Introduction
Sirtuin 1 (Sirt1), a member of class III histone
deacetylases, is a nicotinamide adenine dinucleotide
(NAD+)- dependent histone deacetylase. Recently, it has
become clear that Sirt1 deacetylates histone and non-histone
proteins to participate in multiple cellular process, including
apoptosis, autophagy, calorie restriction, energy metabolism, cell
differentiation, anti-aging and DNA damage repair (1-4).
Of note, Sirt1 helps cells to cope with environmental stress by
targeting abundant transcription factors, such as the Forkhead box
O (FOXO) proteins, tumour protein p53, nuclear factor (NF)-κB,
peroxisome proliferator-activated receptor-γ coactivator (PGC)-1α,
and E2F transcription factor 1 (3,5-7).
Previous evidence indicates that Sirt1 exerts a protective role in
cardiovascular diseases. Hariharan et al (3) have reported that Sirt1 mediates
glucose starvation-induced autophagy by deacetylating FOXO in
cardiomyocytes. In addition, Sirt1 overexpression protects the
heart from ischemia reperfusion injury by inhibiting proapoptotic
molecules (8). These studies
demonstrate that Sirt1 is involved in cardioprotection.
Hypoxia is the fundamental and inevitable
pathophysiological process of cyanotic congenital heart disease
(CHD), such as the Tetralogy of Fallot (TOF). Although the
underlying mechanism of CHD pathogenesis remains unclear, cardiac
apoptotic cell death is vitally important in CHD (9). Of note, previous studies have
demonstrated that Sirt1 promoted cellular survival under hypoxic
conditions by deacetylating hypoxia-inducible factor (Hif)-1α
(10) and Hif-2α (11), implying that Sirt1 may have a
critical role in hypoxic environment. Therefore, it is possible
that Sirt1 may serve a role in protecting cardiomyocytes from
hypoxic injury.
Autophagy is a catabolic process of intracellular
degradation in which cytoplasmic materials are recycled through
autophagosomal sequestration and subsequent lysosomal degradation
(12). Autophagy exists under
stress-free conditions to maintain cellular homeostasis.
Cardiac-specific deficiency of the autophagy related 5 (ATG5) gene
under physiological conditions induces heart failure in mice,
demonstrating that autophagy is required to maintain basal heart
function (13). Autophagy has a
pivotal role in energy metabolism and protein quality control and
has been found to be beneficial to cardiac function in harsh
environments, including during ischemia-reperfusion injury
(14).
Our group has previously demonstrated that
AMP-activated protein kinase (AMPK) protects cardiomyocytes from
hypoxia-induced injury through mitophagy (15). In addition, AMPK has been
demonstrated to promote autophagy via unc-51 like autophagy
activating kinase 1 (ULK1) activation and mammalian target of
rapamycin (mTOR) 1 suppression (16). Whether Sirt1 modulates autophagy
in hypoxic cardiomyocytes via AMPK has not been fully
investigated.
The most conserved endoplasmic reticulum
(ER)-resident unfolded protein response (UPR) regulator, the
inositol requiring kinase enzyme 1α (IRE1α), functions as a cell
fate executor. In response to mild ER stress, the kinase domain of
IRE1α is autophosphorylated, subsequently activating its
endoribonuclease activity to splice the X-box binding protein 1
(XBP1) mRNA to re-establish protein folding homeostasis. However,
under excessive or sustained ER stress, continuous engagement of
IRE1α results in events that simultaneously aggravate protein
misfolding and apoptosis (17).
IRE1α inhibition has been demonstrated to attenuate single
prolonged stress-induced neuronal apoptosis in locus coeruleus
(18). Furthermore, IRE1α is of
vital importance for cytokine-induced apoptosis via c-Jun
N-terminal kinase (JNK) activation in human pancreatic beta cells
(19,20). Jain et al (21) have reported that IRE1α is
activated in cardiomyocytes of rats subjected to chronic hypobaric
hypoxia, with an accompanying increase in apoptosis. Therefore, it
can be hypothesized that Sirt1 may inhibit chronic hypoxia-induced
apoptosis through IRE1α.
The present study sought to investigate the role of
Sirt1 in modulating autophagy and apoptosis in cardiac cells under
chronic hypoxic conditions. The target molecules involved in
mediating these effects, such as AMPK and IRE1α, were also
assessed.
Materials and methods
Patients studied and myocardial
biopsies
A total of 20 patients were enrolled in this study
(from January 2015 to January 2017), all of whom underwent surgical
correction for congenital heart diseases with extracorporeal
circulation in the Department of Cardiovascular Surgery of Xinqiao
Hospital (Chongqing, China). Ten patients had cyanotic (4 females
and 6 males; mean age, 22 months; 9-32, arterial SpO2, 72%; 63-76)
and 10 had acyanotic (4 females and 6 males; mean age, 18 months;
8-27, arterial SpO2, 97%; 95-100) cardiac defects. The relatively
normoxic ventricular tissues samples used as control were obtained
from patients with ventricular septal defect combined with right
ventricular outflow tract obstruction. The hypoxic ventricular
tissue samples were obtained from patients with Tetralogy of Fallot
(TOF) as previously described (22).
The investigation conformed with the Declaration of
Helsinki and was approved by the Human Ethical Committee of Xinqiao
Hospital. Informed consent was obtained from all subjects involved
in the study prior to surgery.
Adenoviral and lentiviral constructs
The Ad-Easy adenoviral vector system (Qbiogene,
Inc., Santa Ana, CA, USA) was used to construct the Ad-Sirt1,
Ad-Sh-Sirt1 and Ad-IRE1α, according to the protocol of the
manufacturer. A previously reported Sirt1-RNAi sequence was used
(23). AdLacZ was used as a
control. The adenoviruses were transduced at multiplicity of
infection (MOI) 50. The Sh-IRE1a and Sh-Scramble lentivirus were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). To
knockdown IRE1α, H9C2 cells were infected with Sh-IRE1α, and stable
cell lines were obtained following puromycin selection.
