With the rapid development of research in the field
of cellular death, it is acknowledged that necrosis can also be
regulated in a programmed manner via a specific signal transduction
pathway called necroptosis or programmed necrosis (1,2).
Necroptosis-mediated cell rupture is morphologically characterized
by the loss of cell plasma membrane and the swelling of organelles
(particularly mitochondria). Nevertheless, necroptosis, a form of
programmed cell death (PCD), and its upstream molecular signaling
pathways are under strict control (3,4).
The initiation of necroptosis requires several different stimuli,
as well as the kinase activity of receptor-interacting
serine/threonine kinase 1 (RIP1) and receptor-interacting
serine/threonine kinase 3 (RIP3) (5).
The present review examined the functions of
RIP1/RIP3-regulated necroptosis on multifaceted pathological
mechanisms. Furthermore, the importance of RIP1/RIP3 in determining
the cell outcome was highlighted by the interactive molecular
pathways noted among cell survival, apoptosis and necroptosis.
Then, the pivotal roles of RIP1/RIP3 in disease treatment were
discussed, highlighting their application potential as new drug
targets.
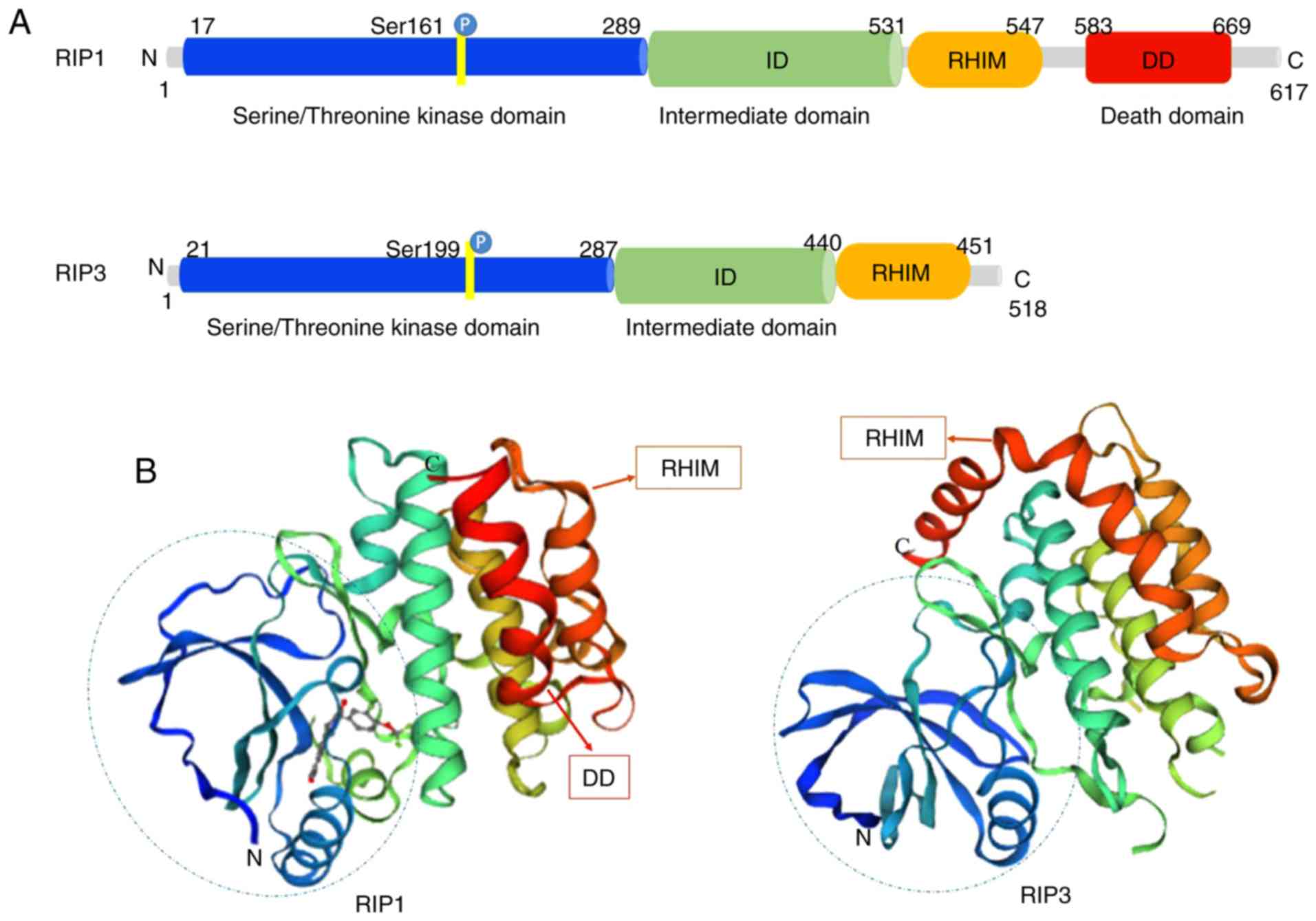
Structurally, RIP1 and RIP3 share nearly half of
their amino acid sequences and have very similar topology features.
The RIP1 protein consists of 671 amino acids (Homo sapiens).
It contains a N-terminal serine/threonine kinase domain, an
intermediate domain (ID), a RHIM and a C-terminal DD (13) (Fig.
1A). RIP3 is composed of 518 amino acids (Homo sapiens),
and contains a N-terminal kinase domain similar to that of RIP1, a
RHIM domain and a unique C-terminus without a DD (14) (Fig.
1A). RIP1 acts as a multifunctional adaptor protein in response
to the activated signal of death receptors (DRs), and its DD binds
to the DRs of TNFR1, Fas and TNF-related apoptosis inducing ligand
(TRAIL) (15,16). It mediates prosurvival NF-κB
activation, caspase-dependent apoptosis and RIP kinase-dependent
necroptosis (17). The ID of RIP1
contains the RHIM that enables the protein to combine with RIP3. In
contrast to RIP1, RIP3 is not directly required for DR-induced cell
survival or death (18). RIP3
binds to RIP1 through its unique C-terminal segment to inhibit RIP1
and TNFR1-mediated NF-κB activation (19) (Fig.
1B). Experiments have revealed that tumor necrosis factor (TNF)
induces the formation of an RIP1/RIP3 complex, indicating that RIP1
interacts with RIP3 through the homotypic RHIM domain (20).
Necroptosis can provide a substitute suicide
mechanism in case of malfunction of the classical apoptosis
machinery (21). Most work on
necroptosis concerns studies of TNF signaling. TNF is a pleiotropic
cytokine that has an essential role in inflammation, tissue injury
and cell death (22). In the TNF
receptor superfamily, researchers have found six human DRs,
including TNFR1, Fas (also known as CD95 or APO-1), DR3 (also known
as TRAMP or APO-3), TRAIL receptor 1 (TRAILR1, also known as DR4),
TRAIL receptor 2 (TRAILR2, also known as DR5, TRICK or KILLER) and
DR6 (also known as CD358) (23-26). However, the most prevalent pathway
is the TNFR1-mediated signal transduction, which can propel cell
survival, apoptosis and necroptosis (27). The present review focused on the
three most dominant of those TNF-mediated pathways.
