ are activated and thus IκB can be
phosphorylated, leading to nuclear factor-κB (NF-κB) release and
MAPK activation. The result of this NF-κB signaling pathway is the
promotion of cell survival. RIP1 polyubiquitylation not only
influences NF-κB activation, but also affects the transition from
complex I to II (26).
Deubiquitylated RIP1 cooperates with its cognate kinase RIP3 for
recruitment to another complex (complex II), which includes TRADD,
FAS-associated protein with a death domain (FADD) and caspase-8. In
this complex, RIP1 and RIP3 are inhibited by caspase-8 and
proteolytic cleavage is induced; therefore, the pro-apoptotic
caspase activation cascade starts. By contrast, when caspase-8 is
inactivated, deficient or deleted, complex II does not initiate the
apoptotic program and the TNFα-mediated necroptotic pathway is used
(27,28). As a consequence, caspase-8 and the
interaction between RIP1 and RIP3 expression could be recognized as
the diverging point of apoptosis and necroptosis (29). Nec-1 could halt the necroptosis
signaling pathways by inhibiting RIPI and RIP3 kinase activity
(14,15). In addition, the formation of
FADD-RIP1-RIP3-NEMO complex has an essential role on TNFα-mediated
necroptotic pathways (30). A
previous study identified the influence of the interaction of mixed
lineage kinase domain-like protein (MLKL) and RIP3 (28). Later studies further supported the
hypothesis that MLKL is indeed required for TNFα-induced