Cell culture
The H9C2 cell line (American Type Culture
Collection, Manassas, VA, USA) was maintained in high glucose
Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), supplemented with 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml
of penicillin, and 100 mg/ml streptomycin at 37°C. After the
indicated treatment, the cells were incubated in serum-free medium
overnight. Then, cells in the hypoxia group were placed in a
modular incubator (Ruskinn Technology Ltd., Bridgend, UK),
containing a gaseous mixture of 94% N2, 5%
CO2 and 1% O2 at 37°C for 24 h. The cells in
normoxia group were placed in a 21% O2 environment.
Unless otherwise stated, the hypoxic cells were subjected to
hypoxia for 24 h. To inhibit AMPK, Compound C (20 µM; Sigma-
Aldrich; Merck KGaA, Darmstadt, Germany) was applied 2 h before
hypoxia treatment.
In vivo hypoxia mouse model
Six to eight weeks old C57BL/6J male mice were
purchased from the Laboratory Animals Center of Third Military
Medical University (Chongqing, China). All animal protocols were
performed in accordance with approved principles of laboratory
animal care and ethical approval was obtained from the Experimental
Animal Committee of The Second Affiliated Hospital of The Third
Military Medical University, Chongqing, China.
A total of 40 C57BL/6J mice were randomly divided
into four groups: control group, mice placed in a normoxic 21%
O2 environment (21% O2+DMSO); hypoxic
untreated group, mice exposed to hypoxia and treated with vehicle
control (10% O2+DMSO); SRT1720 treated group, mice
exposed to hypoxia and treated with SRT1720 (10%
O2+SRT1720); and EX-527 treatment group, mice exposed to
hypoxia and treated with EX-527 (10% O2+EX-527).
O2 control glove boxes and cabinets
(Baker Ruskinn InvivO2 1000; Ruskinn Technology, Ltd.)
were used to maintain a hypoxic environment for the animal studies.
The oxygen concentration was stably maintained at 10% O2
to mimic a hypoxic environment. A carbon dioxide absorbent
(Tiger-sorb, Guangzhou, China) was used to remove the
CO2 produced by the mice in the hypoxic chamber.
Drug administration
The Sirt1 specific activator SRT1720 (Selleck
Chemicals, Houston, TX, USA) and the Sirt1 inhibitor EX-527
(Sigma-Aldrich; Merck KGaA) were dissolved in dimethyl sulfoxide
(DMSO) and diluted to a final concentration with normal saline to
ensure that the final DMSO concentration was 0.5%. To activate or
inhibit Sirt1, mice were weighed and injected daily with SRT1720
(50 mg/kg) or EX-527 (10 mg/kg) intraperitoneally for one week
prior to hypoxia exposure. The mice in the normoxic or hypoxic
control groups received an equivalent volume of diluted DMSO
intraperitoneally at the indicated time points. Next, all of the
mice in the hypoxic groups were housed in the hypoxic environment
(10% O2) for 2 weeks. During the hypoxia exposure, the
mice were administered daily the same drug doses as aforementioned.
The normoxic mice were housed in a normoxic environment as a
control. The mice were euthanized at the end of the hypoxia
experiment and the hearts were harvested for further analysis.
Western blot analysis
Total protein was extracted from the H9C2 cells or
heart samples using cell extraction buffer (Invitrogen; Thermo
Fisher Scientific, Inc.) supplemented with protease and phosphatase
inhibitors (Roche Applied Science, Mannheim, Germany). Protein
concentrations from myocardial tissues and H9C2 cells were measured
by bicinchoninic acid assay, and then separated (50 µg) by
12% SDS-PAGE gel. Then, protein samples were transferred to
polyvinylidene fluoride membranes (Roche Applied Science) and
blocked with 5% BSA for 1 h at room temperature. The membranes were
probed with the indicated primary antibodies at 4°C overnight,
followed by incubation with horseradish peroxidase (HRP)-conjugated
secondary antibodies for 1 h at room temperature, and detection
with enhanced chemiluminescence reagents (Beyotime Institute of
Biotechnology, Shanghai, China). Changes in protein expression were
determined using ImageJ software (National Institutes of Health,
Bethesda, MD, USA). The antibodies against microtubule associated
protein 1 light chain 3b (LC3B; cat. no. 3868), p62 (cat. no.
39749), phosphorylated (p-) JNK (cat. no. 4668), JNK (cat. no.
9252), p- Jun proto-oncogene (c-jun; cat. no. 3270), c-jun (cat.
no. 9165), NF-κB p65 (cat. no. 8242), NF-κB p-p65 (cat. no. 3033),
p53 (cat. no. 2524), cleaved Caspase-3 (cat. no. 9661), AMPK (cat.
no. 5831), p-AMPK (cat. no. 2535), and β-actin (cat. no. 4970) were
used at 1:1,000 dilution and purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). The antibodies against
acetyl-p53 (cat. no. ab61241), IRE1α (cat. no. ab37073),
phospho-IRE1α (cat. no. ab48187), Sirt1 (cat. no. ab110304), and
Bcl2 (cat. no. ab59348) were used at 1:1,000 dilution and obtained
from Abcam. The goat anti-rabbit (cat. no. 7074) and goat
anti-mouse (cat. no. 7076) IgG-HRP antibodies were obtained from
Cell Signaling Technology, Inc.
Flow cytometry
Cell apoptosis was assessed via an Annexin
V-phycoerythrin (PE)/7-aminoactinomycin (AAD) assay. Briefly, after
the indicated treatments, H9C2 cells were digested with trypsin,
washed twice with cold PBS, collected, and suspended in Annexin V
binding buffer. PE-conjugated Annexin V and 7-AAD (BD Biosciences,
San Jose, CA, USA) were added to the cells. Following incubation,
Annexin V binding buffer was added, and the cell suspensions were
analysed by flow cytometry (BD LSRFortessa X-20; BD Biosciences)
and data were analysed by Kaluza 2.0 (Beckman Coulter, Inc., Brea,
CA, USA).
