Introduction
Ischemia induces oxidative stress by inhibiting the
consumption of antioxidants and the activity of antioxidant
enzymes, and increasing the production of toxic free radicals
(1,2). The generation of reactive oxygen
species (ROS) and reactive nitrogen species results in disturbed
Ca2+ homeostasis and the excitotoxicity of neurons in
the ischemic region, which increases the susceptibility of brain
tissues to direct or indirect damage by inflammation and apoptosis
(3,4). In addition, reperfusion leads to the
hydroxylation of nucleotides and peroxidation of phospholipids,
which further perpetuates the ischemic damage to neuronal
membranous structures and DNA integrity (5). An antioxidant defense mechanism has
been suggested to protect the brain from ischemia/reperfusion
injury, by either upregulating endogenous antioxidants in 'at risk'
tissues, or by reducing the oxidative damage via the scavenging of
free radicals overproduced in the ischemic tissues (3,5,6).
Magnolol (5,5′-diallyl-2,2′-dihydroxydiphenyl), a
phenolic constituent of magnolia bark, is a potent antioxidant and
demonstrates depressive effects on the CNS (7,8).
In previous studies, magnolol protected limbs from
ischemia-reperfusion damage in a rat model (9), and protected against the brain
damage induced by experimental heatstroke (10). In addition, magnolol effectively
blocked voltage-dependent Ca2+ channels and reduced cell
necrosis in a mixed neuron-astrocyte culture exposed to chemical
hypoxia (11–14). Furthermore, magnolol reduced
glutamate-induced excitotoxicity in cultured neurons and
ameliorated the brain tissue damage caused by permanent middle
cerebral artery occlusion up to 4 h post-insult (15). However, it remains unclear whether
magnolol is able to protect the brain against further ischemic
insult. Magnolol has been demonstrated to effectively reduced
oxidative stress in neuron cultures (7–9,16,17), and may be protective against
cerebral ischemic-reperfusion injury in animal models. In
particular, it is necessary to determine whether magnolol-mediated
neuroprotection is provided by the intravenous administration of
magnolol, as this mode of administration is most closely associated
with the clinical treatment of patients with stroke (18).
In the present study, the neuroprotective effects of
magnolol in an oxygen-glucose deprived rat model were
characterized. In addition, post-treatment responses and
appropriate neuroprotective dosing were explored through the
intravenous administration of magnolol to rats subjected to
transient focal cerebral ischemia. The underlying mechanisms of
action contributing to the neuroprotective properties of magnolol
were also explored using in vitro and in vivo
assays.
Materials and methods
Reagents and chemicals
Chemicals, including synthetic (±)-α-tocopherol
(cat. no. T-3251) and ascorbic acid (cat. no. A4544), were
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany) and
the highest grade available was used. Hank's balanced salt solution
(HBSS) was used, comprising 8.0 g/l NaCl, 0.4 g/l KCl, 1.0 g/l
glucose, 0.06 g/l KH2PO4 and 0.09 g/l
Na2PO4-7H2O, pH 7.1; glucose-free
HBSS was prepared by omitting glucose and adding an additional
half-molar amount of NaCl to adjust the osmolarity (8.162 g/l
NaCl). When used in the present study, magnolol (Wako Pure Chemical
Industries, Ltd., Osaka, Japan) was dissolved in dimethylsulfoxide
(DMSO) or polyethylene glycol 400 (PEG 400; Merck KGaA).
Lipid peroxidation and radical scavenging
assays in vitro
A malondialdehyde (MDA) assay was performed as
previously described (19–21).
The rat brain tissue was homogenized in 20 mM Tris-HCl buffer,
centrifuged at 1,000 x g for 10 min at 4°C. To 30 µl brain
homogenate supernatant was added 10 µl vehicle (0.1% DMSO),
magnolol (0.01 µM - 1 mM), α-tocopherol (0.1–100 µM),
ascorbic acid (1–5 mM) or β-estradiol (1–150 µM) and 5
µl freshly prepared ferric chloride hexahydrate. The
absorbance was measured using a plate reader at 532 nm. The
2,2-diphenyl-1-picrylhydrazyl (DPPH) assay was conducted by adding
100 µl freshly prepared DPPH radical solution to 100
µl vehicle (0.1% DMSO) or magnolol (0.01 µM - 1 mM)
as previously described (19–21). The absorbance was measured at 517
nm. The 2,2′-azino-bis (3-ethylbenzothiazoline-6-sulfonic acid)
diammonium salt (ABTS) assay was conducted by adding 100 µl
ABTS radical cation solution to 100 µl vehicle (0.1% DMSO)
or magnolol (0.01 µM - 1 mM) (19–21). The absorbance was measured at 734
nm.
Interleukin (IL) -6, tumor necrosis
factor (TNF) -a and nitrate/nitrite (NOX) assay of BV2
and RAW 264.7 cells
Mouse macrophage cell line RAW 264.7 and microglial
cell line BV2 (American Type Culture Collection, Manassas, VA, USA)
cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) were stimulated with lipopolysaccharide (LPS;
Sigma-Aldrich; Merck KGaA), 10 and 100 ng/ml, respectively, based
on preliminary dose-response experiments, and co-cultured with PBS,
vehicle (0.1% DMSO) or magnolol (0.1–50 µM). Following 6 h
of incubation, the levels of IL-6 and TNF-α in the supernatant were
determined using IL-6 (cat. no. DY-406) and TNF-a (cat. no.
DY410-05) DuoSet ELISA kits (R&D Systems, Inc., Minneapolis,
MN, USA) as previously described (19–21). The levels of nitric oxide (NO) in
the supernatant were measured using a nitrate/nitrite fluorometric
assay kit (cat. no. 780051; Cayman Chemical Company, Ann Arbor, MI,
USA) following 16 h of incubation (19–21).
Organotypic hippocampal slice
cultures
Organotypic hippocampal slices were obtained by
harvesting 300 µm-thick hippocampal slices using a motorized
NVSLM1 Vibroslice (Campden Instruments Ltd., Loughborough, UK) from
6–7-day-old, neonatal Sprague-Dawley rats weighing 20–30 g. The
animals were allowed free access to food and water and housed at
25°C and 60% humidity with a 12-h light/dark cycle (19). The slices were placed on
0.4-µm Millicell culture inserts (EMD Millipore, Billerica,
MA, USA) and transferred to 35 mm Petri dishes. Slices were
cultured with organotypic culture medium consisting of 25% horse
serum (Gibco; Thermo Fisher Scientific, Inc.), 25% HBSS and 50%
minimum essential medium (both MP Biomedicals, LLC, Santa Ana, CA,
USA), 5 mg/ml glucose, 1 mM glutamine, 5 mM KCl and 1.5% Fungizone
(Gibco; Thermo Fisher Scientific, Inc.) at 37°C and the culture
medium was refreshed every 3 days.
Oxygen-glucose deprivation (OGD) model in
vitro
The OGD model was generated by hypoxia combined with
aglycemia, as previously described (19–21). Briefly, the hippocampal slices
were cultured for 8–14 days and incubated with medium (1 ml/well
glucose-free HBSS, bubbled with 95% N2 and 5%
CO2) at 37°C for 20 min prior to OGD. The culture dishes
were incubated in an anaerobic chamber at 37°C with 95%
N2 and 5% CO2 for 120 min. A normal control
group was also established, in which the hippocampal slices were
incubated in HBSS with 95% O2 and 5% CO2 at
37°C. Replenishment of the cells was induced by replacing the
medium and incubating the dishes under normoxic conditions for a
further 24 h. To evaluate cell damage, 5 µg/ml propidium
iodide (PI) was incubated with the culture at 37°C for 20 min, and
the absorbance was measured at 630 nm and analyzed using Image Pro
Plus software, version 5.1 (Media Cybernetics, Inc., Rockville, MD,
USA) with a digital CoolSNAP-Procf camera (Media
Cybernetics, Inc.) and an IX71 epi-fluorescence inverted
micro-scope (Olympus Corporation, Tokyo, Japan). The fluorescence
intensity (Ft) of the slices was obtained from three different
fields of the slices. Regions without the slices were used as the
background intensity (F0). At 48 h after OGD, culture slices were
incubated with 10 mM glutamate to determine the final PI
fluorescence (Ffin). The PI uptake, as an indicator of cell death,
was subsequently obtained from the following equation: PI
(%)=(Ft-F0)/(Ffin-F0) ×100 (19).