necroptosis, as key residues of MLKL mutants cannot be
phosphorylated, which prevents the activation of the necrosome
(31). A controversial point is
whether the internal flow of Ca2+ or Na+ ions
is caused by MLKL-induced increased membrane permeability or by the
formation of non-specific membrane channels. A summary of the
TNFα-TNFR1-related signaling pathway of cell survival, apoptosis
and necroptosis is presented in Fig.
2.
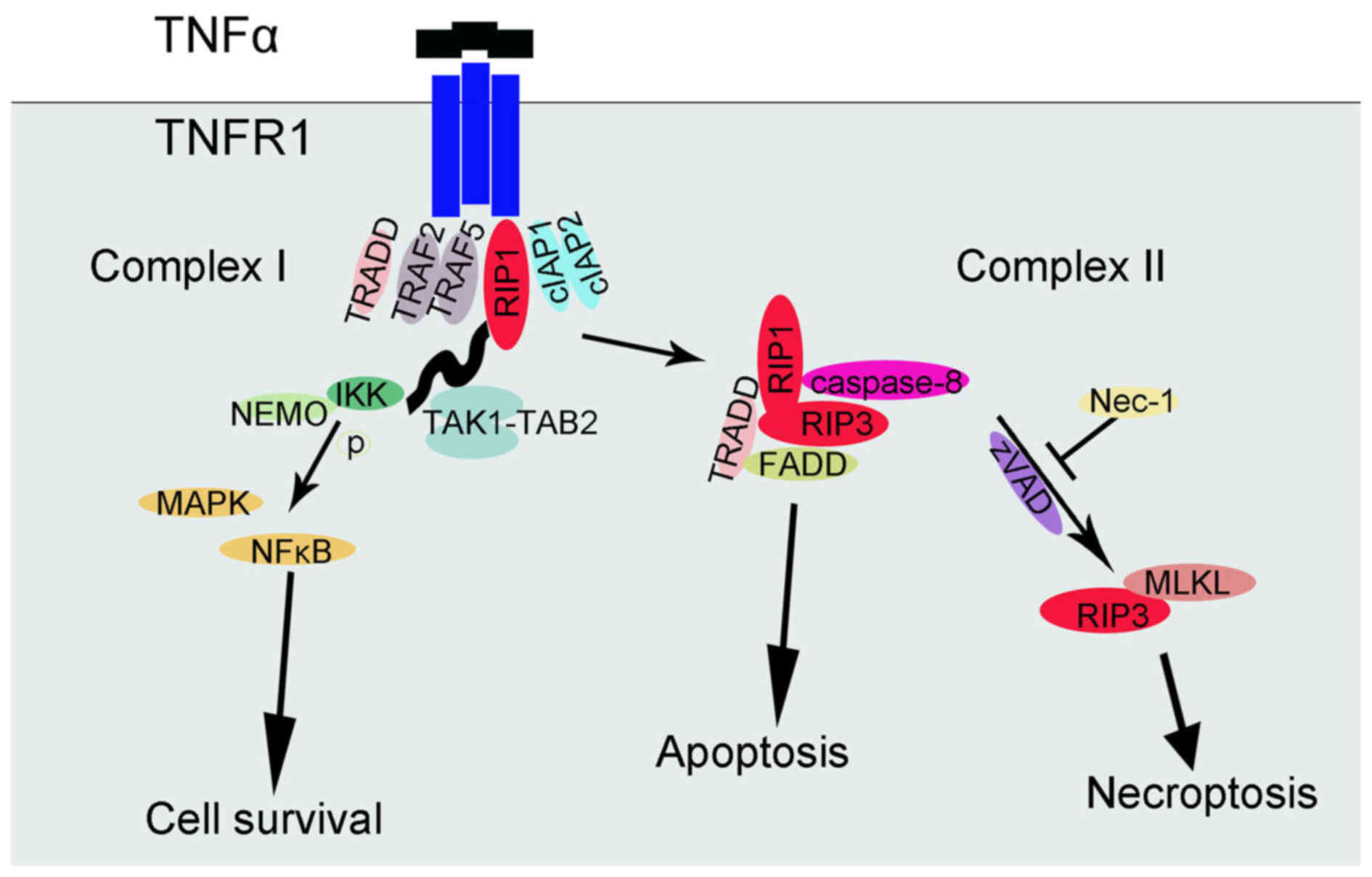 | Figure 2TNFα-TNFR1-related signaling pathway
of cell survival, apoptosis and necroptosis. TNFα, tumor necrosis
factor-α; TNFR1, TNF receptor 1; Nec-1, necroptosis specific
inhibitor-1; RIP1, receptor interacting protein; TRADD, TNFα
receptor-associated death domain; TRAF, TNFR-associated factor;
cIAP, cellular inhibitor of apoptosis; IKK, IκB kinase; NEMO,
nuclear factor-κB essential modulator; NF-κB, nuclear factor-κB;
MAPK, mitogen-activated protein kinase; FADD, Fas-associated
protein with a death domain; MLKL, mixed lineage kinase domain-like
protein; TAK, transforming growth factor β-activated kinase 1;
TAB2, TGF-β activated kinase 1/MAP3K7 binding protein 2. |
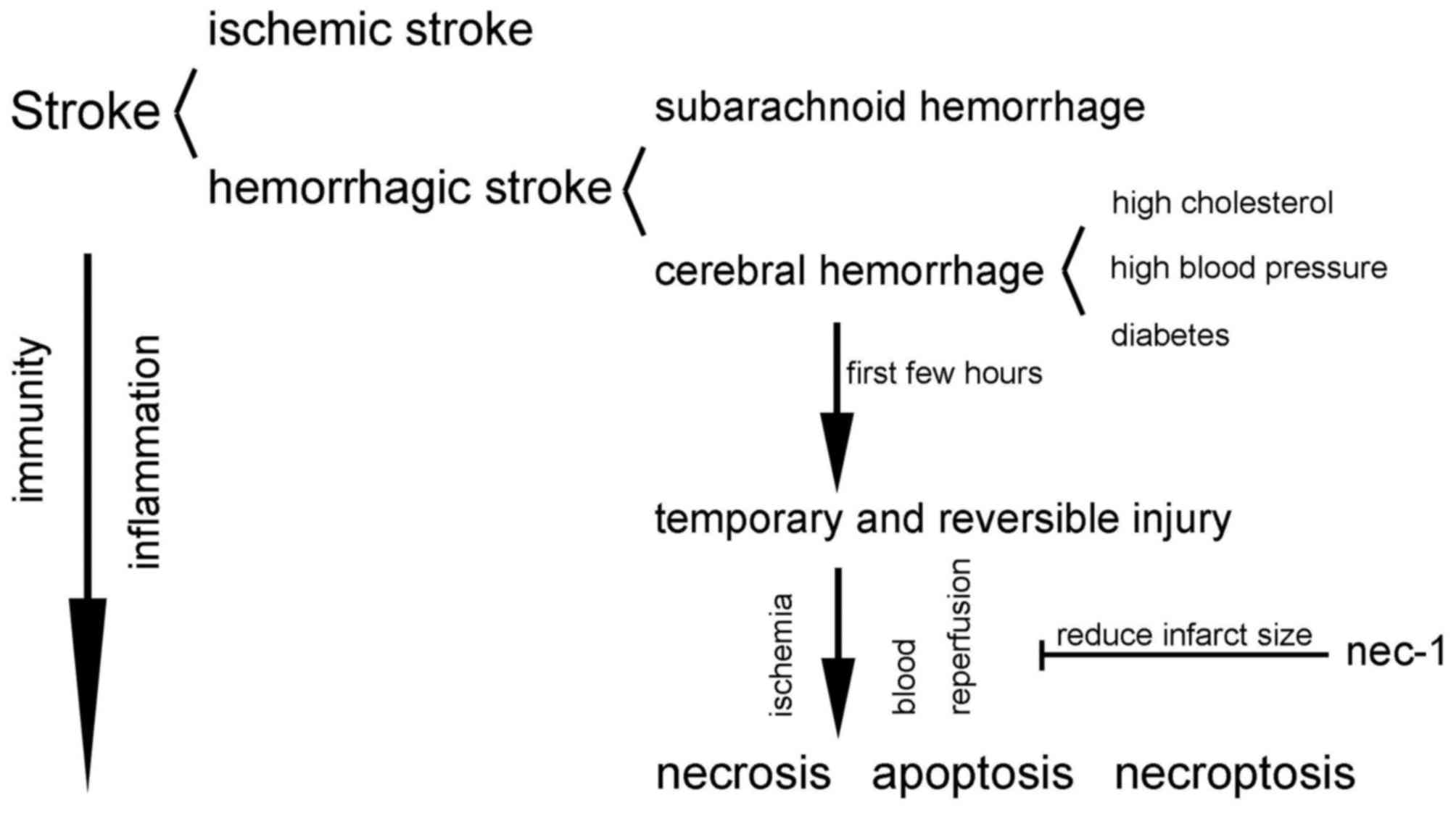
4. Necroptosis in stroke
Stroke is a devastating illness second only to
cardiac ischemia as a cause of mortality worldwide (32). Stroke used to occur mostly in the
elderly, but in recent years patient age and age at mortality
appears to have decreased, garnering attention to the problem.