Determination of apoptosis by TUNEL
staining
The apoptosis rates in tissue sections of mouse
hearts (prepared as described below for the immunofluorescence
experiments) were analysed by TUNEL staining using an in
situ cell death detection kit (Roche Applied Science),
according to manufacturer's protocol. In brief, the slides were
incubated with TUNEL reaction mixture solution to mark the
apoptotic cells, and the total number of cells was determined using
DAPI staining. The slides were visualized under a Leica TCS-SP5
laser-scanning confocal microscope (Leica Microsystems GmbH,
Wetzlar, Germany). The apoptotic rate was calculated as a % of the
number of TUNEL-positive cells over the total number of
DAPI-stained cells.
Immunofluorescence staining
Frozen mouse hearts were embedded in optimal cutting
temperature (OCT) compound (Sakura Finetek Inc., Torrance, CA, USA)
and 6 µm cryosections were cut. The slides were fixed in 4%
paraformaldehyde for 30 min, washed five times with PBS and
incubated with 10% goat serum (Boster Biological Technology, Ltd.,
Wuhan, China) at room temperature. The tissues were immunostained
with Hif1α (cat. no. ab1; Abcam; 1:200), α-actin (cat. no.
SAB4503474; Sigma-Aldrich; 1:400), cardiac troponin T (TNT; cat.
no. ab8295; Abcam; 1:400), or LC3B (cat. no. L7543; Sigma-Aldrich;
1:200), and washed five times with PBS. The samples were then
stained with Cy3-labeled goat anti-mouse IgG (cat. no. A0521;
1:500) or Alexa Fluor 488-labeled goat anti-rabbit IgG (cat. no.
A0423; 1:500) secondary antibodies (Beyotime Institute of
Biotechnology, Shanghai, China) for 1 h at room temperature. The
nuclei were counterstained with DAPI. The cells were imaged with
confocal laser scanning microscopy (Leica Microsystems GmbH).
Statistical analysis
Data are expressed as the mean ± standard error of
the mean. Statistical significance was analysed with GraphPad Prism
5.0 software (GraphPad Software, Inc. La Jolla, CA, USA).
Comparisons between two groups were performed by using Student's
t-test. Differences among multiple groups were analysed using
one-way analysis of variance (ANOVA) followed by Tukey's post hoc
test or, two-way ANOVA followed by Bonferroni correction. P<0.05
was considered to indicate a statistically significant
difference.
Results
Autophagy, apoptosis and Sirt1 are
upregulated in cyanotic patients
Specimens of the right ventricular outflow tract
were isolated from patients diagnosed with cyanotic or acyanotic
congenital heart diseases. Western blot analysis was used to detect
the autophagy markers LC3-II and p62, and apoptosis-associated
proteins, namely the pro-apoptotic cleaved Caspase-3 and the
anti-apoptotic protein Bcl2. The results demonstrated that
expression of LC3-II, an indicator of autophagosome accumulation,
was increased and while the levels of p62, which is degraded via
autophagy, were decreased in the myocardial samples from patients
with cyanotic congenital heart disease (Fig. 1A and C). Additionally, the cleaved
Caspase-3 levels were increased while the Bcl2 levels were
decreased in the cyanotic group (Fig.
1B and C). These results indicated that autophagy and apoptosis
were activated in the heart samples of patients with cyanotic
congenital heart disease.
Furthermore, to examine whether hypoxia affected
cardiac Sirt1 levels, its expression was detected by western
blotting. The results revealed that Sirt1 protein expression was
increased in the myocardia of cyanotic patients compared with
acyanotic controls (Fig. 1B and
C).
Sirt1 enhances autophagy in
cardiomyocytes
To examine the role of Sirt1 in the autophagy
process, Sirt1 was overexpressed in H9C2 cells by use of the
Ad-Sirt1 adenovirus, as well as silenced by use of the Ad-Sh-Sirt1
adenovirus. Ad-LacZ was used as control. Fig. 2A illustrates that Ad-Sirt1
successfully increased Sirt1 expression and Ad-Sh-Sirt1 suppressed
Sirt1 expression compared with the control group. Ad-Sirt1
stimulated a significant accumulation in baseline levels of LC3-II
and a decrease in p62 compared with the control group, indicating
that autophagy was activated when Sirt1 was overexpressed (Fig. 2A and B). By contrast, Sirt1
knockdown resulted in p62 accumulation and LC3-II reduction
compared with the control group (Fig.
2A and B). To analyse the potential role of Sirt1 in
hypoxia-induced autophagy, Ad-Sh-Sirt1-transduced H9C2 cells were
subjected to 24 h hypoxia treatment. Knockdown of Sirt1
significantly decreased LC3-II expression and increased p62
expression in hypoxic cardiomyocytes, compared with the hypoxic
control group, indicating that autophagy is impaired when Sirt1 is
silenced (Fig. 2C and D).
Sirt1 promotes autophagy in hypoxic
cardiomyocytes by activating AMPK
To determine whether Sirt1 modulates AMPK in H9C2
cells exposed to hypoxia, the levels of p-AMPK and AMPK were
determined. Western blotting revealed that the p-AMPK levels were
increased significantly in the Ad-Sirt1 + hypoxia group compared
with the hypoxia control group (Fig.
2E and F). Compared with the hypoxia group, the p-AMPK/AMPK
ratio was significantly decreased in the Ad-Sh-Sirt1 + hypoxia
group (Fig. 2E and F). To
investigate whether the pro-autophagy effect of Sirt1 was
associated with AMPK, the cells were pretreated with the AMPK
inhibitor Compound C (20 µM) along with Ad-Sirt1 prior to
the hypoxia treatment. The results demonstrated that the
pro-autophagy effect of Sirt1 was reversed when Compound C was
added (Fig. 2G and H), suggesting
that the ability of Sirt1 to promote autophagy in hypoxic
cardiomyocytes is mediated by AMPK.