An OGD duration of 120 min induces 70% of the maximal PI-uptake in
CA1 pyramidal neurons (19).
Experimental treatment groups were established as follows: i)
Pre-treatment group, treated with magnolol (10 or 100 µM) or
vehicle (0.1% DMSO) for 1 h prior to the OGD period, with no
additions to the medium during the following 24 h; ii)
post-treatment group, treated with magnolol (100 µM) or
vehicle (0.1% DMSO) at 2, 4 or 6 h post OGD.
Animal anesthesia and monitoring
Male Sprague-Dawley rats (n=120; 8 weeks old,
weighing 260–300 g), were obtained from the National Cheng Kung
University Animal Center (Tainan, Taiwan). The animals were allowed
free access to food and water and housed at 25°C, 60% humidity on a
12-h light/dark cycle. Rats were anesthetized with halothane, 3–4%
for induction and 1–2% for maintenance. The right femoral artery
was cannulated for the measurements of arterial blood gases,
hematocrit, glucose, heart rate and blood pressure. During surgery
the core temperature was maintained at 37.0±0.5°C by the use of a
heating blanket (Harvard Apparatus, Holliston, MA, USA).
All procedures within the present study were
performed in accordance with the recommendations of the Guide for
the Care and Use of Laboratory Animals of the National Institutes
of Health (eighth edition, 2011). The protocol for the present
study was approved by the Committee on the Ethics of Animal
Experiments of the National Cheng Kung University Hospital (permit
no. 102249; Tainan, Taiwan).
Experimental model
A stable ischemia animal model was generated as
previously described, by the occlusion of the right proximal middle
cerebral artery (MCA) with an intra-arterial suture for 90 min
(19–21). The local cortical cerebral
perfusion (local cerebral blood flow, LCBF) was measured using
Laserflo BMP2 Laser-Doppler flowmetry (Vasamed Inc., Eden Prairie,
MN, USA) to ensure the quality of the ischemia and reperfusion
induction (19–23).
The investigators were blinded to the treatment
paradigms and the rats were randomly separated into Magnolol and
vehicle groups. In a first series of experiments, magnolol (0.01,
0.1, 1 or 5 mg/kg; n=10, 10, 13 and 9, respectively) or vehicle
(PEG 400, 10 ml/kg; n=12) was administered intravenously (i.v.) 30
min prior to the onset of ischemia. Rats were euthanized 72 h
following the onset of ischemia and their brains examined using
standard 2,3,5-triphenyltetrazolium chloride (TTC)-stained
histological sections. In a second series of experiments, rats were
treated with magnolol (1 mg/kg, i.v.; n=10) or vehicle (i.v.; n=10)
2 h after the onset of ischemia. Following ischemia for 72 h, rats
were euthanized using histological sections and immunofluorescence
staining.
Another set of rats were treated with magnolol
(0.01, 0.3 or 1 mg/kg, i.v.; n=8 per group) or vehicle (PEG 400; 10
ml/kg, i.v.; n=8) at 2 h following the onset of ischemia, and
euthanized at 24 h post-insult, to assess the levels of
myeloperoxidase (MPO), malondialdehyde (MDA), NOX and
the ratio of reduced glutathione/oxidized glutathione (GSH/GSSG) in
the brain tissue. The NOX level was accessed using the
aforementioned nitrate/nitrite fluorometric assay kit.
Lipid peroxidation and MPO activity in
vivo
MDA and MPO activity in brain tissue were determined
as previously described (19,20). Absorbance was measured using a
plate reader at 532 and 492 nm, respectively.
Histology and immunofluorescence of
8-hydroxy-2′- deoxyguanosine (8-OHdG) and 4-hydroxynonenal
(4-HNE)
Rat brains were sectioned coronally as previously
described (20,22,23), into 40-µm sections at 1-mm
intervals from the bregma AP (4.2 to −6.8 mm) on a cryostat. Two
sets of sections were used for the immunofluorescence staining of
4-HNE and 8-OHdG. The expression of 4-HNE and 8-OHdG was used to
identify membranous lipid peroxidation and DNA damage, respectively
(5). Sections were washed in PBS
(Molecular Probes; Thermo Fisher Scientific, Inc.) and incubated in
3% H2O2 with 50% methanol/PBS at room
temperature for 30 min, then placed in PBS containing 0.3% Triton
X-100 and blocked with 1% normal sheep serum (Millipore, Merck
KGaA) at room temperature for 30 min. Sections were incubated with
monoclonal antibodies directed against 8-OHdG (1:750; MOG-100P) and
4-HNE (1:1,000; MHN-100P; both JaICA, Fukuroi, Japan) overnight at
4°C. Goat anti-rabbit IgG secondary antibodies conjugated with
biotin (1:100; cat. no. 111-065-144; Jackson ImmunoResearch
Laboratories, Inc., West Grove, PA, USA) were subsequently
incubated with the sections at room temperature for 1 h, followed
by fluorescein (DTAF)-conjugated streptavidin (1:150; cat. no.
016-010-084; Jackson ImmunoResearch Laboratories, Inc.).
Immunopositive cells were traced and measured using a Zeiss
Axioskop 2 Mot microscope (Zeiss AG, Oberkochen, Germany) equipped
with a digital CoolSNAP-Procf camera and
computerized image analyzer (MCID Elite 6.01.4; Imaging Research
Inc., St Catharines, ON, Canada). Fluorescence was measured at
450–490 nm and emission >515 nm for DTAF detection.
For the assessment of ischemic damage to the
neuronal perikarya, another set of sections was stained with 0.5%
cresyl violet. Using light microscopy (Zeiss Axioskop 2 Mot
micro-scope), the areas of neuronal perikarya displaying typical
morphologic features of ischemic damage were evaluated.
Detection of free radicals using in situ
dihydroethidium (DHE)
Sections 40 µm in thickness were stained with
5 mM DHE at 37°C for 30 min as previously described (5), and subsequently incubated with DAPI
(2 µM) in PBS at room temperature for 15 sec in a dark
chamber. Section fluorescence was evaluated at an excitation
wavelength of 510–550 nm and emission wavelength of >580 nm for
oxidized DHE (HEt) detection, and at excitation and emission
wavelengths of 365 and >420 nm for DAPI detection. The density
and intensity of the HEt in the ischemic brain tissue was compared
between the magnolol-treated animals and the controls.
Determination of reduced GSH/GSSG
In the ischemic brain tissues, the total GSH and
GSSG levels were determined as previously described by Anderson
(24). Briefly, GSH was oxidized
by 5,5′-dithio-bis(2-nitrobenzoic acid) to generate GSSG with
stoichiometric formation of 5-thio-2-nitrobenzoic acid (TNB). GSSG
was subsequently reduced to GSH by the action of the highly
specific glutathione reductase and NADPH. The rate of TNB formation
was measured at 412 nm and was proportional to the sum of GSH and
GSSG. For GSSG determination, 2-vinylpyridine was used to destroy
the reduced form of GSH, followed by the same procedure as the GSH
assay.
Euthanasia and quantification of ischemic
damage
An5imals were euthanized at 72 h following the onset
of ischemia. A rat brain matrix (RBM 4000C; ASI Instruments Inc.,
Warren, MI, USA) was used to cut 2-mm coronal sections. These
sections were stained using TTC at room temperature for 30 min in a
dark chamber (15). In the
delayed treatment paradigm, ischemic brain damage was measured
using TTC and cresyl violet-stained brain sections as previously
described (20,22,23). Coronal section images were
captured using a DXC-390 3-CCD color camera (Sony, Tokyo, Japan)
equipped with a Micro-Nikon 55 mm f2.8 lens. Areas of ischemic
damage were analyzed using a computerized image analyzer (MCID
Elite 6.01.4). The cerebral ischemic infarction volume was
expressed as a percentage of the contralateral hemisphere volume as
previously described (20,22,23).
