Introduction
Benign prostatic hypertrophy (BPH) is the most
common benign tumor in males, characterized by an increased number
of glandular and connective cells in the prostate and (1). BPH typically causes prostate
enlargement and the secondary condition of chronic bladder outlet
obstruction (2). In addition,
urinary retention, gross hematuria, renal insufficiency and bladder
cancer may arise. BPH may also lead to prostate cancer, which is
the leading cause mortality due to cancer in males (3,4). A
previous study demonstrated that the pathogenesis of BPH is
multifactorial, with factors including age, diabetes, obesity and
ethnicity influencing its development (5). Reports on the association between
diabetes and BPH are conflicting; some have suggested that there is
a positive association (6),
whereas others have reported no association (7).
Type 2 diabetes mellitus (T2D), characterized by
persistent hyperglycemia, affects >370 million people worldwide
and is a major cause of morbidity and mortality (8). T2D is a disease in which tissues are
resistant to the effects of insulin and/or insulin production is
insufficient to meet requirements (1). The consequence of this is an
inability to regulate blood sugar, resulting in chronic
hyperglycemia. For many decades, therapeutic strategies aimed at
decreasing glucose levels have focused on agents that stimulate the
release of insulin from the pancreas or improve insulin-stimulated
glucose uptake in peripheral tissues (9).
Little data is available regarding the association
between diabetes and BPH; however, statistical analysis suggests
that there is an overlap in males who suffer from BPH and diabetes,
indicating that there may be an existing association between these
diseases (10-12). Human genetic studies allow for the
investigation of novel molecules and pathways that contribute to
natural variation in specific traits, including blood glucose
levels or the risk of disease. According to numerous studies, the
number of genes involve in the pathogenic processes of T2D have
been reported. For example, phosphoinositide 3-kinase/protein
kinase B signaling activates forkhead box Os (FoxOs), which
protects against oxidative stress to prevent the death of β
pancreatic cells (13-15). FoxO1 has been reported to control
various cellular processes, including the stress response,
proliferation and differentiation (16,17). As nutrient and FoxO factors are
closely associated, FoxO may also serve an important role in
cell-secreted hormones that regulate glucose homeostasis. These
genes are important for regulating glucose homeostasis and may
therefore be associated with diabetes and potentially BPH. However,
few findings have validated the genes involved in diabetes and BPH.
In our previous study (18), a
cDNA microarray analysis was performed to identify the effects of
diabetes on gene expression profiles in prostate samples. A cell
culture model (human normal prostatic RWPE-1 cell line) was also
employed to study the direct effects of high glucose (14). The results indicated that high
glucose was associated with the downregulation of antioxidative
enzymes and DNA damage repair genes, including MRE11.
In the present study, an in vitro experiment
was conducted using prostatic cell lines RWPE-1 and HPr-1 to verify
the effects of high-glucose solution. Transcriptome sequencing was
subsequently performed to screen the candidate genes that may be
involved in the pathogenic process of prostatic enlargement.
Following validation of the candidate genes by transfection of an
overexpression vector, an in vivo experiment was performed
using rats with streptozotocin (STZ)-induced T2D to further
invetsigate the role of these genes in diabetes.
Materials and methods
Cell culture
Normal prostatic epithelial RWPE-1 and HPr-1 cells
(Institute of Biochemistry and Cell Biology, Chinese Academy of
Sciences, Shanghai, China) were cultured in complete keratinocyte
serum-free medium (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany),
supplemented with 1% penicillin/streptomycin/amphotericin B, 50
μg/ml bovine pituitary extract (Absin Bioscience, Inc.,
Shanghai, China) and 5 ng/ml epidermal growth factor (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The cells were incubated at
37°C in an atmosphere containing 5% CO2. When cells
reached 70-80% confluence they were washed with PBS and detached
using 0.25% trypsin/0.2% EDTA. Cells were viewed under a light
microscope (magnification, ×200; Eclipse T1, Nikon Corporation,
Tokyo, Japan), and subsequently suspended at a concentration of
1×106 cells/ml.
MTS and bromodeoxyuridine (BrdU)
proliferation activity assays
Cell proliferation was analyzed via an MTS assay.
Cells were seeded at a density of 4×103 cells/well in
96-well plates. The high-glucose solution was diluted with culture
medium at 0, 5.5, 25, 50 and 75 mM, which was added to each well
and incubated for 48 h at 37°C. Following determination of the
optimal concentration, cells were incubated with 50 mM high glucose
solution (Sigma-Aldrich; Merck KGaA) for 0, 24, 48 and 72 h.
For the MTS assay, 20 μl MTS reagent (Promega
Corp., Madison, WI, USA) was added to the cells and incubated for 3
h at 37°C in an atmosphere containing 5% CO2. The plates
were then read at 490 nm with a microplate reader (Thermo Fisher
Scientific, Inc.). Each sample was analyzed in triplicate.
A BrdU Cell Proliferation Assay kit was used
according to the manufacturer's protocols (BioVision, Inc.,
Milpitas, CA, USA). Briefly, 10X BrdU solution was added to the
wells and incubated for 3 h at 37°C in an atmosphere containing 5%
CO2. The culture medium was removed and 100 μl
fixing/denaturing solution was added to each well at room
temperature for 30 min. The supernatant was removed and 100
μl 1X BrdU detection antibody solution was added. Cells were
incubated at room temperature for a further 1 h. Prior to washing
with 1X wash buffer twice, the supernatant was removed. A total of
100 μl 1X anti-mouse horseradish-peroxidase-linked antibody
solution (provided in the kit) was added to each well and incubated
at room temperature for 1 h. The supernatant was removed and cells
were washed three times with wash buffer.
3,3′,5,5′-tetramethylbenzidine substrate (100 μl) was added
into each well and absorbance was measured at 650 nm for 5-30 min.
To terminate color development, 100 μl stop solution was
added into each well and the absorbance at 450 nm was measured.
Apoptosis and cell cycle assay
Flow cytometric analysis was performed to assess
apoptosis and cell cycle distribution in RWPE-1 and HPr-1 cells.
The cells were seeded at a density of 1×106 cells/well
in 6-well plates and allowed to attach for 48 h at 37°C prior to
the onset of treatments. Following treatment, cells were detached
using EDTA-free trypsin for 5 min at room temperature. Apoptosis
was assessed using an Annexin V assay kit (FITC Annexin V Apoptosis
Detection Kit with 7-AAD, BD Biosciences, Franklin Lakes, NJ, USA)
according to manufacturer's protocols. For the cell cycle assay,
cells were centrifuged at 500 × g for 10 min at 4°C and
subsequently incubated in 500 μl hypotonic DNA staining
buffer (1 g/l sodium citrate, 0.3% Triton-X 100, 0.1 g/l propidium
iodide, 0.02 g/l Ribonuclease A in distilled water; BD Biosciences)
for 30 min at 4°C. Analyses were performed with a BD Accuri C6 Flow
Cytometer (BD Biosciences). Cell cycle data were analyzed with
FlowJo software v 7.6 (Tree Star, Inc., Ashland, OR, USA).