Cerebral vasospasm, a cerebral blood circulation obstacle triggered
by a sudden blockage or rupture of the cerebral vessels is the main
cause of stroke. On this basis, brain tissues are damaged, and
cranial nerve functions are partially or full disordered. Following
the occurrence of stroke, immunity is a key element of the
pathobiology, while the immune system participates in the brain
damage, exerting a powerful immunosuppressive effect (33). Inflammatory signaling runs
throughout all stages, from early damaging events triggered by
arterial occlusion, to the late post-ischemic tissue repair
(34).
Stroke is divided into two clinical categories:
Ischemic stroke and hemorrhagic stroke. Ischemic stroke refers to
the local brain tissues, including neural cells, glial cells and
vasculature, becoming necrotic due to a lack of blood supply. The
fundamental reason for this is that the blood system of the brain
does not receive timely and sufficient collateral circulation after
external and intracranial artery stenosis or with occluding
lesions, thus decreasing the oxygen and energy supply, which the
surrounding brain tissue metabolism requires. Hemorrhagic stroke
refers to cerebral hemorrhage causing compression and necrosis of
the brain tissue. This type of stroke is also known as a
hemorrhagic cerebrovascular accident, and is generally divided into
two categories: Cerebral hemorrhage and subarachnoid hemorrhage
(33).