Additionally, individual striatal and cortical infarct sizes were
calculated.
Sections were obtained between the Bregma AP-0.22
and −0.78 mm. Six non-overlapping regions (500×400
µm2) were randomly selected for cell counting of
the surviving neurons in the ischemic brain, as previously
described (20,22,23). Surviving neurons were expressed as
the mean number of viable neurons per mm2.
Neurobehavioral testing
Neurologic evaluation was conducted 1 and 3 days
following stroke induction by a qualified observer unaware of the
treatment protocol. Two different neurologic grading systems were
used in the present study. The first was a sensorimotor grading
scale with five categories (0–4)
for forward and sideways visual placing tests of the affected
forelimb, and five categories (0–4) for motor outcome, which is a
modification of previously described methods (20,22,23,25). The second was a neurobehavioral
grading scale of 0 to 28 for rodents, as previously described
(26).
Statistical analysis
Data are expressed as the mean ± standard deviation
and differences between groups were evaluated using an unpaired
Student's t-test or one-way analysis of variance with Fisher's
protected least significant difference post hoc test. To evaluate
the response in changing conditions a paired Student's t-test was
used. The data for neurobehavioral testing are expressed as the
median (95% confidence interval) and were evaluated using a
nonparametric test for independent groups, including the
Kruskal-Wallis and Mann-Whitney U tests. SPSS software, version
17.0 (SPSS, Inc., Chicago, IL, USA) was used to conduct the
analysis. P<0.05 was considered to indicate a statistically
significant difference.
Results
Radical scavenging and antioxidant
assays
Magnolol attenuated Fe3+-induced lipid
peroxidation in brain tissue homogenate with an IC50
value of 0.8±2.8 µM (Table
I), which was lower than the IC50 values for
α-tocopherol, ascorbic acid and β-estradiol (45.9±7.4,
2,770.0±110.0 and 65.6±7.9 µM, respectively). However, in
the DPPH radical scavenging assay, the IC50 value of
magnolol was 531.9±1.0 µM, which was greater than the
IC50 value for a-tocopherol and ascorbic acid (84.4±2.8
and 47.5±2.4 µM, respectively), but lower than that for
β-estradiol (202.4±18.0 mM). In the ABTS radical cation scavenging
assay, the IC50 value of magnolol was 22.3±0.5
µM, which was similar to the IC50 values of the
antioxidants α-tocopherol, β-estradiol and ascorbic acid (35.3±7.3,
16.9±3.2 and 31.7±6.8 µM, respectively).
 | Table IAntioxidant and radical scavenging
potency (IC50) of magnolol and reference antioxidant
compounds. |
Table I
Antioxidant and radical scavenging
potency (IC50) of magnolol and reference antioxidant
compounds.
| Substance | Inhibition of lipid
peroxidation induced by Fe3+ (10 mM) in rat brain
homogenate (µM) | DPPH radical
scavenging assay | ABTS radical
scavenging assay (µM) |
|---|
| Magnolol | 0.8±2.8 | 531.9±1.0
µM | 22.3±0.5 |
| α-tocopherol | 45.9±7.4 | 84.4±2.8
µM | 35.3±7.3 |
| Ascorbic acid | 2,770.0±110.0 | 47.5±2.4
µM | 31.7±6.8 |
| β-estradiol | 65.6±7.9 | 202.4±18.0 mM | 16.9±3.2 |
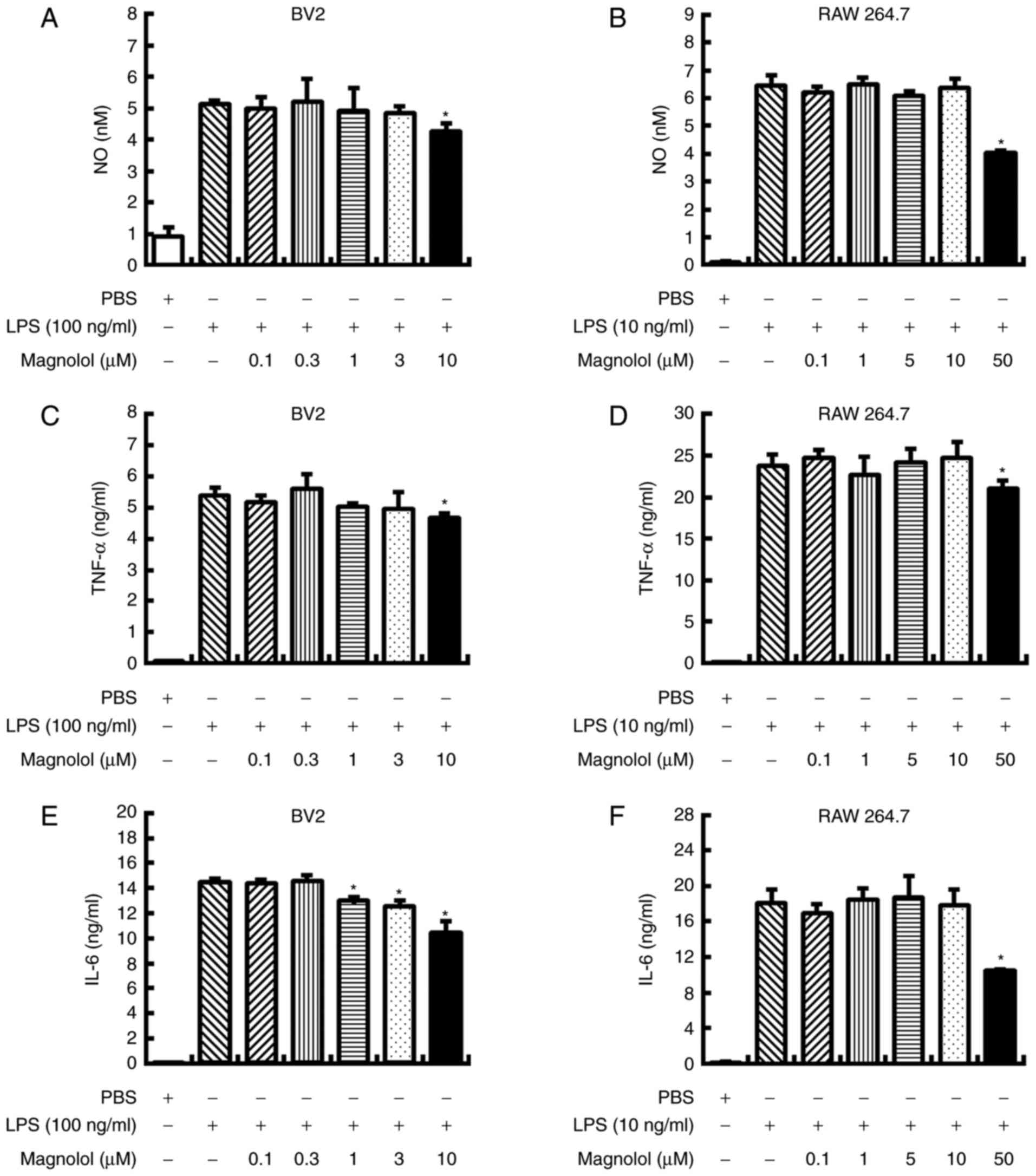
LPS stimulates proinflammatory cytokines
and NO production in vitro
Magnolol significantly reduced NO production when
applied to LPS-stimulated BV2 and RAW 264.7 cells at concentrations
of 10 and 50 µM, respectively (P<0.05; Fig. 1A and B). Similarly, magnolol
significantly reduced TNF-α production in LPS-stimulated BV2 and
RAW 264.7 cells when administered at concentrations of 10 and 50
µM, respectively (P<0.05; Fig. 1C and D). Additionally, magnolol
significantly inhibited IL-6 production when administered at 1–10
µM in LPS-stimulated BV2 cells and 50 µM in
LPS-stimulated RAW 264.7 cells (P<0.05; Fig. 1E and F).