Transcriptome sequencing
RNA was extracted from cells using TRIzol
(Invitrogen; Thermo Fisher, Scientific, Inc.) following the
manufacturer's protocols. RNA was quantified using a Qubit RNA
assay (Thermo Fisher Scientific, Inc.) and the quality was
evaluated by Agilent bioanalyzer 2100 (Agilent Technologies, Inc.,
Santa Clara, CA, USA). A total of ~1,000 ng RNA was used for
library preparation with the VAHTST mRNA-seq v2 Library Prep kit
(Illumina, Inc., San Diego, CA, USA). Briefly, RNA with a polyA
tail was captured using Dynaloligo beads (Invitrogen; Thermo Fisher
Scientific, Inc.). The RNA was fragmented and reverse transcribed
into double-strand cDNA using SuperScript III cDNA Synthesis kit at
70°C for 5 min (Thermo Fisher Scientific, Inc.). Thermocycling
conditions were as follows: 30 cycles of 94°C for 15-30 sec,
55-65°C for 30 sec and 72°C for 1 min. The ends of the cDNA were
repaired by the End-It DNA EndRepair kit (End-It DNA END Repair
kit; Epicentre; Illumina, Inc.) and treated via A-addition. The
modified cDNA was ligated to adapters and subjected to quantitative
polymerase chain reaction (qPCR) amplification (described later).
The library quality was determined using the Agilent bioanalyzer
2100. The libraries were sequenced with an Illumina HiSeq 4000
system. Reads were aligned to the reference genome (GRCH37/hg19) by
TOPHAT v.2.1.0 (19). The reads
per kilobase of exon model per million mapped reads of each gene
were calculated. The differential expression signals were
calculated and P-values were adjusted using Benjamini and
Hochberg's approach (20).
Enrichment of the differentially expressed genes in Kyoto
Encyclopedia of Genes and Genomes pathway was tested using KOBAS
2.0 software (KOBAS, London, UK).
Lentivirus transfection
The full length of MRE11 cDNA was cloned into
pCDNA3.1 vector (Thermo Fisher Scientific, Inc.). 293T cells
(Thermo Fisher Scientific, Inc.) were seeded in a 10 cm plate and
transfected with the pCDNA3.1 vector using RNAi-Mate transfection
reagent (Thermo Fisher Scientific, Inc.). Cells were cultured at
37°C until they covered 80-90% of the plate, at which point they
were then harvested with trypsin and amplified in 15 cm plate at
37°C in an atmosphere containing 5% CO2 overnight. The
pCDNA3.1-MRE11 vector and package vectors (pGag/Pol, pRev and
pVSV-G; GenePharm, Inc., Sunnyvale, CA, USA) were mixed in 1.5 ml
Dulbecco's modified Eagle's medium (DMEM) without serum (GenePharm,
Inc.) and incubated at room temperature for 5 min. A total of 300
μl of RNAi-Mate (GenePharm, Inc.) was prepared and diluted
in 1.5 ml DMEM, which was then mixed with the vectors mixture and
incubated for 20-25 min at room temperature to form the
transfection complex. The cell culture medium was removed and
replaced with 8 ml DMEM. The transfection complex was added to the
cells and incubated for 4-6 h at 37°C in an atmosphere containing
5% CO2. The culture medium was removed and replaced with
RPMI-1640 medium with serum (Thermo Fisher Scientific, Inc.), and
incubated for 72 h at 37°C in an atmosphere containing 5%
CO2. The cell culture supernatant was collected and
centrifuged at 1,788 × g for 4 min at 4°C. The supernatant was
filtered through a 0.45 μm filter and subsequently
centrifuged at 4,472 × g for 2 h at 4°C. The viral solution was
stored at −80°C prior to use. 293T cells were seeded in 96-well
plates at a density of 3×104 cells/well. The viral
solution was diluted 10× with RPMI-1640 medium to give five
different concentrations and the final titer was 2×108
TU/ml. The culture medium was removed from each well and
supplemented with 100 μl of the viral solution. Normal
saline was used as control. Cells were incubated at 37°C in an
atmosphere containing 5% CO2 for 24 h. The cell culture
supernatant was removed and replaced with 100 μl RPMI-1640
medium. Cells were incubated for 72 h at 37°C. Fluorescence
microscopy (magnification, ×200) was used to detect fluorescence
and the titer of virus was calculated according to the dilution
ratio. RWPE-1 cells were then transfected with the lentivirus to
generate MRE11-overexpressing cells when the cell density reached
60-70%.
Animal experiment
All animal experiments were approved by the
Institutional Animal Care and Use Committee of Kunming Medical
University. A total of 20 male Sprague Dawley rats (12 weeks old;
weight, 230-280 g) were obtained from Sun Yay-sen University
Experimental Animal Center (Guangzhou, China). The rats were
maintained at a constant temperature of 24°C, 45-55% humidity with
a 12 h light/dark cycle and were fed standard food pellets with
free access to sterile water. Rats were randomly divided into two
groups (n=6-12). Diabetes was induced by intraperitoneal injection
with STZ (60 mg/kg, Sigma Aldrich; Merck KGaA). Blood glucose
levels were monitored by harvesting blood samples at weeks 2 and 4
to confirm the presence of diabetes. The rats were sacrificed by an
intraperitoneal injection of pentobarbital (160 mg/kg, Sigma
Aldrich; Merck KGaA) at 4 weeks following the induction of
diabetes. Prostate tissues were harvested and stored at −80°C for
qPCR assay or fixed in formalin for 24-48 h at room temperature and
sectioned (2.5 μm) for immunohistochemical staining.
Immunohistochemical staining
The formalin-fixed paraffin-embedded tissues were
deparaffinized and washed twice with PBS. The tissue was blocked
with 3% H2O2 for 5-20 min at room temperature
and subsequently washed three times with PBS. Tissues were fixed
via water incubation at 95°C for 10 min and washed with PBS. The
tissue was then blocked with 5% BSA for 10 min at room temperature
(Sigma Aldrich; Merck KGaA) and incubated with primary antibodies
(Anti-Mre11, ab109623, 1:1,000, Abcam, Cambridge, MA, USA;
anti-PCNA, sc-25280, 1:500, Santa Cruz Biotechnology, Inc., Dallas,
TX, USA) at 4°C overnight. Following three washes with PBS, the
tissue was incubated with a horseradish peroxidase-conjugated
secondary antibody secondary antibody (SV0002; 1:1,000; Wuhan
Boster Biological Technology, Ltd., Wuhan, China) for 1 h at room
temperature. Following washing with PBS, the sections were
developed in freshly prepared 3,3′-diaminobenzidine solution for 30
sec at room temperature and then counterstained with hematoxylin
for 45 sec at room temperature, rehydrated with a graded series of
ethanol, washed with xylene and mounted at room temperature. The
expression of MRE-11 and PCNA was estimated by light microscopy
(magnification, ×200).