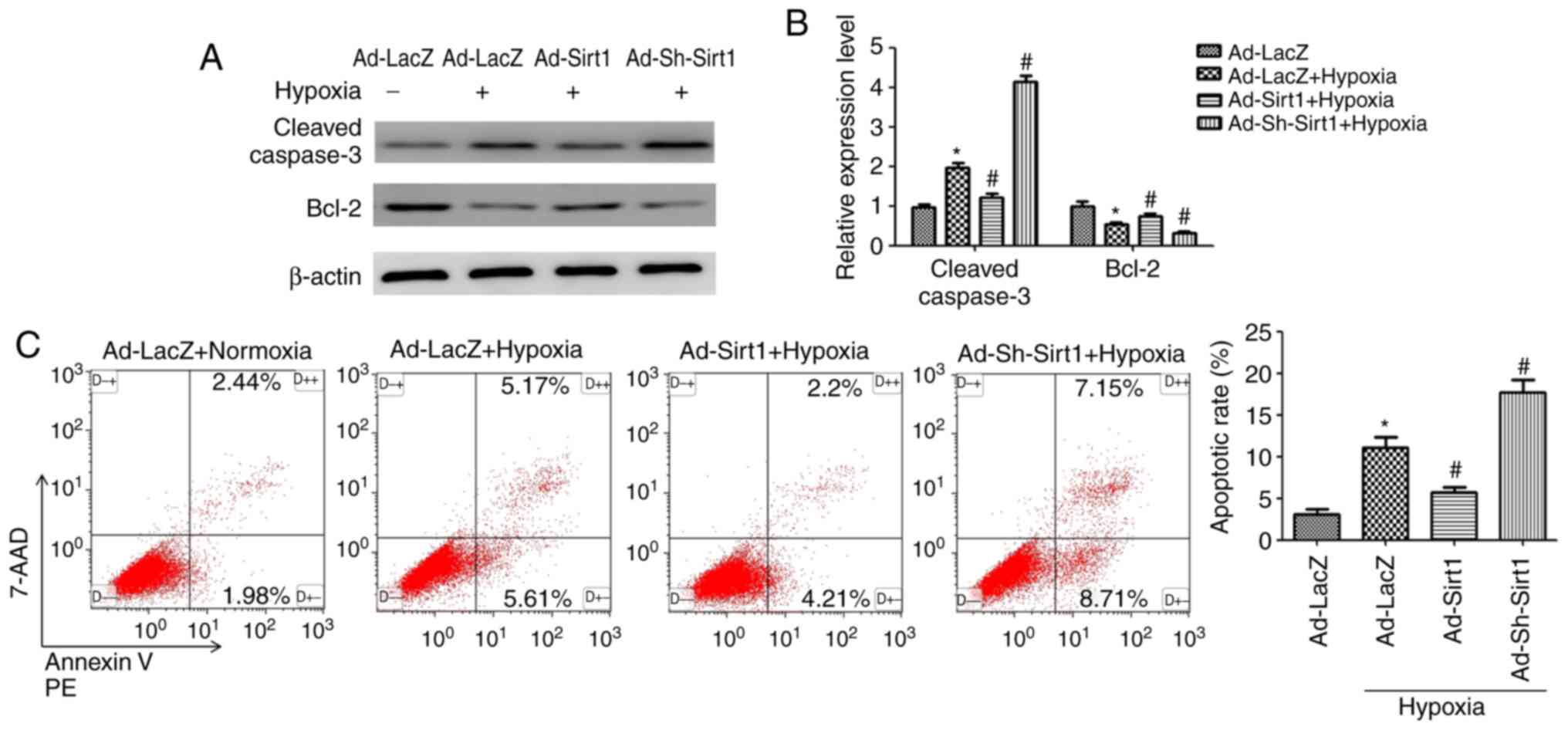
Sirt1 alleviates hypoxia-induced
apoptosis
To determine the effects of Sirt1 on apoptosis in
the context of hypoxia, several methods were employed. Western
blotting results demonstrated that Sirt1 overexpression attenuated
the levels of cleaved Caspase-3 and increased anti-apoptotic Bcl2
expression in hypoxic H9C2 cells (Fig. 3A and B). Furthermore, flow
cytometry analysis revealed that the cell apoptosis rate (early
apoptotic + late apoptotic) was increased following 24 h induction
of hypoxia (Fig. 3C); however,
Sirt1 overexpression resulted in a significant decrease in the % of
apoptotic cells (Fig. 3C).
Collectively, these results revealed that Sirt1 alleviated
apoptosis in hypoxic H9C2 cells.
Sirt1 alleviates apoptosis via IRE1α
suppression
First, the effects of Sirt1 on the IRE1α pathway
were examined by analysing the protein expression levels of IRE1α
and its phosphorylated form. As illustrated in Fig. 4A, hypoxia activated IRE1α, as
demonstrated by an increase in the p-IRE1α/IRE1α ratio. Ad-Sirt1
transduction significantly reduced the activation of IRE1α, whereas
Sirt1 silencing significantly enhanced the activation IRE1α
(Fig. 4A and B). These findings
demonstrated that Sirt1 regulated hypoxia-induced IRE1α
activation.
 | Figure 4Effect of IRE1α on apoptosis in
hypoxic H9C2 myocytes. (A) Western blot analysis of IRE1α, p-IRE1α
and β-actin following the indicated treatments. (B) Densitometry
analysis of the p-IRE1α/IRE1α ratio levels. n=3.
*P<0.05 vs. Normoxia + Ad-LacZ group;
#P<0.05 vs. Hypoxia + Ad-LacZ group. (C) Protein
levels of IRE1α, cleaved Caspase-3, Bcl2, p-JNK, JNK, p-cjun, cjun,
NF-κB p-p65, NF-κB p65, and β-actin in H9C2 cells infected with
Ad-IRE1α or Lv-Sh-IRE1α in response to normoxia or hypoxia. (D)
Densitometric analysis for the western blot data of panel B. n=3.
*P<0.05 vs. Lv-Sh-Scramble + Normoxia group;
#P<0.05 vs. Lv-Sh-Scramble + Hypoxia group;
&P<0.05 vs. Ad-LacZ + normoxia group;
@P<0.05 vs. Ad-IRE1α + Hypoxia group. IRE1α, inositol
requiring kinase enzyme 1α; p-, phosphorylated; JNK, c-Jun
N-terminal kinase; c-jun, Jun proto-oncogene; NF, nuclear factor;
sh, short hairpin. |
Next, the role of IRE1α in hypoxia-induced apoptosis
in H9C2 cells was investigated. IRE1α was overexpressed in the
cells via Ad-IRE1α transduction. To inhibit IRE1α expression in
H9C2 cells, stable cell lines were established by transduction with
the Lv-Sh-IRE1α lentivirus and subsequent puromycin selection. The
protein expression levels of IRE1α were markedly increased or
decreased following Ad-IRE1α or Lv-Sh-IRE1α transduction,
respectively (Fig. 4C). Western
blot analysis was performed to assess changes in the levels of
apoptosis-related proteins following IRE1α modulation. After IRE1α
overexpression in H9C2 cells exposed to hypoxia for 24 h, the
levels of cleaved Caspase-3 were upregulated, while Bcl2 was
drastically decreased. However, IRE1α inhibition exerted the
opposite effects on the expression levels of these
apoptosis-related proteins (Fig. 4C
and D).