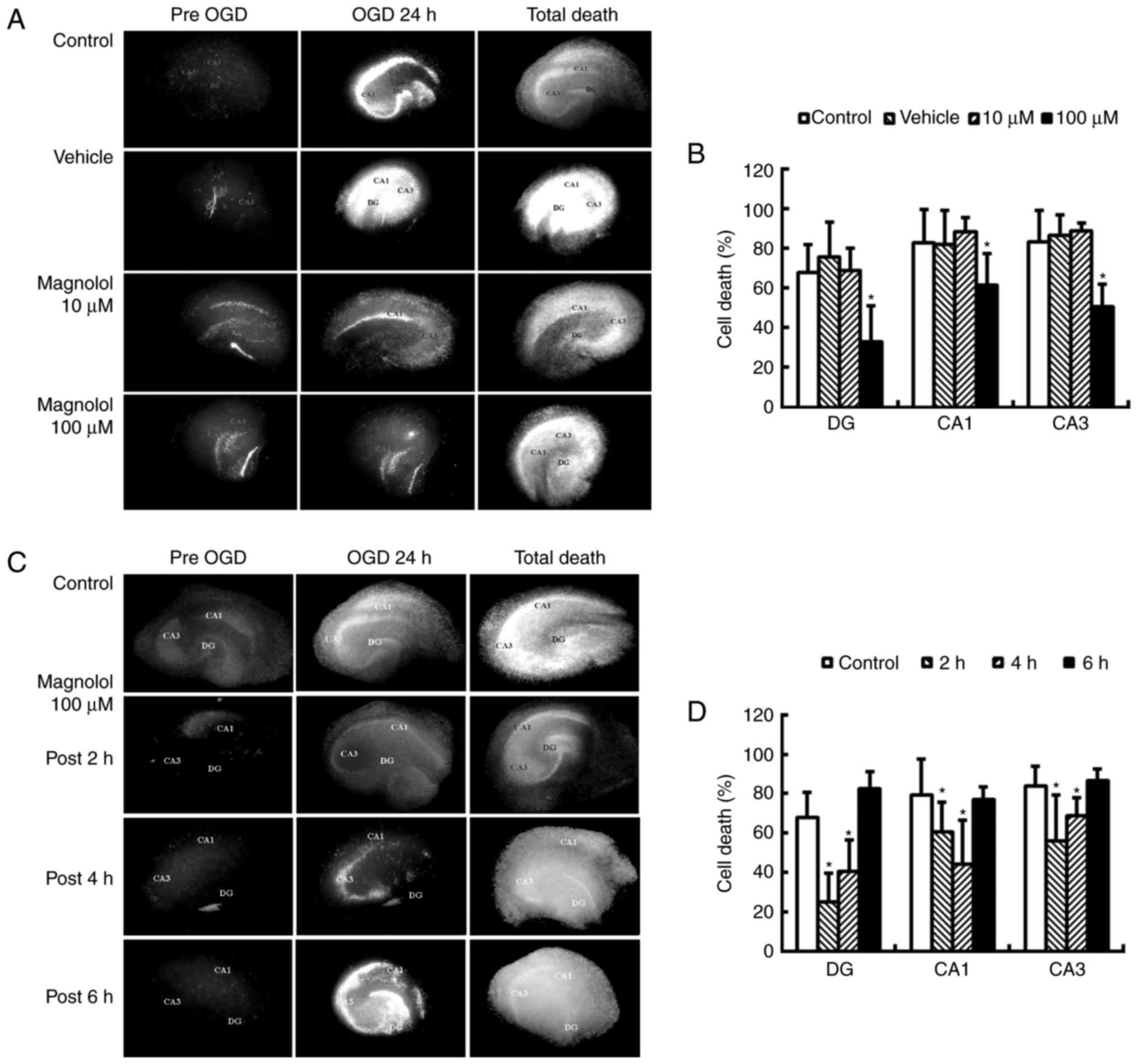
OGD of hippocampal slice cultures
No significant differences were identified in the PI
uptake in cultures treated with 1–100 µM magnolol compared
with the control cultures (data not shown). Pre-treatment with
magnolol at 100 µM 1 h prior to OGD led to a significant
reduction in cell death at 24 h compared with the control
(P<0.05; Fig. 2A and B). The
PI uptake was reduced by 25.0, 41.6 and 56.6% in the CA1, CA3 and
DA regions, respectively compared with the OGD control. Treatment
with magnolol (100 µM) at 2 or 4 h following the onset of
OGD significantly reduced the OGD-induced increases in PI uptake in
the CA1, CA3 and DA sub-regions of the brain compared with the
control (P<0.05; Fig. 2C and
D). The PI uptake was reduced by 23.8–44.1% in CA1, 17.7–33.0%
in CA3 and 40.0–62.9% in DA.
Transient focal cerebral ischemia in
rats
A total of 7 animals (6.0%) died during the course
of the experiment and were excluded. Of these animals, 3 (7.9%)
were in the vehicle-injected group and 4 (5.1%) were in the
magnolol-treated group. As described previously (20,21), animals subjected to transient MCA
occlusion invariably exhibit spontaneous hyperthermia. The
post-insult core temperatures and LCBF were not notably changed by
the intravenous administration of magnolol at 0.01–5 mg/kg (data
not shown). The physiological parameters blood hematocrit, arterial
blood pressure, heart rate and glucose of the experimental animals
were not notably different from those of the control animals over
the course of the experiment (data not shown).
Neuroprotective actions of magnolol
against ischemic-reperfusion insult in vivo
Pre-treatment with magnolol at doses of 0.01, 0.1 or
1 mg/kg 30 min prior to ischemia, significantly reduced the
infarction size in the cortex compared with the control (P<0.05;
Fig. 3A). Treatment with magnolol
at 0.01, 0.1 and 1 mg/kg reduced the infarction volume in the
cortex by 30.2, 24.3 and 22.7%, respectively, compared with the
control. Treatment with magnolol (0.01–5 mg/kg) also significantly
improved the sensory neurologic scores at 72 h post ischemic injury
compared with those in the vehicle group (P<0.05; Table II).