Reverse transcription (RT)-qPCR
assay
Total RNA from sampled cells and tissues was
extracted using TRIzol according to the manufacturer's protocols.
Genomic DNA-free RNA was then converted into cDNA using the M-MLV
Reverse Transcriptase at 42°C for 60 min (Promega Corp.). qPCR was
performed using SYBR Green PCR Master Mix (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). β-actin was used as the reference gene.
Primers sequences were as follows: MRE11, forward
5′-AACGGGAACGTCTGGGTAAT-3′ and reverse
5′-GGCTAAAGCGAAGAACACTGAA-3′; β-actin, forward
5′-CATGTACGTTGCTATCCAGGC-3′ and reverse
5′-CTCCTTAATGTCACGCACGAT-3′. The thermo cycling conditions were as
follows: 95°C for 2 min followed by 30 cycles of 95°C for 30 sec,
60°C for 30 sec and 72°C for 1 min and 72°C for 1 min with a final
extension step at 72°C for 10 min. Fold changes in expression were
calculated using the 2-∆∆Cq method (21) using iCycler iQ software v.3.0
(Bio-Rad Laboratories, Inc.).
Western blot analysis
Protein was extracted from cells and tissues and the
concentration was determined using a BCATM-Protein Assay kit
(Guangzhou Fulengen, Co., Ltd., Guangzhou, China). A total of 10
μl protein was subjected to 10% SDS-PAGE. Protein was then
transferred onto nitrocellulose membranes, which was blocked with
5% non-fat milk in phosphate-buffered saline containing 0.1%
Tween-20 for 1 h at room temperature. Subsequently, the membranes
were incubated overnight at 4°C with the following primary
antibodies: Anti-Mre11, anti-phosphorylated checkpoint kinase 2
(p-CHK2; CST Biological Reagents Co., Ltd., Shanghai, China),
anti-p-M-phase inducer phosphatase 1 (CDC25A; ab75743; Abcam),
anti-CDC25A (BA3149; Wuhan Boster Biological Technology)
anti-p-cyclin dependent kinase (CDK2; 2561; CST Biological Reagents
Co., Ltd.), anti-CDK2 (ab32147; Abcam) and anti-GAPDH (HC301;
TransGen Biotech, Inc., Beijing, China). All primary antibodies
were used at a dilution of 1:1,000. Following three washes with
TBST, membranes were incubated with a horseradish
peroxidase-conjugated secondary antibody (1:2,000; ab6789; Abcam)
for 2 h at room temperature. GAPDH was used as the loading control.
The complexes were detected by enhanced chemiluminescence
(Forevergen, Guangzhou, China). ImageJ v.1.6.0 (National Institutes
of Health, Bethesda, MD, USA) was used for densitometry.
Statistical analysis
All data are presented as the mean ± standard
deviation and at least three replicates of each assay were
performed. Normally distributed data were compared using
independent t-tests, one-way analysis of variance (ANOVA) or
two-way ANOVA and Student Newman Keul's post hoc test. For
abnormally distributed data, the non-parametric Mann-Whitney
U-test, Kolmogorov-Smirnov test, Kruskal-Wallis test and Wilcoxon
test were performed. P<0.05 was considered to indicate a
statistically significant difference. SPSS 18.0 (SPSS, Inc.,
Chicago, IL, USA) and GraphPad Prism software 6.0 (GraphPad
Software, Inc., La Jolla, CA, USA) were used for statistical
analysis.
Results
High glucose concentration induces the
proliferation of RWPE-1 and HPr-1 cells
Cell viability was determined using MTS and BrdU
methods (Fig. 1). Incubation in
high glucose medium significantly induced the proliferation of
RWPE-1 (Fig. 1A–D) and HPr-1
(Fig. 1E–H) cells in a
dose-dependent manner. A concentration of 50 mM of glucose was
determined to be the optimal concentration, as treatment with 75 mM
did not markedly increase cell viability.
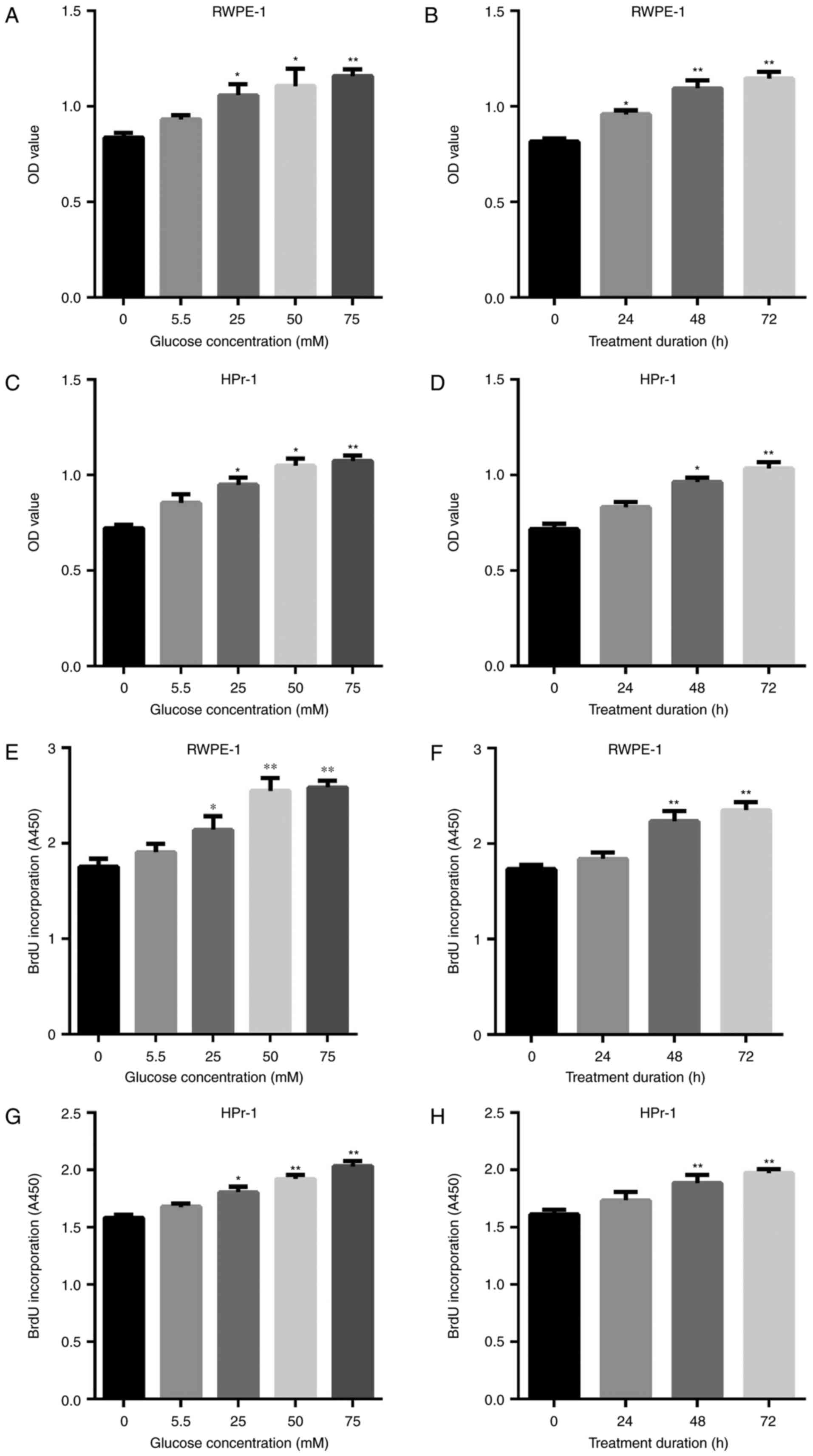 | Figure 1Cell viability was assessed using MTS
and BrdU assays to determine the optimal concentration and duration
of treatment with high glucose solution. (A) RWPE-1 cells were
treated with 0, 5.5, 25, 50 or 75 mM glucose for 48 h or (B) for 0,
24, 48 or 72 h with 50 mM glucose and assessed using an MTS assay.