To explore how IRE1α modulates apoptosis, we
investigated whether IRE1α promoted apoptosis by targeting the
pro-apoptotic JNK/c-jun pathway in hypoxic H9C2 cells. p-JNK and
p-cjun were activated after 24 h hypoxia treatment (Fig. 4C). In addition, the p-JNK/JNK and
p-c-jun/c-jun ratios were significantly increased in the
Ad-IRE1α+hypoxia group compared with its hypoxia control group
(Fig. 4D). Silencing of IRE1α by
Lv-Sh-IRE1α decreased the p-JNK/JNK and p-c-jun/c-jun ratios,
compared to the Lv-Sh-scramble +hypoxia group (Fig. 4C and D). These results indicated
that IRE1α silencing blocked JNK and c-jun activation, leading to
decreased cellular apoptosis.
Next, we explored whether IRE1α regulates apoptosis
through NF-κB p65. IRE1α overexpression further activated NF-κB p65
in hypoxic H9C2 cells, as demonstrated by a significant increase in
the phosphorylation of NF-κB p65 compared with its phosphorylation
state in the hypoxic control (Fig. 4C
and D). Conversely, IRE1α silencing reversed the regulatory
effect of hypoxia on NF-κB p65 activation (Fig. 4C and D). Taken together, these
data suggest that IRE1α activation may exert its pro-apoptotic
effect through the JNK/c-jun pathway and NF-κB p65.
To determine whether Sirt1 mitigated hypoxia-induced
apoptosis through IRE1α, Ad-Sirt1 was coexpressed with Ad-IRE1α.
Western blot results demonstrated that Sirt1 and IRE1α coexpression
significantly mitigated the IRE1α-dependent increase in apoptosis,
as demonstrated by a decrease in cleaved Caspase-3 and an increase
in Bcl2 expression compared with the IRE1α + hypoxia group
(Fig. 5). In addition, the
p-JNK/JNK, p-c-jun/c-jun, and NF-κB p-p65/p65 ratios were decreased
in the Ad-Sirt1 + Ad-IRE1α group compared with the ratios in the
Ad-IRE1α group (Fig. 5). Taken
together, these findings suggest that Sirt1 exerted its
anti-apoptotic effects in hypoxic cardiomyocytes by inhibiting
IRE1α activation.
 | Figure 5IRE1α promotes apoptosis in hypoxic
H9C2 cells, and this effect is reversed by Sirt1 overexpression.
Representative blots and densitometry analysis are shown for the
protein expression levels of cleaved Caspase-3, Bcl2, p-JNK, JNK,
p-cjun, c-jun, NF-κB p-p65 and NF-κB p65, following the indicated
treatments. n=3. *P<0.05 vs. Ad-LacZ + Hypoxia group;
#P<0.05 vs. Ad-IRE1α + Hypoxia group. IRE1α, inositol
requiring kinase enzyme 1α; Sirt1, sirtuin 1; p-, phosphorylated;
JNK, c-Jun N-terminal kinase; c-jun, Jun proto-oncogene; NF,
nuclear factor. |
Effects of SRT1720 and EX-527 on Sirt1
expression in hypoxic mice
The expression of Hif1α, a marker of hypoxia, was
assessed by immunofluorescence staining in control and hypoxic
mice. To examine the effects of hypoxia exposure, Hif1α was
co-immunolabeled with α-actin, and the nuclei were stained with
DAPI, in the heart samples of normoxic and hypoxic mice. The
results revealed that the immunofluorescence intensity of Hif1α was
markedly increased in the hypoxic group compared with the control
mice (Fig. 6A).
To study the functional role of Sirt1 in cardiac
protection in hypoxic mice, hypoxia-exposed mice were treated with
SRT1720, a Sirt1 activator, or EX-527, a selective Sirt1 inhibitor.
As illustrated in Fig. 6B and C,
SRT1720 administration increased Sirt1 expression whereas EX-527
treatment suppressed Sirt1 expression in the hearts of hypoxic
mice. As p53 is a widely accepted Sirt1 substrate, the acetylation
levels of p53 were evaluated in order to determine the deacetylase
activity of Sirt1 in the heart samples from the four groups. The
acetyl-p53/p53 ratio was increased in the EX-527-treated group,
while the ratio was significantly decreased in the SRT1720-treated
group, compared with the hypoxia untreated group (Fig. 6B and C). These results
demonstrated that SRT1720 and EX-527 administration is effective in
modulating the expression and deacetylase activity of Sirt1.
Sirt1 enhances autophagy and inhibits
apoptosis in cardiomyocytes of hypoxic mice
To examine whether Sirt1 induced autophagy in the
heart, the LC3-II and p62 expression levels were assessed.
Long-term treatment with SRT1720 increased cardiac autophagy in
hypoxic mice, as indicated by an increase in LC3-II abundance and a
decrease in p62 accumulation (Fig.
7A). EX-527 treatment inhibited autophagy in hypoxic mice
(Fig. 7A). In addition, AMPK was
significantly activated in the heart tissues of mice housed under
hypoxic conditions. SRT1720 treatment enhanced the AMPK activation.