Cerebral hemorrhage is defined as a hemorrhage
caused by non-traumatic brain parenchyma vascular rupture whose
origin is mainly associated with a cerebrovascular lesion, such as
caused by high cholesterol, diabetes and high blood pressure
(34,35). During the first few hours after a
cerebral hemorrhage occurs, injury of the neurons in the ischemic
penumbra and infarcted surrounding areas is temporary and
reversible; apoptosis or necrosis is only an end result if no
intervention occurs (36).
Ischemic death of neurons exhibits mixed characteristics of
apoptosis and necrosis in terms of biochemistry and ultrastructure
(36). Ischemic cell damage is
common in the clinic and can be divided into two stages, namely the
reversible damage (can be repaired after injury if blood flow is
reinstated) and irreversible damage (due to continuous ischemia,
hypoxia and lack of basic metabolites). Cells mainly die by a
necrosis-like process via dysfunction of the mitochondria in the
irreversible lesion, but not all cell death is triggered by
ischemia. Blood reperfusion, including some infiltration of
neutrophils, cytokines and ROS, etc., can exacerbate tissue damage
(36). At present, studies on
ischemia/reperfusion in vivo are performed on animal models
by blocking one side of the terminal artery of a certain organ or
tissue (36). Furthermore, Nec-1
is the first found molecule that has been found to significantly
reduce infarct size in the rat model of middle cerebral artery
occlusion (3). The similar
effects of Nec-1 can also be found in the study of
ischemia-reperfusion damage in myocardial, retinal and kidney
tissues (37-39). In addition,
intracerebroventricular injection of Nec-1 can significantly reduce
the infarction area in the middle cerebral artery occlusion model
(3), and the protective effect of
7-Cl-O-Nec-1 injection can be detected 6 h after injury. However,
the effect of zVAD-fmk (a caspase inhibitor) injection is extremely
small at this point. That is to say the nerve protection effect of
7-Cl-O-Nec-1 can expand the therapeutic time window, and reveals
the backwardness of necroptosis in cerebral ischemic injury
(40). Furthermore, inflammatory
factors increase significantly after cerebral hemorrhage, as
detected via the immunohistochemical method. Activation of
microglia can be used as another indicator to determine the
occurrence and development of neuroinflammation (32). Microglia cells are inherent immune
cells in the brain, maintaining a static phenotype in the normal
state. As brain injury occurs, microglia cells are activated and
migrate to the damaged regions prior to a response by any other
cells in the body. At the same time, the quiescent branching shape
of the microglia cells translates to an operative amoeba-like
shape. Microglia cells swallow pathogens, apoptotic cells and
cellular debris through proliferation, although their activation
and concrete mechanisms remain unclear (32,36). In Fig. 3, the associations between the
three main forms of cell death and stroke are abstractly
exhibited.
5. Necroptosis in other diseases
N-methyl-D-aspartic acid (NMDA)
receptor-associated diseases
Glutamic acid is an important excitatory
neurotransmitter in the central nervous system, and the activation
of its receptor plays a significant role in physiological
processes. Meanwhile, glutamic acid can also produce a neurotoxic
effect via the excessive activation of NMDA receptor (41). A growing body of research has
suggested that in numerous acute or chronic neurological diseases,
including Huntington's disease, Parkinson's disease and Alzheimer's
disease, this excitatory neurotoxicity is one of the main
mechanisms of neuron death (42,43). It has been proved that Nec-1 can
suppress the decrease in cell vitality and the release of lactate
dehydrogenase, and decrease the number of living cells induced by
NMDA receptor (44). The results
indicate that necroptosis is involved in NMDA-induced cellular
damage.
Inflammatory diseases
Studies have shown the vital role of caspase-8 in
the regulation of necroptosis in intestinal epithelial cells and
terminal ileum cells (45).
Spontaneous formation of inflammatory damage can be observed in
caspase-8-defective mice, and these mice are highly susceptible to
colonitis (45). In patients with
Crohn's disease (which exhibits characteristic necrosis of terminal
ileum epithelial cells), as expression of RIP3 of intestinal
epithelium goblet cells increases, the necroptosis of terminal
ileum cells is enhanced, and this can be blocked by inhibitors of
necroptosis, suggesting that RIP3-mediated necroptosis may take
part in the genesis and development of inflammatory diseases
(45).