(C) HPr-1 cells were treated with 0, 5.5, 25, 50 or 75 mM glucose
for 48 h or (D) for 0, 24, 48 or 72 h with 50 mM glucose and
assessed using an MTS assay. (E) RWPE-1 cells were treated with 0,
5.5, 25, 50 or 75 mM glucose for 48 h or (F) for 0, 24, 48 or 72 h
with 50 mM glucose and assessed using a BrdU assay. (G) HPr-1 cells
were treated with 0, 5.5, 25, 50 or 75 mM glucose for 48 h or (H)
for 0, 24, 48 or 72 h with 50 mM glucose and assessed using a BrdU
assay. *P<0.05 and **P<0.01 vs. control
(0 mM or 0 h). BrdU, bromodeoxyuridine; OD, optical density. |
To further assess the effect of high glucose, flow
cytometry was performed (Fig. 2).
Incubation with high glucose solution markedly increased the number
of cells at G2 phase and decreased the cell number at G1 phase. No
significant differences were observed in the number of the cells at
S phase. For both cell lines, the effect was significantly induced
with 50 mM glucose solution and higher concentrations did not
markedly increase the number of cells at G2 phase. Therefore, 50 mM
of glucose solution may be the optimal concentration to enhance
cell proliferation. In the present study, 50 mM glucose solution
was used to verify the optimal duration of treatment. The results
of flow cytometry analysis demonstrated that cell number peaked at
48 h and did not markedly increase with 72 h treatment.
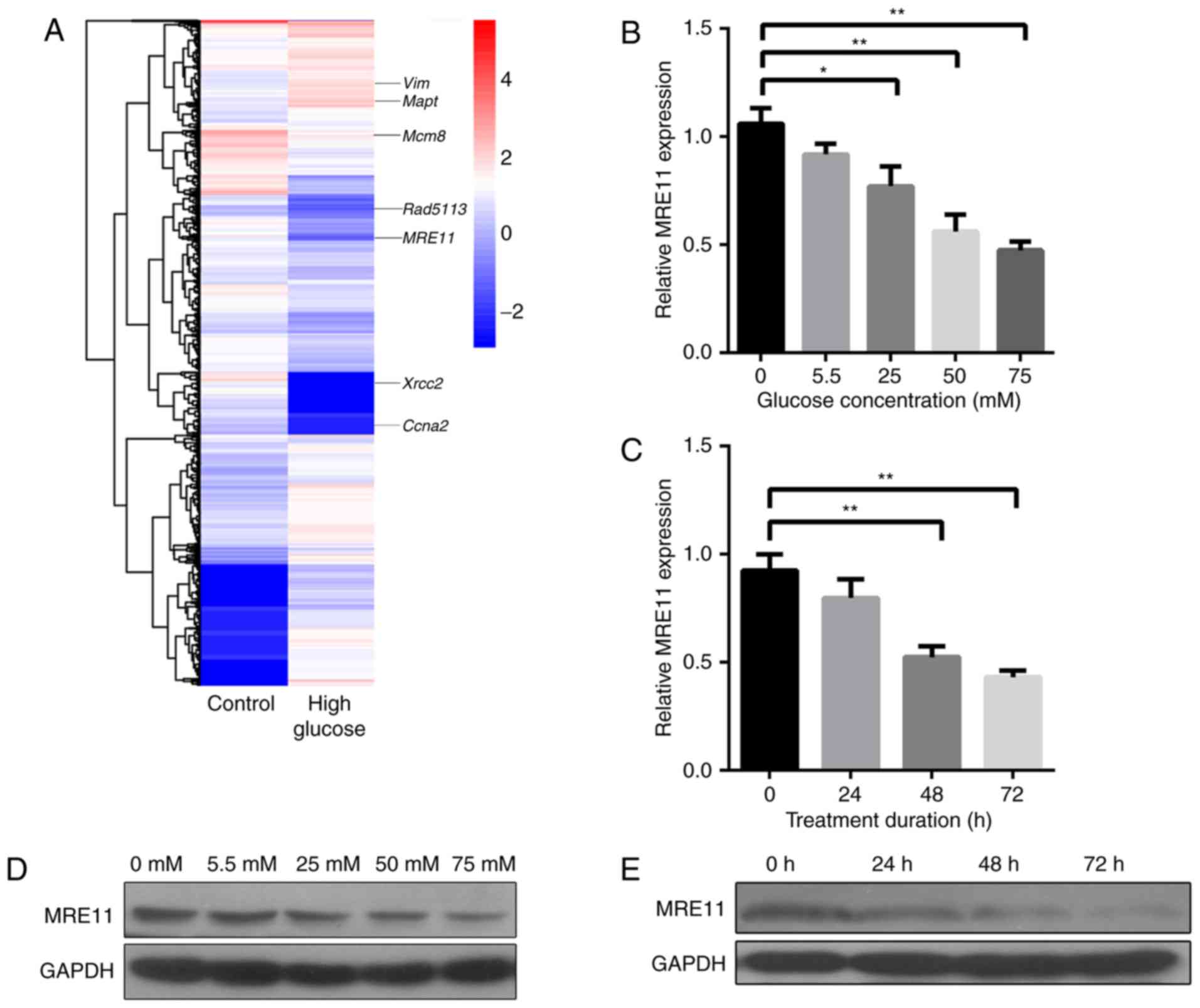
MRE11 is downregulated under high glucose
conditions
To verify the underlying mechanism of high-glucose
induced cell proliferation, transcriptome sequencing was performed
(Fig. 3). Experimental RWPE-1
cells were treated with 50 mM glucose solution for 48 h and control
cells were treated with saline. A hierarchical cluster graph was
generated for the two groups of cells (Fig. 3A). According to the results, MRE11
was significantly downregulated in the experimental group,
indicating that high concentration glucose solution may enhance
cell proliferation by downregulating MRE11.