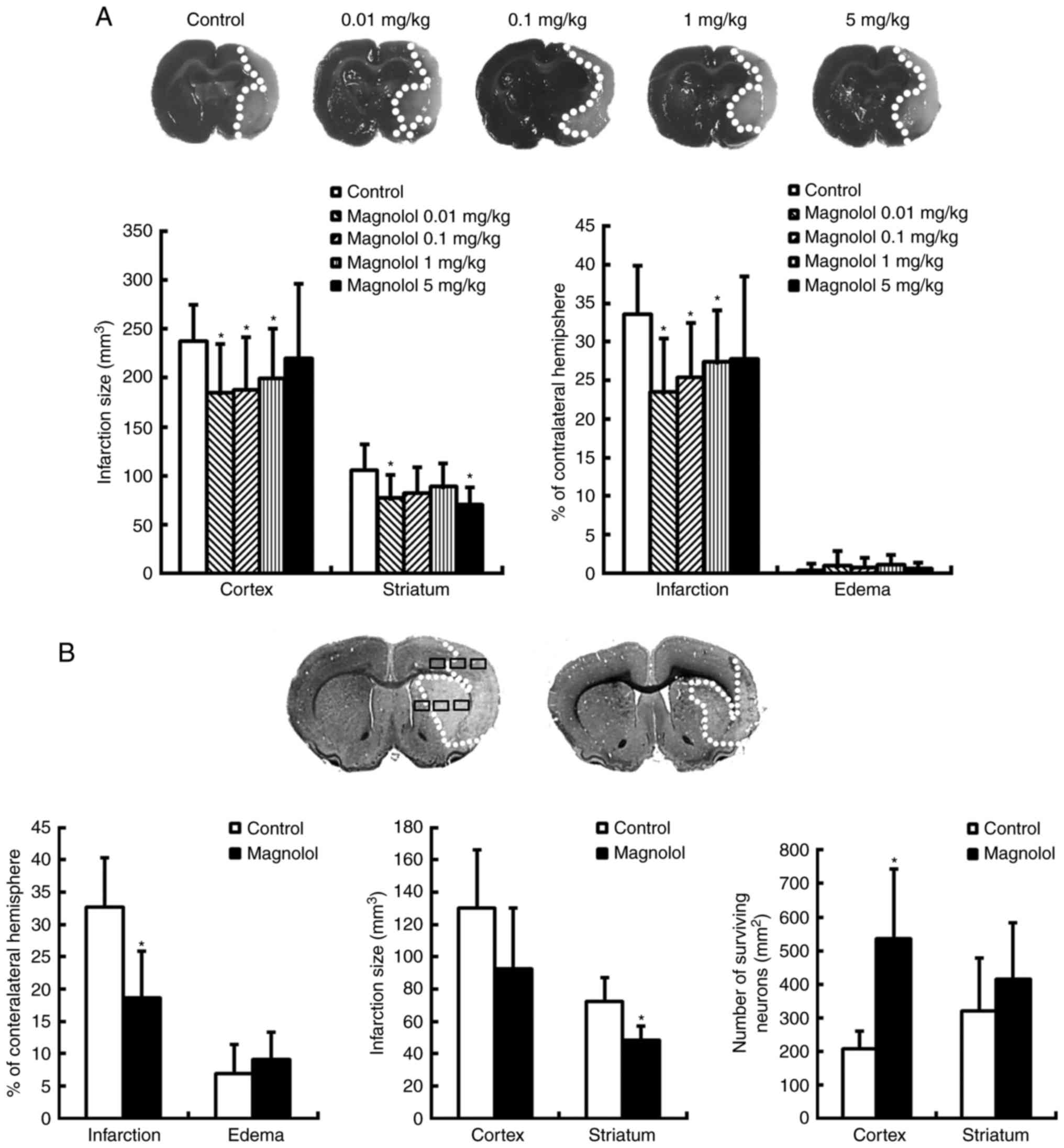 | Figure 3Magnolol treatment reduces cerebral
infarction and increases the number of surviving neurons. (A)
Coronal sections of animals treated with PEG 400 (vehicle, control)
or magnolol (0.01, 0.1, 1, or 5 mg/kg) 30 min prior to the onset of
middle cerebral artery occlusion were obtained 72 h following the
onset of ischemia and stained with 2,3,5-triphenytetrazolium
chloride. The infarction (pale region) was observed in the striatum
and cortex, and the infarction volume was significantly decreased
in the magnolol-treated animals. (B) Coronal sections from animals
treated with PEG 400 (vehicle) or magnolol (1 mg/kg) at 2 h
following the onset of ischemia were stained with cresyl violet at
72 h. In the cortex and striatum, 6 random non-overlapping regions
(500×400 µm2) were selected in which to count the
surviving neurons. In the delayed treatment paradigm, magnolol (1
mg/kg) significantly attenuated the infarction volume and
significantly increased the number of surviving neurons in the
cortex compared with the vehicle group. *P<0.05 vs.
the vehicle group. PEG 400, polyethylene glycol 400. |
| Table IIPre-treatment with magnolol improves
sensorimotor neurobehavioral scores following cerebral
ischemia-reperfusion. |
Table II
Pre-treatment with magnolol improves
sensorimotor neurobehavioral scores following cerebral
ischemia-reperfusion.
| Treatment | Neurologic
behavioral score
|
|---|
| Motor | Sensory | 28-point clinical
scale |
|---|
| Day 1 post
ischemia | | | |
| Vehicle | 2 (1.8–2.2) | 4 (3.9–4.1) | 18.5
(17.4–19.6) |
| Magnolol
(mg/kg) | | | |
| 0.01 | 1 (0.7–1.3)a | 2 (1.4–2.6)a | 9.5
(7.5–11.5)a |
| 0.1 | 1 (0.7–1.3)a | 2 (1.4–2.6)a | 9.5
(8.2–10.8)a |
| 1.0 | 1 (0.8–1.2)a | 2 (1.6–2.4)a | 10
(8.7–11.3)a |
| 5 | 1 (0.7–1.3)a | 2.5
(2.0–3.0)a | 9
(7.6–10.4)a |
| Day 3 post
ischemia | | | |
| Vehicle | 2 (1.7–2.3) | 4 (3.5–4.5) | 18 (16.5–19.5) |
| Magnolol
(mg/kg) | | | |
| 0.01 | 1 (0.7–1.3)a | 2 (1.4–2.6)a | 9
(7.7–10.3)a |
| 0.1 | 1 (0.7–1.3)a | 2 (1.4–2.6)a | 10
(8.6–11.4)a |
| 1 | 1.5
(1.3–1.7)a | 2 (1.6–2.4)a | 11
(9.9–12.1)a |
| 5 | 2 (1.7–2.3)a | 2 (1.3–2.7)a | 9
(7.5–10.5)a |
When magnolol (1 mg/kg) was administered at 2 h post
ischemic onset, the infarction volume in the striatum as a
percentage of the volume of the contralateral hemisphere was
significantly reduced by 43.3% at 72 h post ischemia compared with
the control (P<0.05; Fig. 3B).
The infarction volume in the striatum was reduced by 31.2%, and a
significant improvement in the number of surviving neurons in the
penumbral cortical area was also observed (both P<0.05; Fig. 3B). In addition, treatment with
magnolol 2 h post ischemic onset significantly increased the
neurologic and sensorimotor scores determined 3 days following the
onset of ischemia, compared with the vehicle group (P<0.05;
Table III).
| Table IIIDelayed treatment with magnolol
improves sensorimotor neurobehavioral scores following cerebral
ischemia-reperfusion. |
Table III
Delayed treatment with magnolol
improves sensorimotor neurobehavioral scores following cerebral
ischemia-reperfusion.