Cancer
Necroptosis serves an indispensable role in the
pathogenesis of cancer, as this process usually fails during
tumorigenesis and tumor development (46). For instance, cells in chronic
lymphocytic leukemia cannot undergo necroptosis when stimulated by
TNFα and zVAD, and the key component of necroptosis, RIP3 is
markedly downregulated (47).
Besides, in non-Hodgkin lymphoma, single nucleotide polymorphisms
in the RIP3 gene can be detected and are correlated with an
increased risk of morbidity, indicating that genetic variations in
the RIP3 gene may contribute to this cancer type (48). In addition, more and more
compounds and anticancer agents have been discovered to induce
necroptosis of cancer cells. Shikonin, a small molecule, was the
first compound discovered to induce necroptosis in cancer (49) and a number of studies proved that
shikonin and its analogs were highly antineoplastic without drug
resistance (50-52).
Other diseases
In the mouse myocardial ischemia model, injecting
Nec-1 after reperfusion can markedly shrink the volume of focal
necrosis overall and in vitro (53). Additionally, in the mouse
permanent ligation of coronary artery model, Nec-1 can
significantly shrink the volume of necrosis, inhibit myocardial
tissue fibrosis after necrosis and improve heart function. In the
acute pancreatitis model, RIP3-knockout mice exhibit less necrosis
of pancreatic cells, leakage of pancreatic enzyme and inflammation
of pancreatic tissue (17,28).
Another study has reported that the PCD of capillary endothelial
cells is composed of classic apoptosis and necroptosis. The classic
apoptosis occurs from the original stage and throughout the whole
growing phase, while necroptosis takes place only after appearance
of capillary lumina and filtration barriers (37). Thus, we speculate that its
significance lies in the fact that the collapsed contents of
necroptosis cells can be taken away by the blood flow, preventing
inflammation. Increasing numbers of studies are focusing on this
aspect at present.
6. Conclusions
With the rapid progress of science and technology, a
growing number of studies have shown that, other than the classic
necrosis and apoptosis, cell death consists of another form, known
as necroptosis. The occurrence and progression of necroptosis is
based on specific signaling pathways (such as TNF-α-TNFR1-related
signaling pathway) and regulated by specific biological molecules
(including RIP1, RIP3 and caspase-8). At the same time as the
occurrence of necroptosis, inflammation occurs due to release of
cellular inclusions causing infiltration and activation of
inflammatory cells. Thus far, the specificity of a terminal target,
which is similar to DNA fragmentation when apoptosis occurs, has
not been found in the process of necroptosis. Currently, the basic
judgment of necroptosis can be divided into the morphological
method (electron microscopy and PI staining) and biochemical
methods (Nec-1, RIP1-knockout or RIP3-knockout). The formation of
necrosomes and the phosphorylation of RIP1 and RIP3 can serve as a
complementary judgment indicator.
In clinical diseases, necroptosis is hypothesized to
be associated with autophagy, apoptosis, necrosis and the
inflammation of cells, providing a great possibility for future
clinical combination drug therapy. These studies not only improve
scientific literacy and technology, but also enrich the research of
pathological mechanism in stroke and other diseases. More
importantly, it may provide a knowledge base for clinical studies
as well. In addition, due to the multiple signal regulatory
pathways involved in necroptosis, the mechanism of necroptosis is
extremely complicated and requires further study. Moreover, further
research is also required to investigate the reasons for the
inflammation caused by necroptosis and the varying functions of
associated cells, including the activation and function of
microglia cells.