By contrast, EX-527 administration inhibited AMPK phosphorylation
in the myocardium of mice housed in a hypoxic environment compared
with the hypoxia untreated group (Fig. 7A).
 | Figure 7Sirt1 activation enhances autophagy
and alleviates apoptosis in the myocardium of hypoxic mice. (A)
Representative western blots and densitometric analysis of LC3-II,
p62, p-AMPK, AMPK, cleaved Caspase-3 and Bcl2 protein levels in
heart samples from mice treated as indicated. (B)
Immunofluorescence analysis of LC3B (green) was performed in heart
samples from the experimental mice. The cardiomyocytes were
costained with c-TNT (red) and the nuclei were stained with DAPI
(blue). Scale bar, 20 µm. (C) Representative western blots
and densitometric analysis of the p-IRE1α, IRE1α, cleaved Caspase-3
and Bcl2 levels in heart samples of the experimental mice. (D)
Representative TUNEL staining images of cardiac tissues from the
different groups. The cardiomyocytes were stained with c-TNT (red)
and the nuclei were stained with DAPI (blue). The TUNEL staining
(green) ratio was counted as the % of apoptotic cardiomyocytes over
the total myocytes (Scale bar, 50 µm). n=10.
*P<0.05. vs. Normoxia + DMSO group;
#P<0.05 vs. Hypoxia + DMSO group. Sirt1, sirtuin 1;
LC3, microtubule associated protein 1 light chain 3; p-,
phosphorylated; AMPK, AMP-activated protein kinase; c-TNT, cardiac
troponin T; IRE1α, inositol requiring kinase enzyme 1α; TUNEL,
terminal deoxynucleotidyl-transferase-mediated dUTP nick end
labelling. |
The immunofluorescence intensity of the LC3B
staining in the heart samples was significantly increased in the
hypoxic mice, compared with the control group (Fig. 7B). As expected, treatment with
SRT1720 induced a significant increase in the LC3B intensity in the
hearts from hypoxic mice, whereas EX-527 administration led to a
reduced elevation of this marker (Fig. 7B), indicating that Sirt1 increases
cardiac autophagic flux in vivo.
Next, the expression levels of apoptosis-related
proteins were evaluated. Exposure to hypoxia for two weeks resulted
in a significant increase in the levels of cleaved Caspase-3, as
well as a robust decrease in Bcl2, compared with the control group
(Fig. 7C). There was a
significant decrease in cleaved Caspase-3 and significant increase
in Bcl-2 levels in the hearts of the SRT1720-treated hypoxic mice
compared with hypoxic untreated mice. EX-527 treatment resulted in
the opposite effects (Fig. 7C).
In addition, IRE1α was significantly activated in hypoxic mice,
while SRT1720 treatment decreased IRE1α activation. By contrast,
EX-527 administration enhanced IRE1α activation in the myocardium
of EX-527-treated mice compared with the hypoxia untreated group
(Fig. 7C).
Finally, TUNEL staining was used to explore whether
Sirt1 activation by SRT1720 in mice could protect cardiomyocytes
from hypoxia-induced apoptosis. Hypoxia exposure resulted in a
significant increase in the % of TUNEL-positive cardiomyocytes in
the wall of the heart (Fig. 7D;
P<0.05). Sirt1 overexpression induced by SRT1720 treatment
significantly ameliorated the myocardial apoptosis compared the
hypoxic untreated mice (Fig. 7D;
P<0.05). By contrast, the EX-527-treated hypoxic mice displayed
a robust increase in the number of apoptotic cardiomyocytes
compared with the hypoxic untreated mice (Fig. 7D; P<0.05). These results
indicated that Sirt1 activation in the heart protects
cardiomyocytes from hypoxia-induced apoptosis.
These in vivo results indicate that Sirt1
enhanced autophagy and protected cardiomyocytes from apoptosis in
the heart of hypoxia-exposed mice.
Discussion
Hypoxia is commonly observed in patients with
cyanotic congenital heart disease (24), and identifying cellular protective
mechanisms that counteract hypoxia-induced injury in cardiomyocytes
is of great importance. Tetralogy of Fallot (TOF) is a common
cyanotic heart lesion that consists of four anatomical
malformations (25). In the
present study, it was demonstrated that autophagy and apoptosis are
activated in heart samples of cyanotic patients in response to
hypoxia. In addition, Sirt1 expression levels were demonstrated to
be significantly higher in the samples from the cyanotic group
compared with the samples from the acyanotic controls, indicating
that Sirt1 might mediate cellular adaptive response to hypoxia.
Owing to the development of ultrasonography used in prenatal
diagnosis, the number of patients with congenital heart disease has
decreased over the recent years. Therefore, a limitation of the
present study is that only ten heart samples were collected for
each group. It has been accepted that mammalian cells have
developed an adaptive mechanism to activate prosurvival signalling
pathways to cope with oxygen level decrease or oxygen deficiency
(25,26). Therefore, it can be speculated
that upregulation of Sirt1 could be a compensatory mechanism by
which cardiomyocytes cope with hypoxic stress.
Accumulating studies have confirmed that Sirt1 has a
crucial role in cardiac protection in various cardiovascular
diseases through a complex signalling network, including autophagy
(3) and apoptosis (23). The present study demonstrated that
Sirt1 protected cardiomyocytes from hypoxia-induced apoptosis, at
least in part, by inhibiting the IRE1α cascade in cardiomyocytes
and by promoting autophagy via AMPK activation. AMPK is a
serine/threonine kinase that senses the cellular energy status and
regulates energy homeostasis (15). It is widely accepted that AMPK
activation inhibits mTOR, leading to enhanced autophagy (16). The present study demonstrated that
Sirt1 activated AMPK in cardiomyocytes under hypoxic conditions,
and that the pro-autophagy effect of Sirt1 was abolished when AMPK
was inhibited. Cells were pre-incubated with Compound C for 2 h
prior to hypoxic exposure to inhibit AMPK. In previous studies, the
timing of the administration of Compound C before treatments varied
from minutes to hours (27-29), suggesting that Compound C has a
long term effect in inhibiting AMPK.