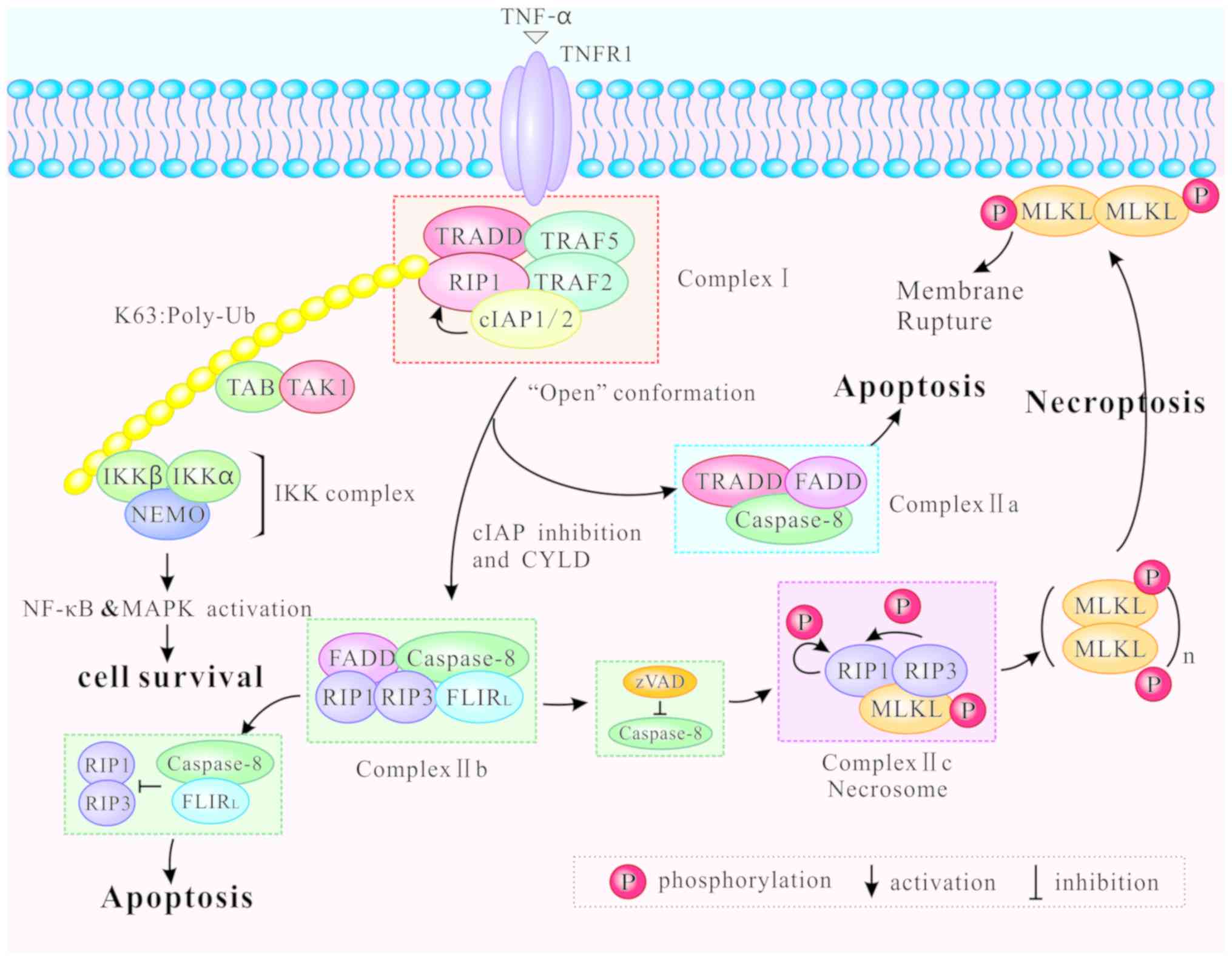
Different modifications of RIP1 can induce distinct
outcomes of cell survival, apoptosis and necroptosis. Following
binding of TNF-α to TNFR1 at the plasma membrane,
TNF-receptor-associated death domain (TRADD) recruits downstream
proteins, namely RIP1, the E3 ubiquitin ligases
TNF-receptor-associated factor (TRAF) 2, TRAF5, and the cellular
inhibitor of apoptosis (cIAP) 1 and cIAP2, to form the complex I
(28,29). Then, the complex I mediates NF-κB
and MAPK signaling, contributing to cell survival or other
non-death functions (4,30,31). The K63-linked ubiquitination of
RIP1 by cIAP1/2 promotes both the formation and activation of the
transforming growth factor-activated kinase 1 (TAK1)-binding
protein (TAB) complex and the inhibitor of NF-κB kinase (IKK)
complex (consisting of NF-κB essential modulator, IKKα and IKKβ),
supporting the NF-κB pathway activation, and ultimately leading to
cell survival (1) (Fig. 2).
Complex I internalizes and transforms into a
death-inducing complex II following caspase-8 activation (29). Two distinct types of complex II
(IIa and IIb) can be distinguished based on their composition and
the activity of their proteins. After dissociating from TNFR1,
TRADD recruits Fas-associated protein with death domain (FADD) and
further promotes recruitment and activation of caspase-8 to form
the complex IIa (29,32). Activation of caspase-8
subsequently induces apoptosis independently of RIP1 or its kinase
activity (32). In certain
circumstances, including upon the absence of cIAP1/2, upon
inhibition of IAP mediated by a small molecule mimic of diablo
IAP-protein mitochondrial protein (33) and upon auto-degradation of
cIAP1/2, RIP1 is released from complex I to form a
caspase-8-activating complex (complex IIb) which mediates apoptosis
(32). Complex IIb consists of
RIP1, RIP3, FADD and the FLICE-like inhibitory protein long form
(FLIPL)/caspase-8 heterodimer, and it promotes RIP1- and
caspase-8-dependent apoptosis (22). Hence, RIP1 is not an indispensable
factor in apoptosis, although the process is favored by its
presence. Under physiological levels, the caspase-8/FLIPL
heterodimer facilitates caspase-8 oligomer assembly to trigger
apoptosis. By contrast, high levels of FLIPL and FLICE-like
inhibitory protein short form (FLIPS) restrict caspase-8/c-FLIPL/S
heterodimer activity, leading to inhibition of apoptosis (34-36). Inactivation of RIP1 and RIP3,
mediated by cleavage via caspase-8/FLIPL heterodimer in Complex
IIb, inhibits necroptosis (37,38) (Fig.
2).
When caspase-8 is inhibited by zVAD, RIP1 and RIP3
are combined via the RHIM, and form complex IIc, also known as the
necrosome (39). Complex IIc is a
crucial cytoplasmic signaling complex, which does not appear in the
TNF-induced cell survival or apoptosis (10,40). Mitochondrial reactive oxygen
species (ROS) oxidize RIP1 at three crucial cysteine sites (C257,
C268 and C586), and promote autophosphorylation of RIP1 at Ser161.
RIP1 autophosphorylation is pivotal for the recruitment of RIP3
(41). In addition, CYLD lysine
63 deubiquitinase (CYLD), as a deubiquitinase removing
polyubiquitin chains from RIP1, facilitates the formation and
activation of RIP1/RIP3 necrosomes by deubiquitylating RIP1. When
CYLD is deficient, necrosomes promote a high level of ubiquitinated
RIP1 and block phosphorylation of RIP1 and RIP3 (32,42). Furthermore, the RHIM is also
required for the RIP1/RIP3 complex formation (19). After RIP1 and RIP3 are combined,
RIP1 gets phosphorylated by RIP3 (10). Intramolecular auto- and
trans-phosphorylation of RIP1/RIP3 promotes recruitment of another
key necroptosis-signaling protein, the mixed lineage kinase domain
like protein (MLKL). MLKL is then phosphorylated by RIP3 to
initiate necroptosis (43). The
phosphorylated MLKL is transferred from the cytosol to the plasma
and intracellular membranes via the four helical bundle-brace
(4HBD-BR) regions of MLKL (44).
The oligomerization of MLKL causes membrane pore formation,
resulting in the destruction of membrane integrity and eventually
leading to necrotic death (45)
(Fig. 2).