Glossary
Abbreviations
Abbreviations:
|
PCD
|
programmed cell death
|
|
DR
|
death receptors
|
|
TNFα
|
tumor necrosis factor-α
|
|
TRAIL
|
TNF-related apoptosis-inducing
ligand
|
|
caspases
|
cysteine aspartyl proteases
|
|
PI
|
propidium iodide
|
|
Nec-1
|
necroptosis-specific inhibitor-1
|
|
RIP
|
receptor interacting protein
|
|
TNFR1
|
tumor necrosis factor receptor 1
|
|
ROS
|
reactive oxygen species
|
|
TRADD
|
TNFα receptor-associated death
domain
|
|
TRAF
|
TNFR-associated factor
|
|
cIAP
|
cellular inhibitor of apoptosis
|
|
IKK
|
IκB kinase
|
|
NEMO
|
nuclear factor-κB essential
modulator
|
|
NF-κB
|
nuclear factor-κB
|
|
MAPK
|
mitogen-activated protein kinase
|
|
FADD
|
Fas-associated protein with a death
domain
|
|
MLKL
|
mixed lineage kinase domain-like
protein
|
|
NMDA
|
N-methyl-D-aspartic acid
|
References
|
1
|
Su Z, Yang Z, Xu Y, Chen Y and Yu Q:
Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol
Cancer. 14:482015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Su Z, Yang Z, Xu Y, Chen Y and Yu Q:
MicroRNAs in apoptosis, autophagy and necroptosis. Oncotarget.
6:8474–8490. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Degterev A, Huang Z, Boyce M, Li Y, Jagtap
P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar
|
|
4
|
Nikoletopoulou V, Markaki M, Palikaras K
and Tavernarakis N: Crosstalk between apoptosis, necrosis and
autophagy. Biochim Biophys Acta. 1833:3448–3459. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schweichel JU and Merker HJ: The
morphology of various types of cell death in prenatal tissues.
Teratology. 7:253–266. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Leist M and Jäättelä M: Four deaths and a
funeral: From caspases to alternative mechanisms. Nat Rev Mol Cell
Biol. 2:589–598. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yuksel S, Tosun YB, Cahill J and Solaroglu
I: Early brain injury following aneurysmal subarachnoid hemorrhage:
Emphasis on cellular apoptosis. Turk Neurosurg. 22:529–533.
2012.PubMed/NCBI
|
|
8
|
Green DR: Apoptotic pathways: Ten minutes
to dead. Cell. 121:671–674. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Adams JM: Ways of dying: Multiple pathways
to apoptosis. Genes Dev. 17:2481–2495. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kroemer G, Galluzzi L and Brenner C:
Mitochondrial membrane permeabilization in cell death. Physiol Rev.
87:99–163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lévy J and Romagnolo B: Autophagy,
microbiota and intestinal oncogenesis. Oncotarget. 6:34067–34068.
2015.PubMed/NCBI
|
|
12
|
Ding WX, Ni HM, Gao W, Hou YF, Melan MA,
Chen X, Stolz DB, Shao ZM and Yin XM: Differential effects of
endoplasmic reticulum stress-induced autophagy on cell survival. J
Biol Chem. 282:4702–4710. 2007. View Article : Google Scholar
|
|
13
|
Galluzzi L and Kroemer G: Necroptosis: A
specialized pathway of programmed necrosis. Cell. 135:1161–1163.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Degterev A, Hitomi J, Germscheid M, Ch'en
IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al:
Identification of RIP1 kinase as a specific cellular target of
necrostatins. Nat Chem Biol. 4:313–321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
You Z, Savitz SI, Yang J, Degterev A, Yuan
J, Cuny GD, Moskowitz MA and Whalen MJ: Necrostatin-1 reduces
histopathology and improves functional outcome after controlled
cortical impact in mice. J Cereb Blood Flow Metab. 28:1564–1573.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Degterev A, Zhou W, Maki JL and Yuan J:
Assays for necroptosis and activity of RIP kinases. Methods
Enzymol. 545:1–33. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ,
Lin SC, Dong MQ and Han J: RIP3, an energy metabolism regulator
that switches TNF-induced cell death from apoptosis to necrosis.
Science. 325:332–336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cho YS, Challa S, Moquin D, Genga R, Ray
TD, Guildford M and Chan FK: Phosphorylation-driven assembly of the
RIP1-RIP3 complex regulates programmed necrosis and virus-induced
inflammation. Cell. 137:1112–1123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Aggarwal BB: Signalling pathways of the
TNF superfamily: A double-edged sword. Nat Rev Immunol. 3:745–756.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Orrenius S: Reactive oxygen species in
mitochondria-mediated cell death. Drug Metab Rev. 39:443–455. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jin Z and El-Deiry WS: Distinct signaling
pathways in TRAIL-versus tumor necrosis factor-induced apoptosis.