Cardiomyocyte apoptosis has a detrimental role in
the progression of CHD (9). In
the present study, expression of phosphorylated IRE1α was
demonstrated to be increased in H9C2 cells exposed to 24 h hypoxia
and in heart samples from mice that were exposed to 2 weeks
hypoxia. IRE1α over-expression increased the rate of
hypoxia-induced apoptosis in H9C2 cells exposed to hypoxia for 24
h, whereas IRE1α inhibition limited apoptosis in H9C2 cells under
the same conditions. These results demonstrated that sustained
IRE1α activation increases apoptosis in hypoxic cardiomyocytes. It
is widely accepted that activated JNK translocates from the
cytoplasm to the nucleus, and consequently phosphorylates ser63 and
ser73 of transcription factor c-jun, and promotes apoptosis by
regulating apoptosis-related proteins (9). NF-κB p65, a member of the NF-κB
family, is involved in immune responses and inflammation, and
activation of the NF-κB signalling pathway can induce the
production of inflammatory cytokines. A recent study revealed that
persistent activation of NF-κB was pro-apoptotic in H9C2
cardiomyocytes (30). The current
study demonstrated that the IRE1α branch of the UPR promoted
apoptosis in chronically hypoxic cardiomyocytes by activating the
pro-apoptotic JNK/c-jun signalling pathway and the NF-κB p65
pathway, and that these effects could be reversed by activation of
Sirt1. Collectively, the present findings suggest that Sirt1 may
exert its protective effects in hypoxic H9C2 cells by decreasing
apoptosis via IRE1α inhibition and promoting autophagy through AMPK
activation.
To further elucidate the protective role of Sirt1 in
hypoxia-induced cardiotoxicity in vivo, a hypoxic mouse
model was established. SRT1720 and EX-527 were used to modulate the
cardiac Sirt1 expression in mice. SRT1720 is a specific and
effective Sirt1 agonist, and has been reported to be 1,000-fold
more potent than resveratrol (31). EX-527 inhibits Sirt1 by closing
the NAD+ binding site of Sirt1, and it has been widely
used as a selective Sirt1 inhibitor (32). Consistent with the in vitro
results, the in vivo study demonstrated that treating
hypoxic mice with the Sirt1 agonist SRT1720 limited hypoxia-induced
apoptosis and upregulated autophagy in mice housed in hypoxic
conditions. EX-527 treatment exhibited the opposite trends. The
in vivo results support the beneficial pharmacological
effect of SRT1720.
Recent studies have revealed that autophagy is
closely related to apoptosis (33). Autophagy could block the induction
of apoptosis by inhibiting the activation of apoptosis-associated
caspases. It is widely accepted that autophagy exhibits a
protective role by inhibiting apoptosis (34). Therefore, it is possible that
Sirt1 inhibits hypoxia-induced apoptosis in cardiomyocytes
partially by promoting autophagy, which needs to be investigated in
future studies. The present study has certain limitations. Firstly,
only one cell line, H9C2, was used for the in vitro
experiments. Secondly, further studies will be needed to fully
elucidate how Sirt1 regulates AMPK and IREα. The current results,
however, suggest that Sirt1 has the potential to favourably
modulate autophagy and decrease apoptosis in the context of hypoxic
cardiomyopathy, and that the beneficial effects of Sirt1 depend at
least partially on AMPK activation and IRE1α inhibition. Therefore,
Sirt1 may be a promising protective target to prevent
hypoxia-induced cardiac damage.
Funding
This study was supported by the National Natural
Science Foundation of China (grant nos. 81770248, 81370004,
81270228 and 81471408).
Availability of data and materials
The datasets used and/or analysed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
GL and YX designed the study. ZJ, BC, and YZ
collected and analyzed the data. YZ, RM, and FT edited the language
of the manuscript, and were involved in data analysis. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
All protocols involving animals were approved by the
Experimental Animal Committee of The Second Affiliated Hospital of
The Third Military Medical University, Chongqing, China.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Anastasiou D and Krek W: SIRT1: Linking
adaptive cellular responses to aging-associated changes in
organismal physiology. Physiology (Bethesda). 21:404–410. 2006.
|
|
2
|
Liu L, Liu C, Zhang Q, Shen J, Zhang H,
Shan J, Duan G, Guo D, Chen X, Cheng J, et al: SIRT1-mediated
transcriptional regulation of SOX2 is important for self-renewal of
liver cancer stem cells. Hepatology. 64:814–827. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hariharan N, Maejima Y, Nakae J, Paik J,
Depinho RA and Sadoshima J: Deacetylation of FoxO by Sirt1 plays an
essential role in mediating starvation-induced autophagy in cardiac
myocytes. Cir Res. 107:1470–1482. 2010. View Article : Google Scholar
|
|
4
|
Liu B, Ghosh S, Yang X, Zheng H, Liu X,
Wang Z, Jin G, Zheng B, Kennedy BK, Suh Y, et al: Resveratrol
rescues SIRT1-dependent adult stem cell decline and alleviates
progeroid features in laminopathy-based progeria. Cell Metab.
16:738–750. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang Y, Duan W, Li Y, Jin Z, Yan J, Yu S
and Yi D: Novel role of silent information regulator 1 in
myocardial ischemia. Circulation. 128:2232–2240. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alcendor RR, Gao S, Zhai P, Zablocki D,
Holle E, Yu X, Tian B, Wagner T, Vatner SF and Sadoshima J: Sirt1
regulates aging and resistance to oxidative stress in the heart.
Circ Res. 100:1512–1521. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ding C, Zou Q, Wang F, Wu H, Wang W, Li H
and Huang B: HGF and BFGF secretion by human adipose-derived stem
cells improves ovarian function during natural aging via activation
of the SIRT1/FOXO1 signaling pathway. Cell Physiol Biochem.
45:1316–1332. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hsu CP, Zhai P, Yamamoto T, Maejima Y,
Matsushima S, Hariharan N, Shao D, Takagi H, Oka S and Sadoshima J:
Silent information regulator 1 protects the heart from
ischemia/reperfusion. Circulation. 122:2170–2182. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
He S, Liu P, Jian Z, Li J, Zhu Y, Feng Z
and Xiao Y: miR-138 protects cardiomyocytes from hypoxia-induced
apoptosis via MLK3/JNK/c-jun pathway. Biochem Biophys Res Commun.
441:763–769. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lim JH, Lee YM, Chun YS, Chen J, Kim JE
and Park JW: Sirtuin 1 modulates cellular responses to hypoxia by
deacetylating hypoxia-inducible factor 1alpha. Mol Cell.