To further assess MRE11 expression, RT-qPCR and
western blotting were performed. The results were consistent with
those of transcriptome sequencing. As presented in Fig. 3B, the expression of MRE11 mRNA was
decreased in response to treatment with high-glucose solution. At
50 mM, the mRNA expression levels of MRE11 were reduced by ~50%
compared with the control group (Fig.
3B). Similarly, the expression of MRE11 decreased to ~50% in
response to treatment with 50 mM glucose solution for 48 h
(Fig. 3C). Protein expression
followed a similar trend to mRNA (Fig. 3D and E). These results suggest
that MRE11 expression is downregulated by high glucose
treatment.
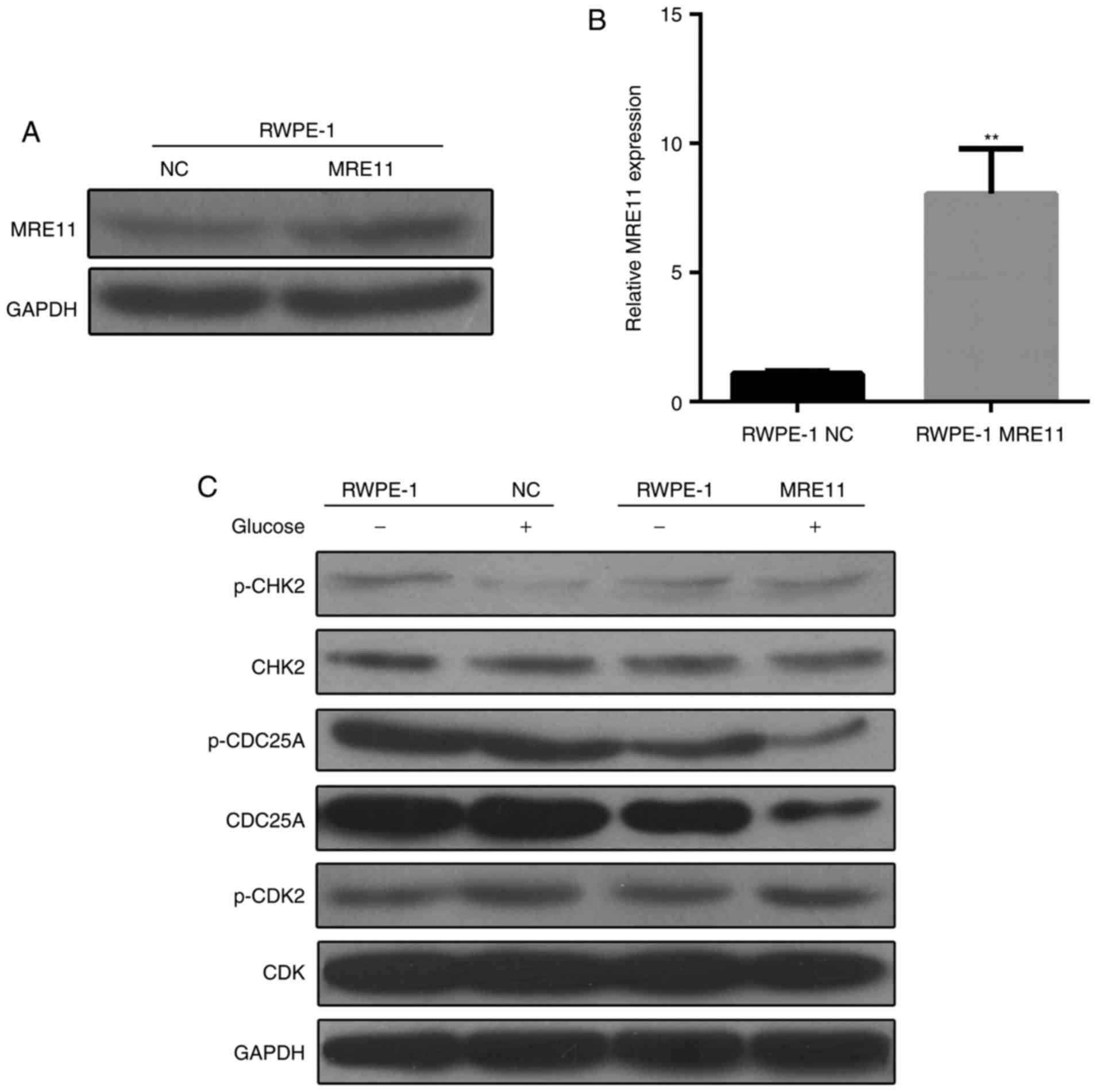
MRE11 induces cell apoptosis following
high glucose treatment
The role of MRE11 in proliferation regulation
remains to be elucidated. In the present study, RWPE-1 cells
underwent transfection to overexpress MRE11. The vector was
successfully transfected and cells expressed ~10-fold move MRE11
compared cells transfected with at empty vector (Fig. 4A and B). Cells were further
analyzed to detect the expression of genes of the MRE11 regulation
network (Fig. 4C). When high
glucose treatment was performed, the expression of p-CHK2 was
markedly downregulated in normal RWPE-1 cells; however, no
significant difference was observed in the MRE11-overexpressing
cells regardless of glucose treatment. These results suggest that
the expression of p-CHK2 is dependent on MRE11 levels, as high
glucose was demonstrated to downregulate MRE11. However, CDC25A
expression was downregulated when MRE11 was overexpressed and high
glucose treatment was applied. The results of the present study
indicate that MRE11 may regulate p-CHK2 and CDC25A expression
levels under high glucose conditions. No obvious differences were
observed in the expression of any other genes between groups
(Fig. 4C).