Mol Cell Biol. 26:8136–8148. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vanden Berghe T, Declercq W and
Vandenabeele P: NADPH oxidases: New players in TNF-induced necrotic
cell death. Mol Cell. 26:769–771. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu YT, Tan HL, Huang Q, Sun XJ, Zhu X and
Shen HM: zVAD-induced necroptosis in L929 cells depends on
autocrine production of TNFα mediated by the PKC-MAPKs-AP-1
pathway. Cell Death Differ. 18:26–37. 2011. View Article : Google Scholar
|
|
24
|
Micheau O and Tschopp J: Induction of TNF
receptor I-mediated apoptosis via two sequential signaling
complexes. Cell. 114:181–190. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ea CK, Deng L, Xia ZP, Pineda G and Chen
ZJ: Activation of IKK by TNFalpha requires site-specific
ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell.
22:245–257. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vandenabeele P, Galluzzi L, Vanden Berghe
T and Kroemer G: Molecular mechanisms of necroptosis: An ordered
cellular explosion. Nat Rev Mol Cell Biol. 11:700–714. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Feng S, Yang Y, Mei Y, Ma L, Zhu DE, Hoti
N, Castanares M and Wu M: Cleavage of RIP3 inactivates its
caspase-independent apoptosis pathway by removal of kinase domain.
Cell Signal. 19:2056–2067. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
He S, Wang L, Miao L, Wang T, Du F, Zhao L
and Wang X: Receptor interacting protein kinase-3 determines
cellular necrotic response to TNF-alpha. Cell. 137:1100–1111. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Deutsch M, Graffeo CS, Rokosh R, Pansari
M, Ochi A, Levie EM, Van Heerden E, Tippens DM, Greco S, Barilla R,
et al: Divergent effects of RIP1 or RIP3 blockade in murine models
of acute liver injury. Cell Death Dis. 6:e17592015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Irrinki KM, Mallilankaraman K, Thapa RJ,
Chandramoorthy HC, Smith FJ, Jog NR, Gandhirajan RK, Kelsen SG,
Houser SR, May MJ, et al: Requirement of FADD, NEMO, and BAX/BAK
for aberrant mitochondrial function in tumor necrosis factor
alpha-induced necrosis. Mol Cell Biol. 31:3745–3758. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sun L, Wang H, Wang Z, He S, Chen S, Liao
D, Wang L, Yan J, Liu W, Lei X, et al: Mixed lineage kinase
domain-like protein mediates necrosis signaling downstream of RIP3
kinase. Cell. 148:213–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Iadecola C and Anrather J: The immunology
of stroke: From mechanisms to translation. Nat Med. 17:796–808.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Engelhardt B and Sorokin L: The
blood-brain and the blood-cerebrospinal fluid barriers: Function
and dysfunction. Semin Immunopathol. 31:497–511. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Konsman JP, Drukarch B and Van Dam AM:
(Peri)vascular production and action of pro-inflammatory cytokines
in brain pathology. Clin Sci (Lond). 112:1–25. 2007. View Article : Google Scholar
|
|
35
|
Jung KH, Yu KH, Kim YD, Park JM, Hong KS,
Rha JH, Kwon SU, Bae HJ, Heo JH, Lee BC, et al: Antithrombotic
management of patients with nonvalvular atrial fibrillation and
ischemic stroke or transient ischemic attack: Executive summary of
the Korean Clinical Practice Guidelines for Stroke. J Stroke.
17:210–215. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang J, Yang W, Xie H, Song Y, Li Y and
Wang L: Ischemic stroke and repair: current trends in research and
tissue engineering treatments. Regen Med Res. 2:32014. View Article : Google Scholar
|
|
37
|
Oerlemans MI, Liu J, Arslan F, den Ouden
K, van Middelaar BJ, Doevendans PA and Sluijter JP: Inhibition of
RIP1-dependent necrosis prevents adverse cardiac remodeling after
myocardial ischemia-reperfusion in vivo. Basic Res Cardiol.