| Treatment | Neurologic
behavioral score
|
|---|
| Motor | Sensory | 28-point clinical
scale |
|---|
| Day 1 post
ischemia | | | |
| Vehicle | 3 (2.6–3.3) | 4 (3.7–4.2) | 20 (19.1–20.9) |
| Magnolol (1
mg/kg) | 1 (0.6–1.4)a | 2 (1.5–2.5)a | 9.5
(8.4–10.6)a |
| Day 3 post
ischemia | | | |
| Vehicle | 3 (2.8–3.2) | 4 (3.8–4.2) | 20 (19.6–20.4) |
| Magnolol (1
mg/kg) | 1.5
(1.1–1.9)a | 2 (1.2–2.8)a | 11
(9.1–12.9)a |
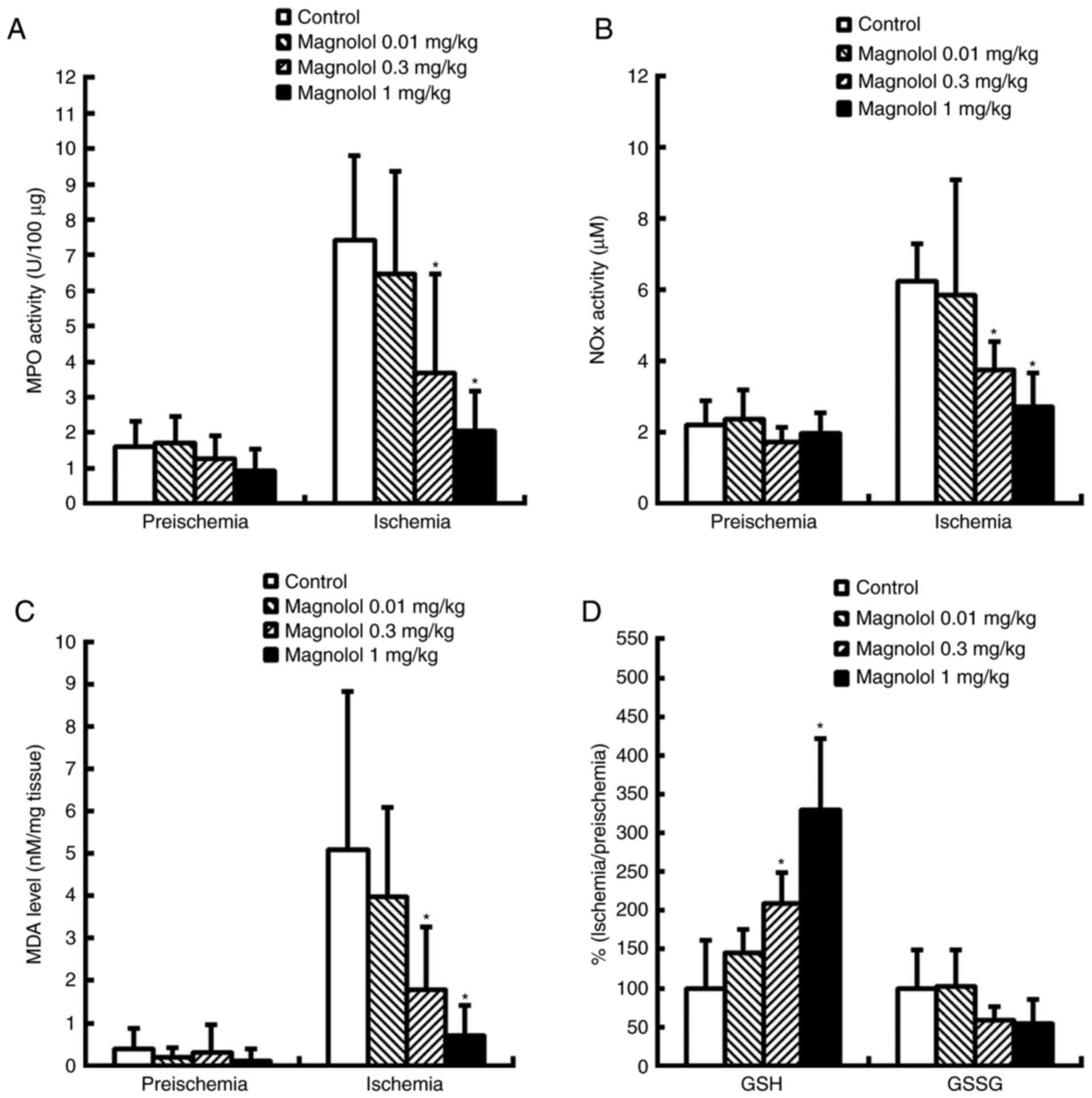
MPO and NOX levels in the
reperfused brain
In the control group, notably increased levels of
MPO and NOX activity were measure in the brain at 24 h
following the onset of ischemia, which is indicative of neutrophil
infiltration (Fig. 4A and B).
Treatment with magnolol (0.3–1 mg/kg) 2 h following the onset of
ischemia resulted in a significant reduction in the levels of MPO
and NOX at 24 h compared with the control group
(P<0.05). MPO and NOX were reduced by 50.4–72.5% and
40.0–56.7% respectively. No significant differences were identified
in MPO and NOX activity following treatment with
magnolol at a dosage of 0.01 mg/kg.
Lipid peroxidation and the GSH/GSSG ratio
are reduced in the reperfused brain
Ischemic-reperfusion insult induced a decrease in
the reduced glutathione level which was accompanied by an increase
in brain malondialdehyde (MDA), indicative of lipid peroxidation,
at 24 h following the onset of ischemia (Fig. 4C and D). Treatment with magnolol
at 0.3 and 1 mg/kg at 2 h after the onset of ischemia significantly
increased the GSH level, which provided protection for the ischemic
brain when measured at 24 h following ischemia onset, compared with
the control group (P<0.05; Fig.
4D); however, GSSG exhibited no significant difference between
the magnolol and control groups. GSH was increased by 109.7–229.8%
and MDA accumulation was reduced by 60.6–80.8% in the magnolol
treatment groups compared with the vehicle-treated controls.
Treatment with magnolol at 0.3 and 1 mg/kg led to significant
reductions in the level of MDA 24 h following ischemia (P<0.05;
Fig. 4C).
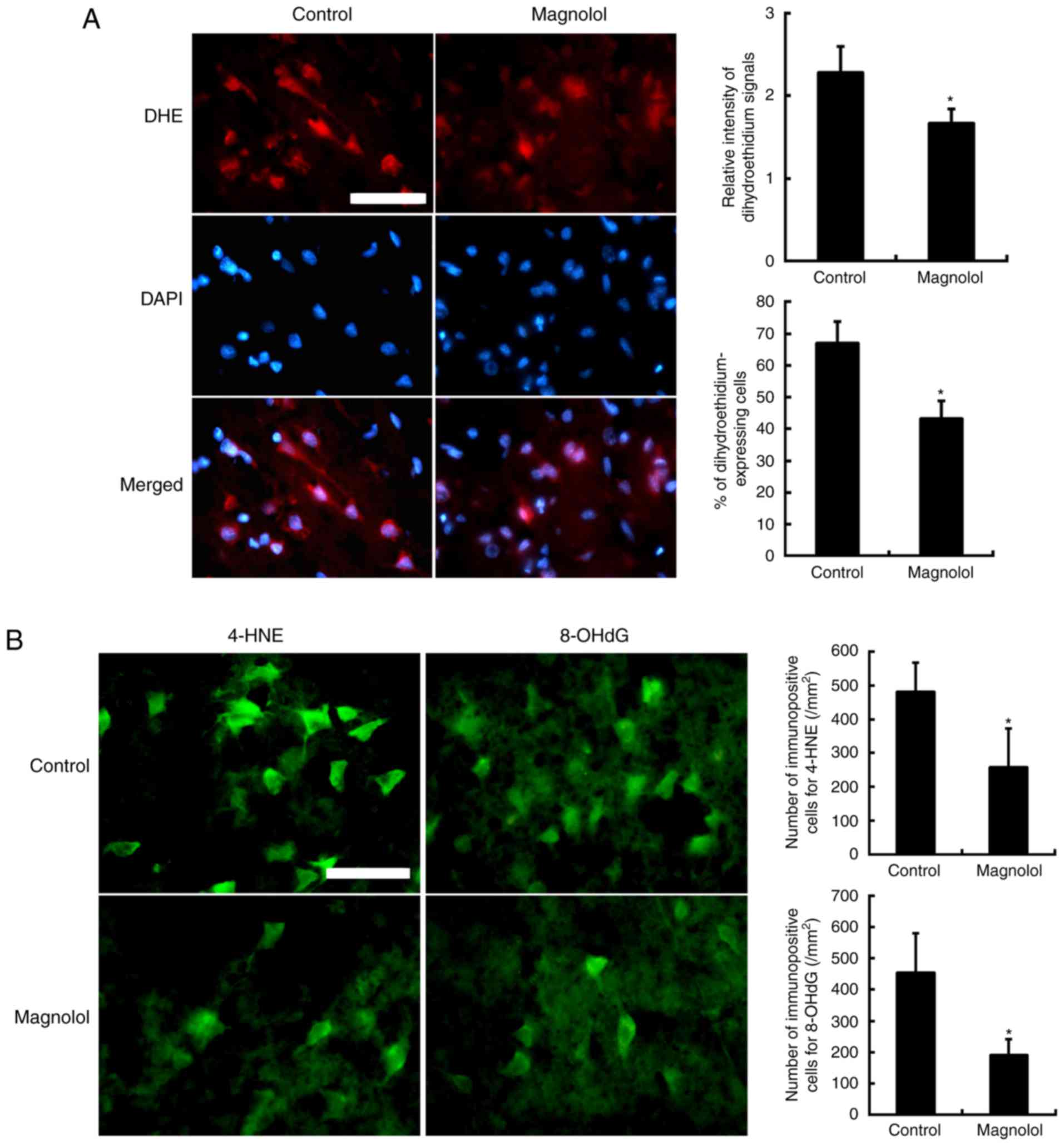
In situ superoxide (O2−) detection
In the fluorescent images, intracellular
O2− production in the perinuclear area
and cytosol was detected via oxidized DHE (HEt) which appeared red
and the nuclei were indicated by blue DAPI staining (Fig. 5A). The HEt signals were measured
in the cytosol. They were primarily localized to the nuclei and
also observed in the cytoplasm of the cells, which had a neuronal
morphology. HEt signals were observed in the axon- and
dendrite-like processes in the vehicle-treated control group.