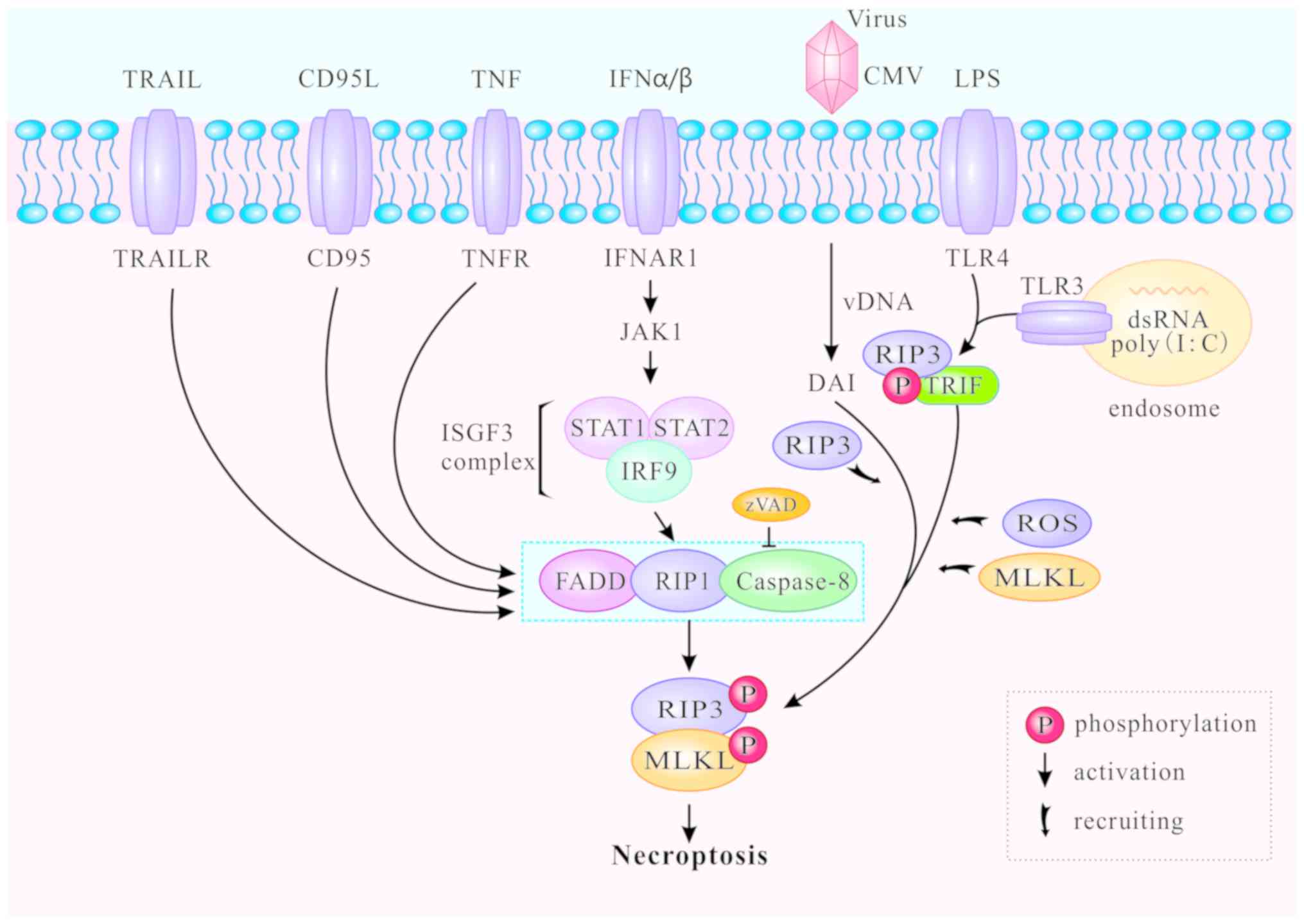
In addition to the classic TNFR1-induced necroptosis
pathway described above, toll-like receptors (TLR) can also mediate
necroptosis. The TLR signaling pathway is generally triggered by
pathogen-associated molecular patterns during viral or microbial
infection (46,47). TLR-mediated necroptosis results in
the destruction of infected cells and is thus beneficial to the
host. The downstream MLKL signaling pathway of RIP3 is
indispensable for both TNFR1- and TLR-induced signaling (46), and caspase-8 can block necroptosis
that is directly initiated by the TIR domain-containing
interferon-β (TRIF)/RIP3/MLKL pathway (48,49). TLR4 and TLR3 are respectively
activated by lipopolysaccharide (LPS) (50) and polyinosine-polycytidylic acid
(I:C), a synthetic double stranded RNA (dsRNA) mimic (51). Thereafter, TLR3 and TLR4 activate
RIP3 and participate in ensuing necroptosis via TRIF or MyD88
(52-54). The C-terminal RHIM motif is
required for RIP3 to interact with TRIF or MyD88. The RIP3/TRIF
signaling complex recruits and phosphorylates MLKL, inducing ROS
accumulation and mediating TLR3- and TLR4-induced necroptosis
(46,47) (Fig.
3).
Increasing numbers of necroptotic stimuli have been
identified and divided into two groups: RIP1-dependent and
RIP1-independent (Fig. 3).
RIP1-dependent stimuli include TNF-α, Fas, TRAIL, interferon
(IFN)-α and IFN-β. The primary death-inducing signaling complex
(DISC) is assembled by stimulation of Fas or TRAILR at the plasma
membrane, thereby activating caspase-8 and triggering apoptosis
independently of RIP1 (55). cIAP
deficiency promotes the recruitment of RIP1 and Fas when caspase-8
is blocked, and enhances the formation of the cytosolic ripoptosome
complex which induces necroptosis (56). In bone-marrow-derived macrophages,
type I IFNα and IFNβ bind to their cognate receptor IFNα/β receptor
subunit 1 (IFNAR1) to activate Janus kinase 1 and form the
IFN-stimulated gene factor 3 (ISGF3) complex (consisting of STAT1,
STAT and IFN-regulatory factor 9). The ISGF3 complex promotes
induction and activation of necrosomes, and triggers necroptosis in
a transcription-dependent pathway (57). RIP1-independent stimuli generally
refer to LPS, dsRNA and viruses. DNA-dependent activator of IFN
regulatory factors (DAI) can identify viral dsRNA, promote the
recruitment of RIP3 to form necrosomes without RIP1, and induce
RIP3-dependent necroptosis (58).
Illuminating the molecular mechanisms involved in necroptosis will
elucidate further the molecular biology underlying the
pathology.
RIP1 and RIP3 were recently delineated as two
important effectors in the cell death network, as they are cascade
proteins responding to complex TNFR signaling and regulating
cellular survival, apoptosis and necroptosis. It is important to
note that RIP3 is indispensable for necroptosis, whereas RIP1 is
not. TNF-α-induced RIP1/RIP3 interaction engages RIP3 recruitment,
leading to RIP3/RIP3 homo-oligomerization and RIP3
autophosphorylation. Phosphorylated RIP3 recruits and
phosphorylates MLKL, which promotes MLKL oligomer-executed
necroptosis (59). However, the
directorial functions of RIP1 are not always positive for RIP3.
Researchers have constructed RIP3 dimers and RIP3 oligomers to
analyze how RIP1 induces the activation of RIP3 (60). The results demonstrated that the
dimerization of RIP3 alone was insufficient to induce the cell
death. The RIP3 dimer seeded a RHIM-dependent complex controlled by
both caspase-8 and RIP1. On the other hand, without TNF stimulation
and RIP1 activity, the oligomerization of RIP3 was sufficient to
induce necroptosis (60). These
results indicated that RIP1 not only activates RIP3 in response to
TNF signaling, but also participates in cytoprotection. TNFR1
promotes the activation of the NF-κB pathway, p38α and its
downstream effector MAPK-activated protein kinase 2 kinase (MK2),
thereby promoting cell survival (61). Recently, Jaco et al
(62) reported that activated MK2
phosphorylated RIP1 at Ser321 to synergize TNF-induced cell rescue.