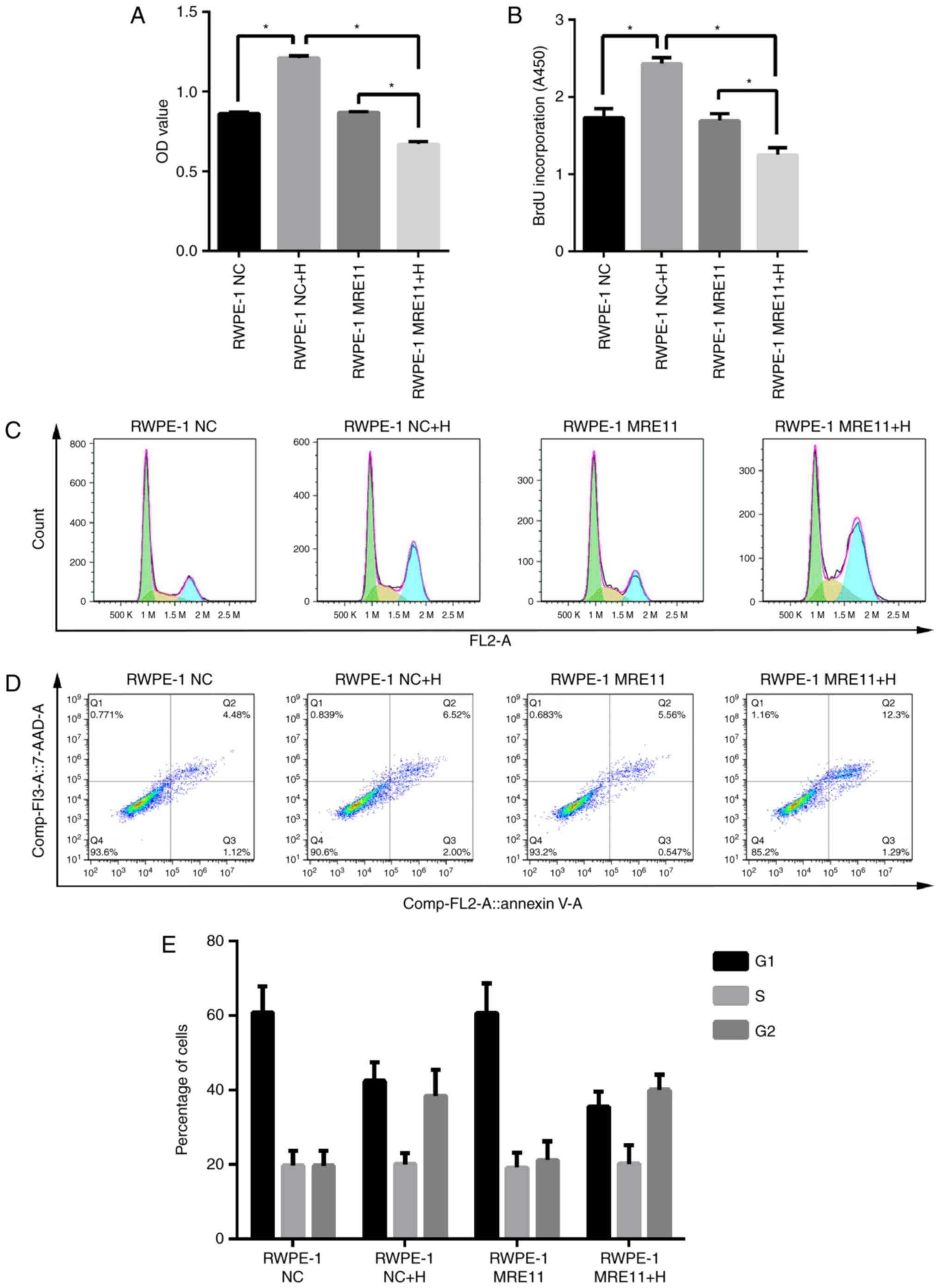
The effect of high glucose on the cell viability of
RWPE-1 cells was investigated and the results demonstrated that
viability significantly increased with high glucose treatment
(Fig. 5A and B). However, when
RWPE-1 cells were transfected with cells overexpressing MRE11, cell
viability decreased when incubated in high glucose solution
(Fig. 5A and B). Flow cytometry
revealed that cell cycle distribution was similar for normal and
MRE11-overexpressing RWPE-1 cells regardless of high-glucose
treatment (Fig. 5E). However,
cell apoptosis analysis indicated that both the number of apoptotic
normal RPEW-1 cells was increased following treatment with
high-glucose solution. However, the ratios were significantly
raised 2-fold when MRE11 was overexpressed and high-glucose
solution was applied. Overexpression of MRE11 had no significant
effect on cell apoptosis compared with normal cells without
high-glucose treatment (Fig.
5D).
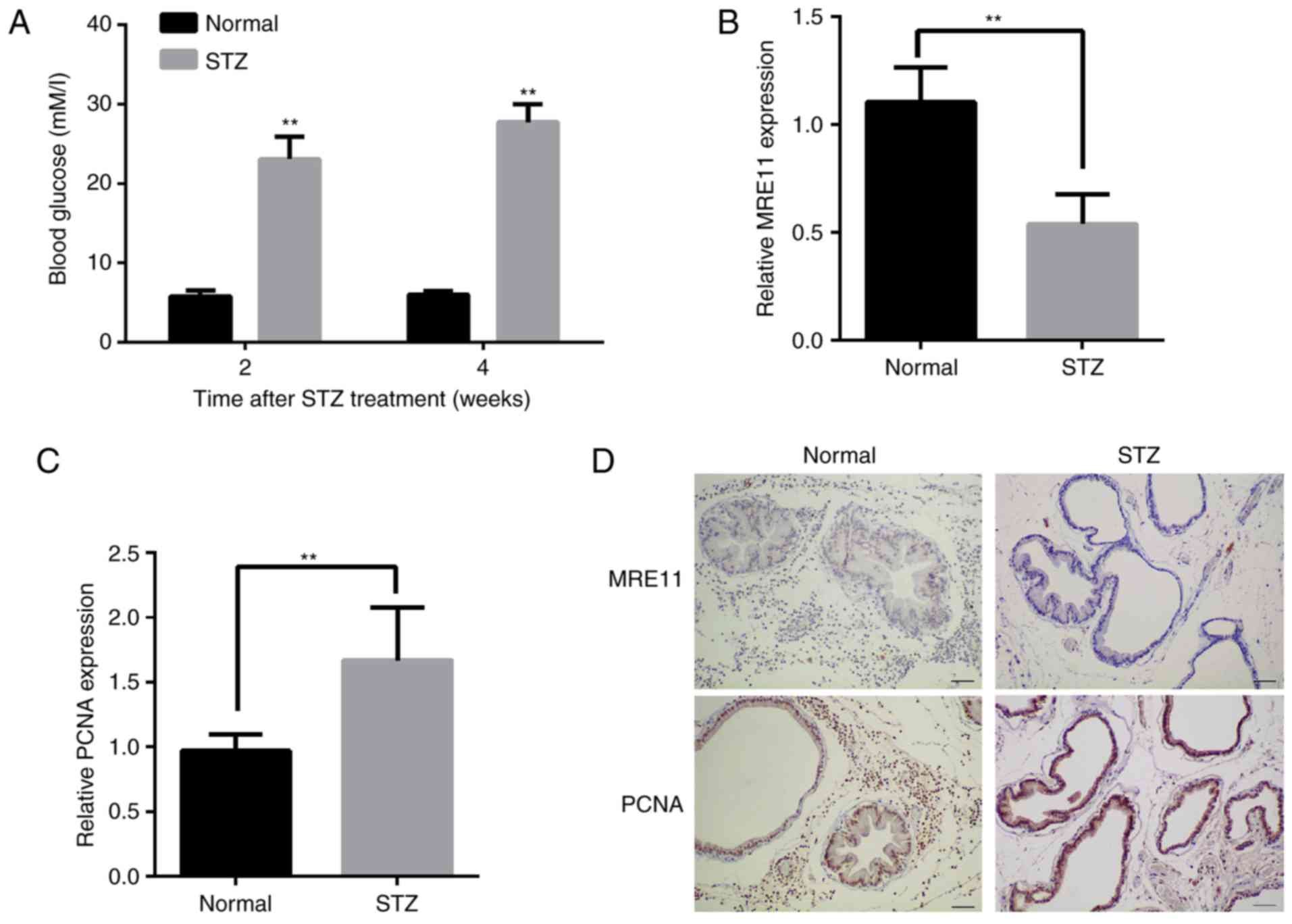
MRE11 expression is downregulated in
diabetic prostate tissue
To measure MRE11 expression in diabetic prostate
tissue, an STZ-induced diabetic rat model was established. The
blood glucose concentration of rats at 2 and 4 weeks following STZ
treatment were 23.07±2.86 and 27.7±2.33 mM/l, respectively, which
indicates that the diabetes model was successfully established
(Fig. 6A). The expression of
MRE11 mRNA was determined by RT-qPCR. MRE11 expression was
significantly downregulated in the STZ group compared with the
control group (Fig. 6B). The
level of proliferating cell nuclear antigen (PCNA), which is
associated with cell proliferation, was also assessed. The results
demonstrated that the level of PCNA in the STZ group was 2-fold
higher compared with the control group (Fig. 6C). Prostate tissue samples were
subjected to immunohistochemical staining, with MRE11 and PCNA
stained in brown. The results of immunohistochemical staining also
suggest that the expression of MRE11 were decreased in the STZ
group, whereas PCNA levels were increased (Fig. 6D).
Discussion
In the present study, RWPE-1 and HPr-1 cell lines
were treated with a high glucose solution to investigate the effect
of T2D on prostatic cell proliferation and its association with
prostatic enlargement and BPH. Transcriptome sequencing indicated
that MRE11 was downregulated under high glucose conditions. The
overexpression of MRE11 in RWPE-1 cells revealed that the
regulation of MRE11 expression is associated with cell
proliferation and apoptosis in a high glucose environment. MRE11
was significantly downregulated in the prostatic tissues of rats
treated with STZ.
MRE11 has been reported to activate
ataxia-telangiectasia-mutated kinase 1 and induce DNA damage repair
reactions (22,23). The present study demonstrated that
high concentrations of glucose are associated with increased
intracellular oxidative stress, DNA damage, antioxidative enzymes
deficiency and impaired DNA damage repair machinery in the
prostate. It is speculated that restoring the DNA repair machinery
may protect prostate cells from oxidative DNA damage induced by
high glucose (24). In the
present study, MRE11 expression was downregulated under high
glucose conditions, which is consistent with the results of a
previous study (24). The results
of the present study revealed that cell viability was significantly
increased under high glucose conditions, and this increased
proliferation may be an indicator of oncogenesis. MRE11 typically
functions alongside other molecules, including Mre11-Rad50-Xrs2
double strand break repair complex (25,26). In the present study, the
expression of CHK2, CDC25A and CDK was assessed to determine
whether the MRE11 overexpression vector was successfully