107:2702012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rosenbaum DM, Degterev A, David J,
Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J and Savitz SI:
Necroptosis, a novel form of caspase-independent cell death,
contributes to neuronal damage in a retinal ischemia-reperfusion
injury model. J Neurosci Res. 88:1569–1576. 2010.
|
|
39
|
Linkermann A, Bräsen JH, Himmerkus N, Liu
S, Huber TB, Kunzendorf U and Krautwald S: Rip1
(receptor-interacting protein kinase 1) mediates necroptosis and
contributes to renal ischemia/reperfusion injury. Kidney Int.
81:751–761. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zheng W, Degterev A, Hsu E, Yuan J and
Yuan C: Structure-activity relationship study of a novel
necroptosis inhibitor, necrostatin-7. Bioorg Med Chem Lett.
18:4932–4935. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Beal MF: Role of excitotoxicity in human
neurological disease. Curr Opin Neurobiol. 2:657–662. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tabrizi SJ, Cleeter MW, Xuereb J, Taanman
JW, Cooper JM and Schapira AH: Biochemical abnormalities and
excitotoxicity in Huntington's disease brain. Ann Neurol. 45:25–32.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Beal MF: Excitotoxicity and nitric oxide
in Parkinson's disease pathogenesis. Ann Neurol. 44(Suppl 1):
S110–S114. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bonde C, Noraberg J, Noer H and Zimmer J:
Ionotropic glutamate receptors and glutamate transporters are
involved in necrotic neuronal cell death induced by oxygen-glucose
deprivation of hippocampal slice cultures. Neuroscience.
136:779–794. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Günther C, Martini E, Wittkopf N, Amann K,
Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath
MF, et al: Caspase-8 regulates TNF-α-induced epithelial necroptosis
and terminal ileitis. Nature. 477:335–339. 2011. View Article : Google Scholar
|
|
46
|
Fulda S: The mechanism of necroptosis in
normal and cancer cells. Cancer Biol Ther. 14:999–1004. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu P, Xu B, Shen W, Zhu H, Wu W, Fu Y,
Chen H, Dong H, Zhu Y, Miao K, et al: Dysregulation of TNFα-induced
necroptotic signaling in chronic lymphocytic leukemia: Suppression
of CYLD gene by LEF1. Leukemia. 26:1293–1300. 2012. View Article : Google Scholar
|
|
48
|
Cerhan JR, Ansell SM, Fredericksen ZS, Kay
NE, Liebow M, Call TG, Dogan A, Cunningham JM, Wang AH, Liu-Mares
W, et al: Genetic variation in 1253 immune and inflammation genes
and risk of non-Hodgkin lymphoma. Blood. 110:4455–4463. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, Luo
J and Hu X: Shikonin circumvents cancer drug resistance by
induction of a necroptotic death. Mol Cancer Ther. 6:1641–1649.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wada N, Kawano Y, Fujiwara S, Kikukawa Y,
Okuno Y, Tasaki M, Ueda M, Ando Y, Yoshinaga K, Ri M, et al:
Shikonin, dually functions as a proteasome inhibitor and a
necroptosis inducer in multiple myeloma cells. Int J oncol.
46:963–972. 2015. View Article : Google Scholar :
|
|
51
|
Fu Z, Deng B, Liao Y, Shan L, Yin F, Wang
Z, Zeng H, Zuo D, Hua Y and Cai Z: The anti-tumor effect of
shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent
necroptosis. BMC Cancer. 13:5802013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xuan Y and Hu X: Naturally-occurring
shikonin analogues - a class of necroptotic inducers that
circumvent cancer drug resistance. Cancer Lett. 274:233–242. 2009.
View Article : Google Scholar
|
|
53
|
Smith CC, Davidson SM, Lim SY, Simpkin JC,
Hothersall JS and Yellon DM: Necrostatin: A potentially novel
cardioprotective agent? Cardiovasc Drugs Ther. 21:227–233. 2007.
View Article : Google Scholar : PubMed/NCBI
|