Therefore, these results suggest that RIP1 serves as a positive
checkpoint within TNF stimulation that integrates cytokine
production and cell survival. RIP1 is therefore thought as an
inhibitor rather than an initiator of RIP3-induced necroptosis
(63).
Furthermore, ROS participate in the regulation of
necroptosis in many cell types, and enhance the formation of
necrosomes induced by Smac mimetic bivalent 6 compound (BV6)/TNFα
(64). BV6/TNFα-stimulated ROS
generation promotes the stabilization of the RIP1/RIP3 feedback
loop. A previous study demonstrated that the metabolic enzymes
glycogen phosphorylase (PYGL), glutamate dehydrogenase 1 (GLUD1),
and glutamate-ammonia ligase (GLUL), are activated by RIP3
(65). Enhancement of aerobic
respiration is mediated by TNF-induced ROS. However, the major
mechanism of ROS and RIP1/RIP3 remains not completely understood. A
recent study demonstrated that mitochondrial ROS activated RIP1
autophosphorylation at Ser161 via oxidation of three crucial
cysteines in RIP1 (41). This
phosphorylation accelerated RIP1 recruitment of the RIP3
aggregation and formed a necrosome, which then resulted in
mitochondrial depolarization and cell necroptosis (66). These studies provided essential
insight into the reciprocal regulation between RIP1/RIP3 and ROS in
necroptosis.
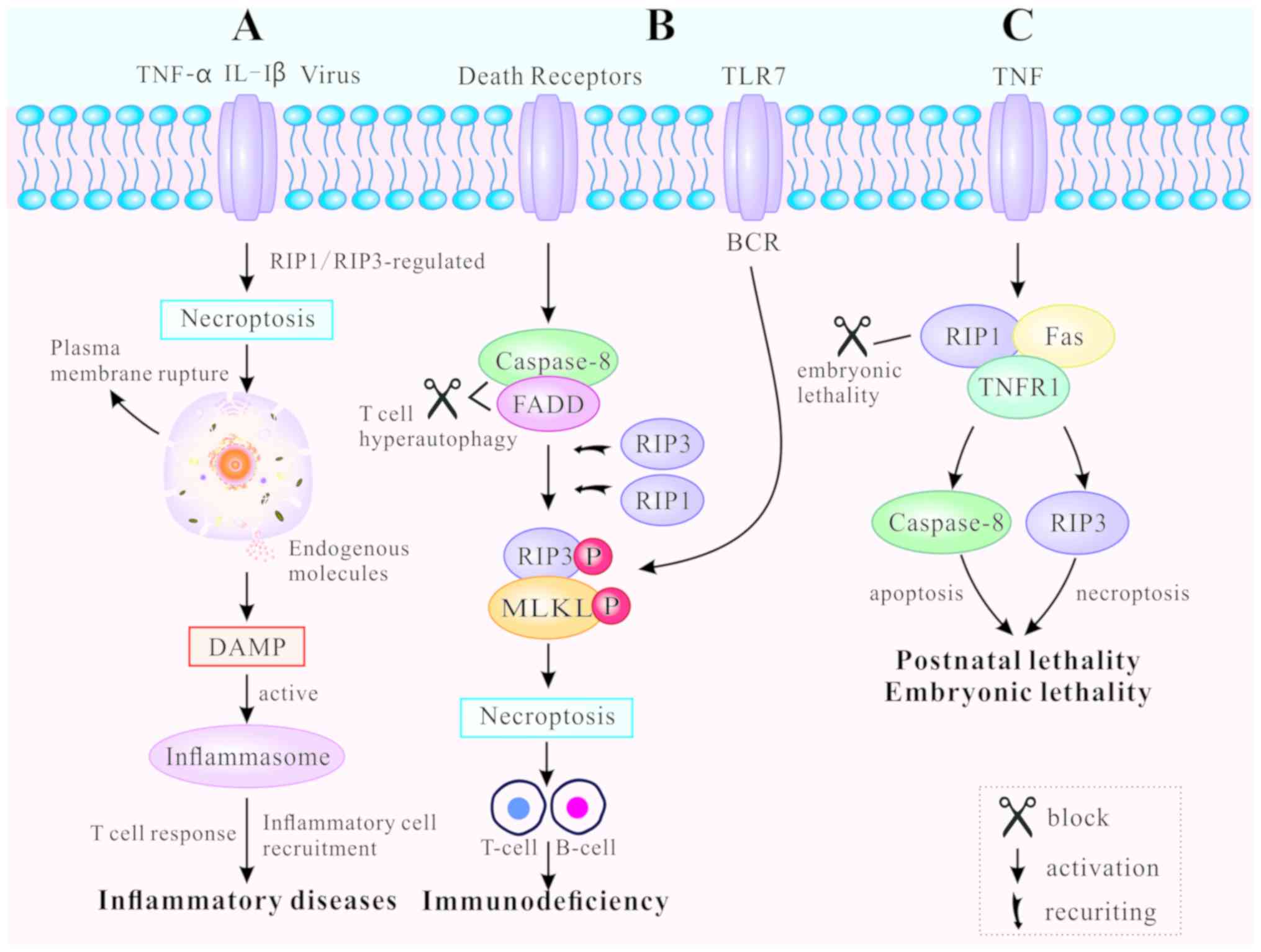
Necroptosis, as a novel pathway of PCD, leads to
release of endogenous moldecules from disrupted dying cells, that
subsequently triggers inflm-mation and immune response (67). Necroptosis-inducing factors
include TLR3 and TLR4 agonists [such as interleukin (IL)-1β)], TNF,
certain viral infections and T cell receptors (22). TNF-mediated and TLR-mediated
signaling pathways are the primary pathways for necroptosis, both
regulated by RIP3 (10).
RIP1/RIP3 or RIP3/TRIF signaling complexes recruit and
phosphorylate downstream MLKL, causing rupture of the plasma
membrane, as well as the release of endogenous molecules (45). These endogenous molecules are
known as damage-associated molecular patterns (DAMPs). They are
also identified as part of the extended IL-1 family (IL-1α, IL-1β,
IL-18, IL-33, IL-36α, β and γ) (68). The leakage of DAMPs can activate
inflammasomes, facilitate inflammatory cell recruitment to the site
of infection, and promote subsequent virus-specific T cell
responses to induce inflammation (69). Thus, regulating the mechanism of
necroptosis can result in both inhibition and promotion of the
immune response (Fig. 4A).
DRs regulate necroptosis in various cell types, such
as B lymphocytes and T lymphocytes, both of which are essential for
immune homeostasis and tolerance (48,70). Blocked DRs fail to clear activated
lymphocytes and unbalanced lymphoid homeostasis, ultimately leading
to autoimmune lymphoproliferative syndrome (ALPS) (71). FADD, caspase-8 and RIP kinases are
indispensable for T cell clonal expansion, contraction and
antiviral responses (48). When
caspase-8 and FADD are deficient, T cells undergo hyper-autophagy
and proliferative inhibition in response to antigenic stimulation.
In addition, T cells generate necrosomes, which induce
caspase-8-independent necroptosis (72). RIP1 and RIP3 are either recruited
by DRs or by other cellular signaling molecules, including DNA
damage and antigen receptor ligation (73). Chronic necroptosis may be the
basis of human fibrotic and autoimmune disorders (74). Likewise, B cell receptor (BCR)
mediates necroptosis via reaction with TLR7 to prevent autoimmune
diseases (75,76). The RIP1 inhibitor necrostatin-1
(nec-1) has been demonstrated to suppress B-cell necroptosis, when
pretreating B cells from patients with systemic lupus erythematosus
(70). Fig. 4B illustrates how programmed death
can promote clonal loss of lymphocytes during infection and protect
patients against autoimmune disease.