transfected into cells. CHK2 is activated by MRE11, which in turn
activates CDC25A via phosphorylation (27,28). CDK2 is an important molecule in
forming the Mre11-Rad50-NBS1 (MRN) complex and is crucial to enable
MRE11 to perform DNA repair functions (29). However, alterations in p-CHK2 and
p-CDC25A expression were observed, whereas there was no marked
change in CDK2/p-CDK2 expression. Therefore, CDK/p-CDK2 does not
appear to be affected by high-glucose conditions. p-CHK2 and
p-CDC25A were downregulated when cells were exposed to high glucose
treatment, which suggests that the DNA repair system was inhibited.
p-CHK2 expression was downregulated in normal cells, whilst
p-CDC25A was decreased within MRE11-overexpressing cells. This
indicates that CHK2 is positively associated with MRE11 expression.
It has been reported that CDC25A is regulated by CHK2, therefore
the association between CDC25A and MRE11 may be indirect (30). According to a recent study, CHK2
deficiency in mice induces a broad spectrum of tumorigenesis
(27). However, an efficient DNA
repair machinery is present in normal cells to resolve DNA damage
(22,23); once the function of this vital
machinery is compromised, cells are unable to maintain genetic
stability and may undergo neoplastic transformation (27). This may explain why cell viability
was increased when cells were exposed to high glucose conditions.
In MRE11-overexpression cells, cell viability was decreased and
cell apoptosis increased under high glucose conditions. This may be
due to the toxicity of the high concentration of glucose. When
MRE11 is overexpressed, the DNA repair system may be maintained in
a high glucose environment; as such, differentiation into cancerous
cells is impaired. The high concentration of glucose increases
oxidative stress, reducing cell viability and cell apoptosis.
Results of the in vivo experiment supported
the in vivo data. PCNA was used as a biomarker to analyze
cell viability in prostatic tissues (31,32). The results demonstrated that MRE11
expression was decreased whereas PCNA was increased. However,
observations of the diabetic tissues also indicated that high
glucose conditions induced cell enlargement, a common indicator of
BPH. These results suggest that diabetes may induce BPH. MRE11 also
serves an important role during the pathogenesis of BPH and may be
considered as a biomarker for the diagnosis of BPH or diabetes, as
well as a novel therapeutic target.
The results of the present study indicate that there
may be an association between BPH and diabetes. MRE11 expression
was downregulated under high glucose conditions and in in a rat
model of diabetes. To further validate the importance of MRE11,
further studies utilizing other diabetic animal models should be
performed. Effective clinical assays to measure the expression of
MRE11 are also required to accurately diagnose and develop
effective treatments for of BPH and diabetes.
Acknowledgments
The present study was supported by the Yunnan
Applied Basic Research Project (grant no. 2014FB058) and the
National Natural Science Foundation of China (grant nos. 81560417
and 81460385).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Gandhi J, Weissbart SJ, Smith NL, Kaplan
SA, Dagur G, Zumbo A, Joshi G and Khan SA: The impact and
management of sexual dysfunction secondary to pharmacological
therapy of benign prostatic hyperplasia. Transl Androl Urol.
6:295–304. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Komninos C and Mitsogiannis I:
Obstruction-induced alterations within the urinary bladder and
their role in the pathophysiology of lower urinary tract
symptomatology. Can Urol Assoc J. 8:E524–E530. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schwartz I, Wein AJ, Malloy TR and Glick
JH: Prostatic cancer after prostatectomy for benign disease.