38:864–878. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dioum EM, Chen R, Alexander MS, Zhang Q,
Hogg RT, Gerard RD and Garcia JA: Regulation of hypoxia-inducible
factor 2alpha signaling by the stress-responsive deacetylase
sirtuin 1. Science. 324:1289–1293. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rabinowitz JD and White E: Autophagy and
metabolism. Science. 330:1344–1348. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nakai A, Yamaguchi O, Takeda T, Higuchi Y,
Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et
al: The role of autophagy in cardiomyocytes in the basal state and
in response to hemodynamic stress. Nat Med. 13:619–624. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu L, Jin X, Hu CF, Li R, Zhou Z and Shen
CX: Exosomes derived from mesenchymal stem cells rescue myocardial
ischaemia/reperfusion injury by inducing cardiomyocyte autophagy
Via AMPK and Akt pathways. Cell Physiol Biochem. 43:52–68. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang H, Liu B, Li T, Zhu Y, Luo G, Jiang
Y, Tang F, Jian Z and Xiao Y: AMPK activation serves a critical
role in mitochondria quality control via modulating mitophagy in
the heart under chronic hypoxia. Int J Mol Med. 41:69–76. 2018.
|
|
16
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shore GC, Papa FR and Oakes SA: Signaling
cell death from the endoplasmic reticulum stress response. Curr
Opin Cell Biol. 23:143–149. 2011. View Article : Google Scholar :
|
|
18
|
Zhao W, Han F and Shi Y: IRE1alpha pathway
of endoplasmic reticulum stress induces neuronal apoptosis in the
locus coeruleus of rats under single prolonged stress. Prog
Neuropsychopharmacol Biol Psychiatry. 69:11–18. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brozzi F, Nardelli TR, Lopes M, Millard I,
Barthson J, Igoillo-Esteve M, Grieco FA, Villate O, Oliveira JM,
Casimir M, et al: Cytokines induce endoplasmic reticulum stress in
human, rat and mouse beta cells via different mechanisms.
Diabetologia. 58:2307–2316. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brozzi F, Gerlo S, Grieco FA, Juusola M,
Balhuizen A, Lievens S, Gysemans C, Bugliani M, Mathieu C,
Marchetti P, et al: Ubiquitin D regulates IRE1α/c-Jun N-terminal
Kinase (JNK) protein-dependent apoptosis in pancreatic beta cells.
J Biol Chem. 291:12040–12056. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jain K, Suryakumar G, Ganju L and Singh
SB: Amelioration of ER stress by 4-phenylbutyric acid reduces
chronic hypoxia induced cardiac damage and improves hypoxic
tolerance through upregulation of HIF-1α. Vascul Pharmacol.
83:36–46. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu B, Zhang HG, Zhu Y, Jiang YH, Luo GP,
Tang FQ, Jian Z and Xiao YB: Cardiac resident macrophages are
involved in hypoxiainduced postnatal cardiomyocyte proliferation.
Mol Med Rep. 15:3541–3548. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zheng W, Lu YB, Liang ST, Zhang QJ, Xu J,
She ZG, Zhang ZQ, Yang RF, Mao BB, Xu Z, et al: SIRT1 mediates the
protective function of Nkx2.5 during stress in cardiomyocytes.
Basic Res Cardiol. 108:3642013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sommer RJ, Hijazi ZM and Rhodes JF:
Pathophysiology of congenital heart disease in the adult: Part III:
Complex congenital heart disease. Circulation. 117:1340–1350. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhu L, Wang Q, Zhang L, Fang Z, Zhao F, Lv
Z, Gu Z, Zhang J, Wang J, Zen K, et al: Hypoxia induces PGC-1alpha
expression and mitochondrial biogenesis in the myocardium of TOF
patients. Cell Res. 20:676–687. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhu Y, Feng Z, Jian Z and Xiao Y: Long
noncoding RNA TUG1 promotes cardiac fibroblast transformation to
myofibroblasts via miR29c in chronic hypoxia. Mol Med Rep.
18:3451–3460. 2018.PubMed/NCBI
|
|
27
|
Yan J, Duan J, Wu X, Guo C, Yin Y, Zhu Y,
Hu T, Wei G, Wen A and Xi M: Total saponins from Aralia taibaiensis
protect against myocardial ischemia/reperfusion injury through AMPK
pathway. Int J Mol Med. 36:1538–1546. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gu Y, Gao L, Chen Y, Xu Z, Yu K, Zhang D,
Zhang G and Zhang X: Sanggenon C protects against cardiomyocyte
hypoxia injury by increasing autophagy. Mol Med Rep. 16:8130–8136.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu MH, Lin XL, Guo DM, Zhang Y, Yuan C,
Tan TP, Chen YD, Wu SJ, Ye ZF and He J: Resveratrol protects
cardiomyocytes from doxorubicin-induced apoptosis through the
AMPK/P53 pathway. Mol Med Rep. 13:1281–1286. 2016. View Article : Google Scholar
|
|
30
|
Hamid T, Gu Y, Ortines RV, Bhattacharya C,
Wang G, Xuan YT and Prabhu SD: Divergent tumor necrosis factor
receptor-related remodeling responses in heart failure: Role of
nuclear factor-kappaB and inflammatory activation. Circulation.
119:1386–1397. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Milne JC, Lambert PD, Schenk S, Carney DP,
Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, et al: Small
molecule activators of SIRT1 as therapeutics for the treatment of
type 2 diabetes. Nature. 450:712–716. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vachharajani VT, Liu T, Brown CM, Wang X,
Buechler NL, Wells JD, Yoza BK and McCall CE: SIRT1 inhibition
during the hypoinflammatory phenotype of sepsis enhances immunity
and improves outcome. J Leukoc Biol. 96:785–796. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Song S, Tan J, Miao Y, Li M and Zhang Q:
Crosstalk of autophagy and apoptosis: Involvement of the dual role
of autophagy under ER stress. J Cell Physiol. 232:2977–2984. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Thorburn A: Apoptosis and autophagy:
Regulatory connections between two supposedly different processes.
Apoptosis. 13:1–9. 2008. View Article : Google Scholar :
|