Apoptosis and necroptosis are closely associated
with embryonic lethality and postnatal development (49). RIP1 determines cell survival or
death by associating with TNFR1, TLRs and Fas (17,77). Embryonic lethality of
RelA-deficient mice has been demonstrated to be mediated by
apoptosis and necroptosis (78).
FADD functions as an adaptor to induce apoptosis by recruiting and
activating caspase-8 (79). The
characteristics of FADD-deficient embryos are high levels of RIP1
production and massive necrosis. RIP1 ablation allows normal
embryo-genesis in FADD-deficient mice, but these mice usually die
around their first postnatal day (80). In addition, different RIP1 kinase
inactivating mutations have distinct effects on the embryogenesis
of FADD-deficient mice. For example, RIP1K45A has been found not to
prevent the embryonic lethality of FADD-deficient mice, while RIP1Δ
(with an altered P-loop in the kinase domain) does (81). If RIP1 was necessary for the
activation of RIP3, as aforementioned, FADD−/−
RIP1−/− and FADD −/− RIP3−/− mice
should survive to adulthood (82). Nevertheless, a recent study
revealed that RIP1-deficient mice die soon after birth, leading to
speculation for the positive role of RIP1 in embryonic development
and postnatal life (83). RIP1
deletion enhances primary cell sensitivity to
FADD/caspase-8-mediated apoptosis induced by TNF. In addition, RIP1
deletion promotes RIP3/MLKL-mediated necroptosis induced by TLR
ligation (via TRIF) or interferon (84). The perinatal lethality of
RIP1−/− mice can be rescued by a combination of
additional mutations. A recent study indicated that haploid
insufficiency of RIP3 improved the survival period of
RIP1−/− FADD−/− double knockout mice beyond
weaning age, while RIP1−/− FADD−/−
RIP3−/− triple knockout (TKO) mice were significantly
smaller in size and weight (85).
Furthermore, complete ablation of RIP3 further prolonged the life
span of TKO mice displaying normal size and weight (85). Therefore, it can be concluded that
RIP1 may prevent postnatal lethality by blocking two different cell
death pathways: FADD/caspase-8-mediated apoptosis and
RIPK3/MLKL-mediated necroptosis. The effect of PCD on animal
development is highly complex and has not yet been thoroughly
elucidated (Fig. 4C).
The increasing discovery of inhibitors and drugs
affecting the RIP1/RIP3 cascade pathway already shows promise
towards the treatment of various diseases (Table I). Nec-1 has been identified as a
specific and potent small-molecule and active inhibitor of RIP1
(2). Nec-1 is widely used in
disease models to examine the contribution of RIP1 to cell death
and inflammation. Other necrostatins, including Nec-3, Nec-4 and
Nec-5, also stabilize RIP1 in an inactive conformation through
interactions with hydrophobic pockets and highly conserved amino
acids (2,86). However, the strongest inhibition
of RIP1 has been observed with Nec-1 stable (Nec-1s) (87). A previous study identified
GSK2982772 (compound 5) as a novel inhibitor of RIP1 (88). GSK2982772 potently binds to RIP1
with exquisite kinase specificity and has high activity in blocking
TNF-dependent necroptosis, as well as inflammation. Based on this
previous study of GSK2982772, Yoshikawa et al (27) designed and synthesized a novel
class of RIP1 kinase inhibitor, the compound 22
[7-oxo-2,4,5,7-tetrahydro-6H-pyrazolo(3,4-c)pyridine], which
possesses moderate RIP1 kinase inhibitory activity and P-gp
mediated efflux. Furthermore, using a mouse model of systemic
inflammatory response syndrome, it was demonstrated that compound
56 (RIPA-56) targeted RIP1 directly, and reduced TNFα-induced cell
mortality and multi-organ damage (89). The pan-Aurora kinase inhibitor
Tozasertib (also known as VX-680 and MK-0457) was recently
demonstrated as a potent compound in inhibition of RIP1-dependent
necroptosis and in the blockage of cytokinesis in cells (90). The food and drug
administration-approved anticancer agents ponatinib and pazopanib
were demonstrated to be submicromolar inhibitors of necroptosis
through the targeting of components upstream of MLKL (91). Ponatinib inhibits both RIP1 and
RIP3, while pazopanib preferentially targets RIPK1. Both drugs have
potential values for the treatment of pathology caused or
aggravated by necroptotic cell death (91). Overall, the aforementioned studies
indicated that RIP1 kinase may serve as a novel target for
therapeutic drug development in human disease therapy.
RIP3 is a critical regulator of necroptosis,
however, very few specific inhibitors have yet been reported. B-Raf
(V600E) inhibitors are generally considered as an important
anticancer drug in metastatic melanoma therapy (92). To date, the B-Raf inhibitor
dabrafenib was demonstrated to be a potent inhibitor of RIP3
(92). Dabrafenib decreased
RIP3-mediated Ser358 phosphorylation of MLKL and disrupted the
interaction between RIP3 and MLKL (93). Results indicated that dabrafenib
could serve as a RIP3 inhibitor and as a potential preventive or
therapeutic agent for RIP3-involved necroptosis-related diseases
(93). As expected, dabrafenib
significantly reduced infarct lesion size and attenuated
upregulation of TNF-α in mouse models of ischemic brain injury
(94). In addition, murine
cytomegalovirus (CMV) M45 contains a RHIM domain, and was confirmed
to be a competitive inhibitor of RIP3 (95). Human CMV blocks TNF-induced
necroptosis following RIP3 activation and MLKL phosphorylation
(95), leading to inhibition of
the host defense mechanism.
As diagnostic and therapeutic strategies, miRNAs
provide a novel perspective for RIP1 inhibition. As aforementioned,
several inhibitors have been demonstrated to suppress the
pro-necroptosis function of RIP1 at the protein level; however,
these do not function at the mRNA level. miR-155 represses
cardiomyocyte progenitor cell necroptosis by targeting RIP1 rather
than activating the Akt pro-survival pathway (96), suggesting that miR-155 might be a
novel approach in improving cell engraftment. Additionally, in a
myocardial ischemia/reperfusion model, it was demonstrated that
miR-103/107, as a necrosis-suppressor miRNA, directly targeted FADD
(97,98). FADD participates in hydrogen
peroxide-induced necroptosis by influencing the formation of
RIP1/RIP3 complex, suggesting that FADD-targeting by miR-103/107
might be a new approach for preventing myocardial necrosis. Recent
research has indicated that miRNA dysregulation is involved in
triple-negative breast cancer (TNBC) (100). Overexpression of miR-182
inhibits the CYLD action on the ubiquitin chains on RIP1, leading
to caspase-8-dependent apoptosis in TNF-α-treated TNBC cells
(99). Additionally, miR-145,
which is downregulated in TNBC, targeted cIAP1 and reduced the
formation of the RIP1/FADD-caspase-8 complex (100). Therefore, miRNAs can be
perceived as a novel approach for RIP1 regulation. However, their
potential effects in disease intervention requires further research
(Table II).
In TNFα-induced necroptosis, as RIP1 and RIP3 form a
protein complex through their common RHIM domain, phosphorylation
and activation of RIP3 and downstream MLKL occur (106). In addition, RHIM-containing
proteins, such as TLR, and interferon regulatory factors (Z-DNA
binding protein 1, also known as DAI or DLM1) are known to activate
RIP3 and further transduce necrosis signals to MLKL (107). TLR3 or TLR4 directly activate
necroptosis through the RHIM-dependent association of TRIF with
RIP3. This pathway proceeds independently of RIP1, but remains
dependent on MLKL downstream of RIP3 kinase (46). Dyngo 4a blocks the internalization
of TLR4 and prevents RIP3-induced necroptosis of macrophages
(108). A previous study
unveiled DAI as the RIP3 partner to interact with RIP3, mediating
virus-induced necrosis analogous to the RIP1/RIP3 complex
controlling TNF-induced necroptosis (109). Table I summarizes the direct and
indirect inhibitors of RIP3.