Treatment with magnolol (1 mg/kg) at 2 h after the onset of
ischemia significantly reduced the relative intensity of the HEt
signals in the axon- and dendrite-like processes and the cytosol
(P<0.05). Data are expressed as the percentage of HEt-positive
cells relative to the number of DAPI-stained cells. Treatment with
magnolol (1 mg/kg) significantly reduced the percentage of
DHE-expressing cells compared with the vehicle-treated control
cells. Data are expressed as the ratio percentage of the cytosolic
HEt fluorescence intensity relative to the back-ground
intensity.
Immunofluorescence staining of 4-HNE and
8-OHdG
In the control group, the immunoreactivity of 8-OHdG
was localized to the nuclei in neurons (Fig. 5B). Clear immuno-reactivity of
4-HNE was observed in the region of ischemic damage in neuronal
dendrites and axons. Compared with the control, magnolol (1 mg/kg)
significantly reduced the number of 4-HNE and 8-OHdG positive cells
visible following immunofluorescence staining (P<0.05). The
proportion of cells stained with 4-HNE was reduced by 46.5% and the
proportion of cells stained with 8-OHdG was reduced by 58.0% in the
ischemic brain.
Discussion
The results of the present study indicate that
magnolol is an effective direct radical scavenger with antioxidant
action, that is able to inhibit the production of pro-inflammatory
cytokines and NOX in LPS-stimulated BV2 and RAW 264.7
cells, consistent with previous studies (7–9,12,15). It was also observed that
pretreatment with magnolol (100 µM) significantly reduced
OGD-induced damage in organotypic hippocampal slices. Additionally,
delayed treatment with magnolol (100 µM) at up to 4 h
post-insult also significantly reduced OGD-induced damage. In
animal models, magnolol (0.3–1 mg/kg) effectively reduced
post-stroke increases in oxidative and nitrosative damage, and
reduced neutrophil infiltration. In addition, the present study
indicated that magnolol upregulated the antioxidant reserves in the
ischemic brain, as measured by the level of GSH. Consequently,
magnolol effectively reduced in situ
O2− accumulation and attenuated
membranous lipid peroxidation and the hydroxylation of nucleotides
following cerebral ischemia-reperfusion. The i.v. administration of
magnolol (0.01–1 mg/kg) significantly improved the neurobehavioral
scores and significantly reduced the area of brain infarction in
the animals following transient MCA occlusion. This neuroprotection
remained when magnolol (1 mg/kg) was administered at 2 h following
the onset of ischemia.
A previous study by the present authors demonstrated
that magnolol administered intraperitoneally (i.p.) at 50–200
mg/kg, conferred neuroprotection in rats subjected to permanent
focal cerebral ischemia (15).
Magnolol administered i.p. at larger dosages of 100–200 mg/kg,
however, also induced spontaneous hypothermia (15,27). In the present study, magnolol
(0.01–1 mg/kg; i.v.) was effective against ischemic-reperfusion
insult in vivo. Notably, these i.v. injections of magnolol
at smaller dosages did not notably affect the rats' core body
temperatures. The difference between the dosages used for i.v. and
i.p. magnolol was ~1:200-fold, and further investigation is
required to determine the reason for this and to identify the most
effective dose and method of administration (17). The results of the present study
demonstrated that magnolol was effective when administered i.v. at
low doses (0.01–1 mg/kg), which is closer aligned with the clinical
treatment of ischemic stroke patients where i.v. tissue-type
plasminogen activator is used.
Magnolol protected the brain against
ischemic-reperfusion insults in vivo and in vitro.
The neuroprotective effect of magnolol in the present study cannot
be accounted for by changes in glucose, heart rate, arterial blood
pressure, hemodilution (as measured by blood hematocrit), or
changes in LCBF, as the values of these parameters exhibited no
significant differences between the vehicle-injected and
magnolol-treated groups. The magnolol-mediated neuroprotection
observed in the present study was also independent of the
magnolol-mediated hypothermic action observed previously when it
was administered at large dosages (200 mg/kg; i.p.) (15,27), as the core temperatures of the
rats did not markedly change. OGD-induced damage in the organotypic
hippocampal slices was also protected against by magnolol, which
had no contributing hypothermic parameters (15,20).
In the present study, it was revealed that magnolol
has a therapeutic window of 2–4 h for ischemic-reperfusion insults
in vitro. However, post-treatment with magnolol at 4 h
post-insult was ineffective following cerebral ischemia-reperfusion
insult in rats (data not shown). Thus, the in vivo and in
vitro therapeutic windows may not coincide with the field of
neuroprotection. A previous study reported that magnolol (100
mg/kg; i.p.) protected against permanent focal cerebral ischemia
when administered up to 4 h following the injury (15). It is possible that although
magnolol at a large dosage (100 mg/kg, i.p.) did not induce
hypothermia within 40 min of administration in the previous study,
it may have caused hypothermia at a later stage of administration,
thereby contributing to the extension of the therapeutic window of
opportunity to 4 h (15,28,29). Another theory is that the
reperfusion damage may have counteracted the magnolol-induced
neuroprotection (30,31). It is possible that a combination
of mild hypothermic therapy with magnolol at low dosages (0.01–1
mg/kg) that do not induce hypothermia may further enhance the
neuroprotection observed in the present study (28,29). However, this requires further
investigation to confirm. Further studies are also required to
clarify the mechanisms underlying the discrepancy between the in
vivo and in vitro data obtained in the present study
concerning the therapeutic windows.
The present study has demonstrated that magnolol at
0.01 mg/kg is able to protect the brain against transient focal
cerebral ischemia without affecting the MDA, MPO, NOX,
and GSH levels in the ischemic brain. These findings suggest that a
different dosing regimen may present with different pharmacologic
mechanisms of action (15,21).
Additional studies are required to investigate other mechanisms,
including the ability to reduce necrosis, autophagy or apoptosis
that may also account for the neuroprotective effect of magnolol
observed in the present study.
In summary, the present study has revealed that
magnolol is a potent radical-scavenger and antioxidant as evaluated
by antioxidant activity assays in vitro and a series of
anti-oxidant reserve, lipid peroxidation and nitrosative assays
in vivo. Magnolol also demonstrated clear anti-nitrosative
and -inflammatory effects by suppressing the NOX and MPO
activity induced by ischemic insults in vivo, and by
reducing the production of NO and proinflammatory cytokines in
LPS-stimulated BV2 and RAW 264.7 cells. Additionally, it was
demonstrated that the therapeutic window of magnolol was up to 2–4
h, and that magnolol protects against ischemic brain damage, as
assessed by the whole animal and organotypic tissue-based assays of
transient ischemic stroke, respectively. Consequently, treatment
with magnolol resulted in a notable reduction in the accumulation
of ROS following ischemia and therefore, attenuated the extent of
lipid peroxidation, oxidative DNA damage and final neuronal deaths
in the ischemic brain tissues. This indicates that the ability of
magnolol to scavenge free radicals may be responsible for its
neuroprotective effects following ischemia.
Acknowledgments
The present study was supported by a grant from the
National Science Council of Taiwan (grant no.
99-2314-B-006-022-MY3).