Cancer. 58:994–996. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hua L, Zhang J, Wu H, Sui Y, Zhang W, Qian
L and Wang Z: Prostate cancer after prostatectomy for benign
prostatic hyperplasia. Zhonghua Nan Ke Xue. 10:612–613. 2004.In
Chinese. PubMed/NCBI
|
|
5
|
Kristal AR, Arnold KB, Schenk JM,
Neuhouser ML, Weiss N, Goodman P, Antvelink CM, Penson DF and
Thompson IM: Race/ethnicity, obesity, health related behaviors and
the risk of symptomatic benign prostatic hyperplasia: Results from
the prostate cancer prevention trial. J Urol. 177:1395–1400. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Qu X, Huang Z, Meng X, Zhang X, Dong L and
Zhao X: Prostate volume correlates with diabetes in elderly benign
prostatic hyperplasia patients. Int Urol Nephrol. 46:499–504. 2014.
View Article : Google Scholar
|
|
7
|
Chen Z, Miao L, Gao X, Wang G and Xu Y:
Effect of obesity and hyperglycemia on benign prostatic hyperplasia
in elderly patients with newly diagnosed type 2 diabetes. Int J
Clin Exp Med. 8:11289–11294. 2015.PubMed/NCBI
|
|
8
|
Kahn SE, Cooper ME and Del Prato S:
Pathophysiology and treatment of type 2 diabetes: Perspectives on
the past, present, and future. Lancet. 383:1068–1083. 2014.
View Article : Google Scholar
|
|
9
|
Raccah D: Insulin therapy in patients with
type 2 diabetes mellitus: Treatment to target fasting and
postprandial blood glucose levels. Insulin. 1:158–165. 2006.
View Article : Google Scholar
|
|
10
|
Breyer BN and Sarma AV: Hyperglycemia and
insulin resistance and the risk of BPH/LUTS: An update of recent
literature. Curr Urol Rep. 15:4622014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fleshner NE and Bhindi B: Metabolic
syndrome and diabetes for the urologist. Can Urol Assoc J. 8(7-8
Suppl 5): S159–S161. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shimizu S, Tsounapi P, Shimizu T, Honda M,
Inoue K, Dimitriadis F and Saito M: Lower urinary tract symptoms,
benign prostatic hyperplasia/benign prostatic enlargement and
erectile dysfunction: Are these conditions related to vascular
dysfunction? Int J Urol. 21:856–864. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Majewski N, Nogueira V, Robey RB and Hay
N: Akt inhibits apoptosis downstream of BID cleavage via a
glucose-dependent mechanism involving mitochondrial hexokinases.
Mol Cell Biol. 24:730–740. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gottlob K, Majewski N, Kennedy S, Kandel
E, Robey RB and Hay N: Inhibition of early apoptotic events by
Akt/PKB is dependent on the first committed step of glycolysis and
mitochondrial hexokinase. Genes Dev. 15:1406–1418. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Medema RH, Kops GJ, Bos JL and Burgering
BM: AFX-like Forkhead transcription factors mediate cell-cycle
regulation by Ras and PKB through p27kip1. Nature. 404:782–787.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Buteau J and Accili D: Regulation of
pancreatic beta-cell function by the forkhead protein FoxO1.
Diabetes Obes Metab. 9(Suppl 2): S140–S146. 2007. View Article : Google Scholar
|
|
17
|
Glauser DA and Schlegel W: The emerging
role of FOXO transcription factors in pancreatic beta cells. J
Endocrinol. 193:195–207. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chunwei YX, Yu W, Baoyi Z, Ya W, Shujun F,
Zebin C and Xingqiao W: Morphological and genetic changes in the
prostate of rats with diabetes. Chin J Exp Surg. 28:1535–1538.
2011.
|
|
19
|
Trapnell C, Pachter L and Salzberg SL:
TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics.
25:1105–1111. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate-a practical and powerful approach to
multiple testing. J R Statistical Soc. 57:289–300. 1995.
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
Czornak K, Chughtai S and Chrzanowska KH:
Mystery of DNA repair: The role of the MRN complex and ATM kinase
in DNA damage repair. J Appl Genet. 49:383–396. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zha S, Boboila C and Alt FW: Mre11: Roles
in DNA repair beyond homologous recombination. Nat Struct Mol Biol.
16:798–800. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ye C, Li X, Wang Y, Zhang Y, Cai M, Zhu B,
Mu P, Xia X, Zhao Y, Weng J, et al: Diabetes causes multiple
genetic alterations and downregulates expression of DNA repair
genes in the prostate. Lab Invest. 91:1363–1374. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Paull TT and Gellert M: A mechanistic
basis for Mre11-directed DNA joining at microhomologies. Proc Natl
Acad Sci USA. 97:6409–6414. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang X and Paull TT: The Mre11/Rad50/Xrs2
complex and non-homologous end-joining of incompatible ends in S.
cerevisiae. DNA Repair (Amst). 4:1281–1294. 2005. View Article : Google Scholar
|
|
27
|
Inagaki A, Roset R and Petrini JH:
Functions of the MRE11 complex in the development and maintenance
of oocytes. Chromosoma. 125:151–162. 2016. View Article : Google Scholar :
|
|
28
|
Zhao H, Traganos F, Albino AP and
Darzynkiewicz Z: Oxidative stress induces cell cycle-dependent
Mre11 recruitment, ATM and Chk2 activation and histone H2AX
phosphorylation. Cell Cycle. 7:1490–1495. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Buis J, Stoneham T, Spehalski E and
Ferguson DO: Mre11 regulates CtIP-dependent double-strand break
repair by interaction with CDK2. Nat Struct Mol Biol. 19:246–252.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Myer DL, Robbins SB, Yin M, Boivin GP, Liu
Y, Greis KD, Bahassi el M and Stambrook PJ: Absence of polo-like
kinase 3 in mice stabilizes Cdc25A after DNA damage but is not
sufficient to produce tumors. Mutat Res. 714:1–10. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bryja A, Dyszkiewicz-Konwinska M, Budna J,
Ciesiółka S, Kranc W, Borys S, Jeseta M, Urbaniak O, Bukowska D,
Antosik P, et al: Expression of cell mitotic progression proteins
and keratinocyte markers in porcine buccal pouch mucosal cells
during short-term, real-time primary culture. J Biol Regul Homeost
Agents. 31:297–309. 2017.PubMed/NCBI
|
|
32
|
Sales CF, Santos KPED, Rizzo E, Ribeiro
RIMA, Santos HBD and Thomé RG: Proliferation, survival and cell
death in fish gills remodeling: From injury to recovery. Fish
Shellfish Immunol. 68:10–18. 2017. View Article : Google Scholar : PubMed/NCBI
|