Necroptosis participates in the development of
several diseases, such as atherosclerosis cardiovascular disease, a
leading cause of mortality worldwide (110). Overexpression of RIP3 during
necroptosis of primary macrophages induced by oxidized LDL (ox-LDL)
facilitates the development of the disease (111). Furthermore, monoclonal
antibodies can be detected in the core of atherosclerotic plaques,
specifically recognizing the phosphorylation form of RIP3 at Ser232
(112). Notably, the mortality
of apolipoprotein E/RIP3 double-knockout mice was delayed
dramatically (112). These
findings indicated that RIP3-mediated necroptosis in
atherosclerotic plaques may release pro-inflammatory cytokines that
exacerbate atherosclerosis. Of note, PS-341, a potent and specific
proteasome inhibitor, was demonstrated to impair macrophage
necroptosis through stabilization of cIAPs and disruption of the
formation of the RIP1/RIP3 complex (113).
RIP1 inhibition leads to a reduction of infarct
size, implying a functional importance of necroptosis in myocardial
ischemia (MI) (114). Luedde
et al (20) analyzed RIP3
expression in murine hearts and highlighted the potential
functional significance of RIP3-dependent necroptosis in the
modulation of post-ischemic adverse remodeling in MI. A previous
study demonstrated that RIP3 was upregulated in murine hearts
subjected to ischemia-reperfusion (IR) injury, as well as in
cardiomyocytes treated with LPS and hydrogen peroxide (115). This study further illustrated
that upregulated RIP3 evoked endoplasmic reticulum (ER) stress,
ultimately resulting in cardiomyocyte necroptosis in the setting of
cardiac IR injury (115). In
addition, in a mouse cardiac hypertrophy model established by
transverse abdominal aortic constriction, both mRNA and protein
expression levels of RIP1 and RIP3 were increased significantly.
Losartan downregulated the expression of RIP1/RIP3, resulting in
the inhibition of necroptosis and to the alleviation of cardiac
hypertrophy (116). RIP1/RIP3
may thus be an attractive target for future therapies that aim to
limit the adverse consequences of cardiac disease.
The quaternary nitrogen herbicide paraquat is a
highly toxic pro-oxidant that triggers oxidative stress and
multi-organ failure, including that of the heart. Recently, Zhang
et al (117) revealed
that Nec-1 pretreatment prevented cardiac contractile dysfunction,
reduced RIP1/RIP3 interaction, downregulated the RIP1/RIP3/MLKL
signaling pathway, and dramatically inhibited the production of ROS
in paraquat-challenged mice. Thus, the RIP1/RIP3/MLKL signaling
cascade may represent an innovative therapeutic direction for
paraquat poisoning-induced cardiac contractile dysfunctions
(Table III).
Abundant research on RIP1/RIP3 has highlighted its
role in cancer, which is due to its necroptosis-inducing function
(118). Chen et al
(119) have noted that
necroptosis is a critical cell-killing mechanism in response to
severe stress and blocked apoptosis, and have proposed that it can
serve as an alternative cell death program to prevent cancer.
Previous studies have indicated that increased RIP3 expression was
correlated with cancer development, including colon and lung
cancers, nasopharyngeal carcinoma and non-Hodgkin lymphoma
(120-122). The topoisomerase inhibitor SN38,
an active metabolite of irinotecan, was demonstrated to mediate
cytotoxicity through the TNF/TNFR signaling pathway in a panel of
colon cancer cells (123). SN38
also promoted the progression of necroptosis, inhibited cell
proliferation and induced DNA damage accumulation (123). This suggested that the
SN38-induced activation of RIP1 and subsequent necroptosis may
exert the therapeutic efficacy on colorectal carcinoma (123). Furthermore, Xin et al
(124) reported that degradation
of suppressor of cytokine signaling 1, a key negative regulator of
IFN-γ signaling, was prevented by TNF through RIP1/RIP3 signaling.
The authors suggested that necroptotic inhibition might be a novel
strategy for the treatment of acute myeloid leukemia through the
combination of RIP1/RIP3 inhibitor with IFN-γ. Recently, bufalin
was demonstrated to increase the expression of necroptosis
mediators RIP1/RIP3 and ROS, leading to poly(ADP-ribose) polymerase
(PARP)-dependent tumor cell death and tumor growth inhibition in
MCF-7 and MDA-MB-231 human breast cancer cells (125). The promising role of
RIP1/RIP3-dependent necroptosis in cancer therapy warrants
attention in future studies (Table
III).
The death of hepatocytes initiates and aggravates
chronic inflammation and fibrosis during liver injury, ultimately
leading to liver cirrhosis and hepatocellular carcinoma. Increasing
evidence indicates that necroptosis has a key role in acute liver
injury and chronic liver injury (129). Deutsch et al (130) indicated that RIP1 and RIP3 have
different roles in drug-induced or immunological acute liver
injuries. In Concanavalin A (ConA)-induced autoimmune hepatitis,
RIP3 deletion delayed hepatic injury, while RIP1 inhibition
markedly exacerbated ConA-induced hepatitis (130). Conversely, in acetaminophen
(APAP)-mediated liver injury, blockade of RIP1 or RIP3 ameliorated
APAP toxicity (130). Zhang
et al (131) demonstrated
that RIP1 was strongly expressed and promoted acute liver failure
in mice treated with APAP (300 mg/kg, intraperitoneally injected).
Further analysis demonstrated that Nec-1, the inhibitor of RIP1,
significantly inhibited APAP-induced hepatic JNK phosphorylation
and mitochondrial Bax translocation (131). Using the APAP model in
RIP3−/− mice, Ramachandran et al (132) found that RIP3 knockout
significantly reduced hepatotoxicity after 6 h of APAP treatment
(200-300 mg/kg), while the protective effect of RIP3 knockout on
the liver was not obvious after 24 h of APAP treatment.
Chronic liver injury usually includes alcoholic
fatty liver disease (AELD), nonalcoholic fatty liver disease
(NAFLD), liver fibrosis and cirrhosis. It has been reported that
the expression of RIP3 in the liver tissue of patients with AFLD is
abnormally elevated, and a series of pathological changes, such as
hepatocyte lipid accumulation and elevated transaminase, are
observed in the liver (133,134). Additionally, Afonso et al
(135) reported that the
expression of RIP3 and phosphorylated MLKL was increased in the
liver tissue of patients with NAFLD, while RIP3 knockout
significantly reduced the steatosis and inflammatory response in
mice with methionine-choline diet. Choi et al (136) found that melatonin reduced the
expression of RIP1, RIP3 and MLKL in rat fibrotic liver induced by
CCl4, and inhibited the expression of high-mobility
group box 1 (HMGB1) and IL-1α. This suggested that melatonin can
alleviate liver fibrosis by inhibiting the inflammatory signaling
associated with programmed necrosis (136). Further studies are required to
fully elucidate the role of RIP1/RIP3-dependent necroptosis in
liver injury (Table III).