Notes
[1] Competing
interests
The authors declare there is no competing
interest.
References
|
1
|
Traystman RJ, Kirsch JR and Koehler RC:
Oxygen radical mechanisms of brain injury following ischemia and
reperfusion. J Appl Physiol (1985). 71:1185–1195. 1991. View Article : Google Scholar
|
|
2
|
Bramlett HM and Dietrich WD:
Pathophysiology of cerebral ischemia and brain trauma: Similarities
and differences. J Cereb Blood Flow Metab. 24:133–150. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Clemens JA: Cerebral ischemia: Gene
activation, neuronal injury, and the protective role of
antioxidants. Free Radic Biol Med. 28:1526–1531. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chan PH: Reactive oxygen radicals in
signaling and damage in the ischemic brain. J Cereb Blood Flow
Metab. 21:2–14. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee EJ, Chen HY, Lee MY, Chen TY, Hsu YS,
Hu YL, Chang GL and Wu TS: Cinnamophilin reduces oxidative damage
and protects against transient focal cerebral ischemia in mice.
Free Radic Biol Med. 39:495–510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee EJ, Lee MY, Chen HY, Hsu YS, Wu TS,
Chen ST and Chang GL: Melatonin attenuates gray and white matter
damage in a mouse model of transient focal cerebral ischemia. J
Pineal Res. 38:42–52. 2005. View Article : Google Scholar
|
|
7
|
Shen YC, Sung YJ and Chen CF: Magnolol
inhibits Mac-1 (CD11b/CD18)-dependent neutrophil adhesion:
Relationship with its antioxidant effect. Eur J Pharmacol.
343:79–86. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang Y, Li CY, Lin IH, Lee AR and Hu MK:
Synthesis and radical scavenging of novel magnolol derivatives. J
Pharm Pharmacol. 54:1697–1703. 2002. View Article : Google Scholar
|
|
9
|
Chen HY, Hung YC, Lee EJ, Chen TY, Chuang
IC and Wu TS: The protective efficacy of magnolol in hind limb
ischemia-reperfusion injury. Phytomedicine. 16:976–981. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chang CP, Hsu YC and Lin MT: Magnolol
protects against cerebral ischaemic injury of rat heatstroke. Clin
Exp Pharmacol Physiol. 30:387–392. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Teng CM, Yu SM, Chen CC, Huang YL and
Huang TF: EDRF-release and Ca+(+)-channel blockade by magnolol, an
antiplatelet agent isolated from Chinese herb Magnolia officinalis,
in rat thoracic aorta. Life Sci. 47:1153–1161. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee MM, Huang HM, Hsieh MT, Chen CS, Yeh
FT and Kuo JS: Anti-inflammatory and neuroprotective effects of
magnolol in chemical hypoxia in rat cultured cortical cells in
hypoglycemic media. Chin J Physiol. 43:61–67. 2000.PubMed/NCBI
|
|
13
|
Lee MM, Hseih MT, Kuo JS, Yeh FT and Huang
HM: Magnolol protects cortical neuronal cells from chemical hypoxia
in rats. Neuroreport. 9:3451–3456. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin YR, Chen HH, Ko CH and Chan MH:
Neuroprotective activity of honokiol and magnolol in cerebellar
granule cell damage. Eur J Pharmacol. 537:64–69. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee WT, Lin MH, Lee EJ, Hung YC, Tai SH,
Chen HY, Chen TY and Wu TS: Magnolol reduces glutamate-induced
neuronal excitotoxicity and protects against permanent focal
cerebral ischemia up to 4 hours. PLoS One. 7:e399522012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Watanabe K, Watanabe H, Goto Y, Yamaguchi
M, Yamamoto N and Hagino K: Pharmacological properties of magnolol
and honokiol extracted from Magnolia officinalis: Central
depressant effects. Planta Med. 49:103–108. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lin SP, Tsai SY, Lee Chao PD, Chen YC and
Hou YC: Pharmacokinetics, bioavailability, and tissue distribution
of magnolol following single and repeated dosing of magnolol to
rats. Planta Med. 77:1800–1805. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chapman SN, Mehndiratta P, Johansen MC,
McMurry TL, Johnston KC and Southerland AM: Current perspectives on
the use of intravenous recombinant tissue plasminogen activator
(tPA) for treatment of acute ischemic stroke. Vasc Health Risk
Manag. 10:75–87. 2014.PubMed/NCBI
|
|
19
|
Lee EJ, Chen HY, Hung YC, Chen TY, Lee MY,
Yu SC, Chen YH, Chuang IC and Wu TS: Therapeutic window for
cinnamophilin following oxygen-glucose deprivation and transient
focal cerebral ischemia. Exp Neurol. 217:74–83. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen TY, Lin MH, Lee WT, Huang SY, Chen
YH, Lee AC, Lin HW and Lee EJ: Nicotinamide inhibits nuclear
factor-kappa B translocation after transient focal cerebral
ischemia. Crit Care Med. 40:532–537. 2012. View Article : Google Scholar
|
|
21
|
Tai SH, Hung YC, Lee EJ, Lee AC, Chen TY,
Shen CC, Chen HY, Lee MY, Huang SY and Wu TS: Melatonin protects
against transient focal cerebral ischemia in both reproductively
active and estrogen-deficient female rats: The impact of
circulating estrogen on its hormetic dose-response. J Pineal Res.
50:292–303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen TY, Tai SH, Lee EJ, Huang CC, Lee AC,
Huang SY and Wu TS: Cinnamophilin offers prolonged neuroprotection
against gray and white matter damage and improves functional and
electrophysiological outcomes after transient focal cerebral
ischemia. Crit Care Med. 39:1130–1137. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Juan WS, Huang SY, Chang CC, Hung YC, Lin
YW, Chen TY, Lee AH, Lee AC, Wu TS and Lee EJ: Melatonin improves
neuroplasticity by upregulating the growth-associated protein-43
(GAP-43) and NMDAR postsynaptic density-95 (PSD-95) proteins in
cultured neurons exposed to glutamate excitotoxicity and in rats
subjected to transient focal cerebral ischemia even during a
long-term recovery period. J Pineal Res. 56:213–223. 2014.
View Article : Google Scholar
|
|
24
|
Anderson ME: Determination of glutathione
and glutathione disulfide in biological samples. Methods Enzymol.
113:548–555. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Belayev L, Alonso OF, Busto R, Zhao W and
Ginsberg MD: Middle cerebral artery occlusion in the rat by
intraluminal suture. Neurological and pathological evaluation of an
improved model. Stroke. 27:1616–1623. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Clark WM, Rinker LG, Lessov NS, Hazel K,
Hill JK, Stenzel-Poore M and Eckenstein F: Lack of interleukin-6
expression is not protective against focal central nervous system
ischemia. Stroke. 31:1715–1720. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hsieh MT, Chueh FY and Lin MT: Magnolol
decreases body temperature by reducing 5-hydroxytryptamine release
in the rat hypothalamus. Clin Exp Pharmacol Physiol. 25:813–817.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Danton GH and Dietrich WD: Inflammatory
mechanisms after ischemia and stroke. J Neuropathol Exp Neurol.
62:127–136. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ishikawa M, Sekizuka E, Sato S, Yamaguchi
N, Inamasu J, Bertalanffy H, Kawase T and Iadecola C: Effects of
moderate hypothermia on leukocyte-endothelium interaction in the
rat pial microvasculature after transient middle cerebral artery
occlusion. Stroke. 30:1679–1686. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nakashima M, Niwa M, Iwai T and Uematsu T:
Involvement of free radicals in cerebral vascular reperfusion
injury evaluated in a transient focal cerebral ischemia model of
rat. Free Radic Biol Med. 26:722–729. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sugawara T and Chan PH: Reactive oxygen
radicals and pathogenesis of neuronal death after cerebral
ischemia. Antioxid Redox Signal. 5:597–607. 2003. View Article : Google Scholar : PubMed/NCBI
|