Increasing evidence has demonstrated that
necroptosis has an important role in the pathogenesis of multiple
types of kidney injury. Linkermann et al (137) first determined the presence of
necroptosis in a murine model of renal ischemia-reperfusion injury
(IRI). The detection of RIP1 and RIP3 in whole-kidney lysates and
freshly isolated murine proximal tubules revealed the contribution
of necroptosis in renal injury (137). A subsequent study demonstrated
that, in a mouse model of IRI, necroptosis lead to primary organ
damage, and RIP3-knockout mice were protected from IRI (138). Another study used MLKL-knockout
mice to investigate the role of necroptosis in acute kidney injury
(139). Their results revealed
the indispensable role of MLKL in the necroptotic pathway (139). Additionally, Xiao et al
(140) reported that the effect
of necroptosis and the RIP1/RIP3/MLKL signaling pathway in renal
inflammation and interstitial fibrosis was associated with
primitive tubulointerstitial injury. Inhibition of necroptosis
reduced the inflammatory response and interstitial fibrosis in
renal tissues (140). Therefore,
the signaling pathways and the main regulators of necroptosis may
serve as potential candidates for therapeutic strategies in kidney
injury (Table III).
Apoptosis was previously demonstrated to be a
significant form of cell loss in photoreceptor death, but
RIP-mediated necrosis was recently discovered to be a crucial mode
of photoreceptor cell loss in an experimental model of retinal
detachment (141). Expression of
RIP3 undergoes a 10-fold increase after retinal detachment
(142). Nevertheless, Nec-1 or
RIP3 deficiency substantially prevent necroptosis and reduce
oxidative stress of apoptosis-inducing factor (142). Additionally, cone photoreceptor
death in retinitis pigmentosa (RP) is widely considered as a
necroptotic mechanism, while rod photoreceptor death is
characterized by apoptotic features (143). Murakami et al (144) reported that RIP3 expression was
elevated in rd10 mouse retinas in the cone phase, but not in rod
degeneration, and thus suggested that RIP3 may be a potential
target in protecting cone photoreceptors. On the other hand, Sato
et al (145) found that
RIP1/RIP3 accelerated both cone and rod photoreceptor degeneration
in interphotoreceptor retinoid-binding protein (Irbp)−/−
mice. It is worth noting that Irbp deficiency displays severe early
and progressive photoreceptor degeneration. Based on these studies,
RIP1/RIP3 may be regarded as a potential therapeutic target to
prevent or delay photoreceptor degeneration in patients with
RP.
In an animal model of retinal detachment (RD),
cleaved IL-1β was reported in infiltrated macrophages undergoing
RIP3-dependent necroptosis, rather than dying photoreceptors
(146). In addition, Ito et
al (147) suggested a direct
connection between amyotrophic lateral sclerosis induced by
optineurin deficiency and RIP1-regulated necroptosis and
inflammation. Because it promotes both inflammation and cell death,
RIP1/RIP3 may be a common mediator of human degenerative diseases
characterized by axonal degeneration (Table III).
Donor organ injury is invariably mentioned in
transplantation, due to immune responses related to IRI and
alloimmune rejection (148). Lau
et al (149) reported
that in caspase-inhibited tubular epithelial cells (TECs),
necroptosis was triggered in vitro, following inhibition by
Nec-1 or RIP3−/− TEC. Following transplantation,
recipients receiving RIP3−/− kidneys had longer survival
and improved renal function (149). These results suggested that
inhibition of RIP3-dependent necroptosis and inflammatory injury in
donor organs may provide clinical benefit. Toxic epidermal
necrolysis (TEN) is a severe adverse drug reaction with a high
mortality rate (150). Kim et
al (151) detected upregulated expression of RIP3 and elevated
phosphorylation of MLKL in skin sections from patients with TEN.
Dabrafenib notably prevented RIP3-mediated MLKL phosphorylation and
decreased necroptosis, by inhibiting RIP3 in a TEN model (151).
Based on these findings, it can be speculated that RIP may
represent a potential target for treatment of TEN. Table III presents a list of existing
studies on the role of RIP1/RIP3-mediated necroptosis in
disease.
It is well-known that necroptosis, a
physiologically relevant form of cell death, is involved in
pathological cell death resulting from IRI. RIP1/RIP3 is a key
cascade involved in necroptosis, which is regulated by caspase
activation and ubiquitination. RIP1 regulates cell death and
survival, while RIP3 mediates apoptosis and necroptosis. Both TLR
and virus-mediated activation are recognized or identified by
specific receptors or sensors placed either inside or on the
surface of cells. RIP1/RIP3 mediates the initiation of the
necroptotic response to different stimuli. Following ligand binding
and receptor activation, RIP3 enters complex II via interaction
with RIP1, which then activates MLKL participation in inflammation,
immune response, embryonic development and metabolic
abnormality.
In conclusion, an improved understanding of the
molecular events regulating necroptotic cell death would be
extremely beneficial for disease therapy, as these mechanisms are
implicated in a variety of human illnesses. Further research in
this area can be expected to provide promising opportunities
regarding therapeutic exploitation of cell death programs. Due to
its crucial roles in organ growth and tumor cell proliferation,
RIP1/RIP3 may be a key potential therapeutic strategy in multiple
severe diseases. Therefore, by using small molecules that
specifically target necroptosis, it may be possible to alleviate
symptoms and prolong the lives of patients. In coming years,
further research on this alternative cell death pathway may include
identification of additional RIP1 and RIP3 substrates. RIP3 appears
to be the foremost promoter of necroptosis. In the future, studies
exploiting RIP3 inhibitors may provide crucial insight into the
diagnosis and treatment of necroptosis-associated diseases.
This study was sponsored by the National Science
Foundation of China (grant no 81802504), the Sichuan National
Science Research Funding (grant no. 2018JY0645), Sichuan Health and
Family Planning Commission Funding (grant no. 16ZD0253), Chengdu
National Science Research Funding (grant no. 2018-YFYF-00146-SN),
and funding from the Sichuan Scientific Research Grant for Returned
Overseas Chinese Scholars for Dr Yi Wang. This study was also
supported by the National Key Research and Development Plan of
China (grant no. 2017YFC0113901), the Key Research and Development
Plan of Sichuan Science and Technology Bureau (grant no.
2019YFS0278), the Health Care for government officials of Sichuan
Province and the Sichuan Health and Family Planning Commission
Funding (grant no. 120094) for Dr Yuping Liu.
The datasets used and/or analyzed during the
present study are available from the corresponding author on
reasonable request.
YW, RT and YL conceived and designed the review.
DZ, TLiu, TLei, SD and LG wrote the manuscript. TLei, DQ and CL
prepared the figures. YW, RT and YL reviewed and edited the
manuscript. All authors read and approved the final manuscript, and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
|
1
|
Vandenabeele P, Galluzzi L, Vanden Berghe
T and Kroemer G: Molecular mechanisms of necroptosis: An ordered
cellular explosion. Nat Rev Mol Cell Biol. 11:700–714. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Degterev A, Huang Z, Boyce M, Jagtap P,
Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar
|
|
3
|
Christofferson DE and Yuan J: Necroptosis
as an alternative form of programmed cell death. Curr Opin Cell
Biol. 22:263–268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ashkenazi A and Salvesen G: Regulated cell
death: Signaling and mechanisms. Annu Rev Cell & Dev Biol.
30:337–356. 2014. View Article : Google Scholar
|
|
5
|
Zhang YY and Liu H: Connections between
various trigger factors and the RIP1/RIP3 signaling pathway
involved in necroptosis. Asian Pac J Cancer Prev. 14:7069–7074.
2013. View Article : Google Scholar
|
|
6
|
Mason AR, Elia LP and Finkbeiner S: The
receptor-interacting serine/threonine protein kinase 1 (RIPK1)
regulates progranulin levels. J Biol Chem. 292:3262–3272. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stanger BZ, Leder P, Lee TH, Kim E and
Seed B: RIP: A novel protein containing a death domain that
interacts with Fas/APO-1 (CD95) in yeast and causes cell death.
Cell. 81:513–523. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun X, Lee J, Navas T, Baldwin DT, Stewart
TA and Dixit VM: RIP3, a novel apoptosis-inducing kinase. J Biol
Chem. 274:16871–16875. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Newton K: RIPK1 and RIPK3: Critical
regulators of inflammation and cell death. Trends Cell Biol.
25:347–353. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cho YS, Challa S, Moquin D, Genga R, Ray
TD, Guildford M and Chan FK: Phosphorylation-driven assembly of the
RIP1-RIP3 complex regulates programmed necrosis and virus-induced
inflammation. Cell. 137:1112–1123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kaiser WJ, Upton JW, Long AB,
Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T and
Mocarski ES: RIP3 mediates the embryonic lethality of
caspase-8-deficient mice. Nature. 471:368–372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou W and Yuan J: Necroptosis in health
and diseases. Semin Cell Dev Biol. 35:14–23. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vandenabeele P, Declercq W, Van Herreweghe
F and Vanden Berghe T: The role of the kinases RIP1 and RIP3 in
TNF-induced necrosis. Sci Signal. 3:pp. re42010, View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu XN, Yang ZH, Wang XK, Zhang Y, Wan H,
Song Y, Chen X, Shao J and Han J: Distinct roles of RIP1-RIP3
hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis.
Cell Death Differ. 21:1709–1720. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wen L, Zhuang L, Luo X and Wei P:
TL1A-induced NF-kappaB activation and c-IAP2 production prevent
DR3-mediated apoptosis in TF-1 cells. J Biol Chem. 278:39251–39258.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ahmad M, Srinivasula SM, Wang L, Talanian
RV, Litwack G, Fernandes-Alnemri T and Alnemri ES: CRADD, a novel
human apoptotic adaptor molecule for caspase-2, and FasL/tumor
necrosis factor receptor-interacting protein RIP. Cancer Res.
57:615–619. 1997.PubMed/NCBI
|
|
17
|
Festjens N, Vanden BT, Cornelis S and
Vandenabeele P: RIP1, a kinase on the crossroads of a cell's
decision to live or die. Cell Death Differ. 14:400–410. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Newton K, Sun X and Dixit VM: Kinase RIP3
is dispensable for normal NF-kappa Bs, signaling by the B-cell and
T-cell receptors, tumor necrosis factor receptor 1, and Toll-like
receptors 2 and 4. Mol Cell Biol. 24:1464–1469. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun X, Yin J, Starovasnik MA, Fairbrother
WJ and Dixit VM: Identification of a novel homotypic interaction
motif required for the phosphorylation of receptor-interacting
protein (RIP) by RIP3. J Biol Chem. 277:9505–9511. 2002. View Article : Google Scholar
|
|
20
|
Luedde M, Lutz M, Carter N, Sosna J,
Jacoby C, Vucur M, Gautheron J, Roderburg C, Brg N, Reisinger F, et
al: RIP3, a kinase promoting necroptotic cell death, mediates
adverse remodelling after myocardial infarction. Cardiovasc Res.
103:206–216. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Günther C, Neumann H, Neurath MF and
Becker C: Apoptosis, necrosis and necroptosis: Cell death
regulation in the intestinal epithelium. Gut. 62:1062–1071. 2013.
View Article : Google Scholar
|
|
22
|
Pasparakis M and Vandenabeele P:
Necroptosis and its role in inflammation. Nature. 517:311–320.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Magnusson C and Vaux DL: Signalling by
CD95 and TNF receptors: Not only life and death. Immunol Cell Biol.
77:41–46. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lan YH, Wu YC, Wu KW, Chung JG, Lu CC,
Chen YL, Wu TS and Yang JS: Death receptor 5-mediated TNFR family
signaling pathways modulate γ-humulene-induced apoptosis in human
colorectal cancer HT29 cells. Oncol Rep. 25:419–424. 2011.
|
|
25
|
Pan G, Bauer JH, Haridas V, Wang S, Liu D,
Yu G, Vincenz C, Aggarwal BB, Ni J and Dixit VM: Identification and
functional characterization of DR6, a novel death domain-containing
TNF receptor. FEBS Lett. 431:351–356. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Andera L: Signaling activated by the death
receptors of the TNFR family. Biomed Pap Med Fac Univ Palacky
Olomouc Czech Repub. 153:173–180. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yoshikawa M, Saitoh M, Katoh T, Seki T,
Bigi SV, Shimizu Y, Ishii T, Okai T, Kuno M, Hattori H, et al:
Discovery of 7-Oxo-2,4,5,7-tetrahydro-6 H-pyrazolo[3,4-c]pyridine
derivatives as potent, orally available, and brain-penetrating
receptor interacting protein 1 (RIP1) kinase inhibitors: Analysis
of structure-kinetic relationships. J Med Chem. 61:2384–2409. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vanlangenakker N, Bertrand MJM, Bogaert P,
Vandenabeele P and Berghe TV: TNF-induced necroptosis in L929 cells
is tightly regulated by multiple TNFR1 complex I and II members.
Cell Death Dis. 2:pp. e2302011, View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Micheau O and Tschopp J: Induction of TNF
receptor I-mediated apoptosis via two sequential signaling
complexes. Cell. 114:181–190. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wong WW, Gentle IE, Nachbur U, Anderton H,
Vaux DL and Silke J: RIPK1 is not essential for TNFR1-induced
activation of NF-kappaB. Cell Death Differ. 17:482–487. 2010.
View Article : Google Scholar
|
|
31
|
Mack C, Sickmann A, Lembo D and Brune W:
Inhibition of proinflammatory and innate immune signaling pathways
by a cytomegalovirus RIP1-interacting protein. Proc Natl Acad Sci
USA. 105:3094–3099. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang L, Du F and Wang X: TNF-alpha induces
two distinct caspase-8 activation pathways. Cell. 133:693–703.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Benetatos CA, Mitsuuchi Y, Burns JM,
Neiman EM, Condon SM, Yu G, Seipel ME, Kapor GS, Laporte MG, Rippin
SR, et al: Birinapant (TL32711), a bivalent SMAC mimetic, targets
TRAF2-associated cIAPs, abrogates TNF-induced NF-κB activation, and
is active in patient-derived xenograft models. Mol Can Ther.
13:867–879. 2014. View Article : Google Scholar
|
|
34
|
Hughes MA, Powley IR, Jukesjones R, Horn
S, Feoktistova M, Fairall L, Schwabe JW, Leverkus M, Cain K,
MacFarlane M, et al: Co-operative and hierarchical binding of
c-FLIP and caspase-8: A unified model defines how c-FLIP isoforms
differentially control cell fate. Mol Cell. 61:834–849. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Oberst A, Dillon CP, Weinlich R, McCormick
LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS and Green DR:
Catalytic activity of the caspase-8-FLIPL complex inhibits
RIPK3-dependent necrosis. Nature. 471:363–367. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ikner A and Ashkenazi A: TWEAK induces
apoptosis through a death-signaling complex comprising
receptor-interacting protein 1 (RIP1), Fas-associated death domain
(FADD), and caspase-8. J Biol Chem. 286:21546–21554. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lin Y, Devin A, Rodriguez Y and Liu Z:
Cleavage of the death domain kinase RIP by Caspase-8 p rompts
TNF-induced apoptosis. Genes Dev. 13:2514–2526. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vanden BT, Linkermann A, Jouan-Lanhouet S,
Walczak H and Vandenabeele P: Regulated necrosis: The expanding
network of non-apoptotic cell death pathways. Nat Rev Mol Cell
Biol. 15:135–147. 2014. View Article : Google Scholar
|
|
39
|
Li J, Mcquade T, Siemer AB, Napetschnig J,
Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, et
al: The RIP1/RIP3 necrosome forms a functional amyloid signaling
complex required for programmed necrosis. Cell. 150:339–350. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
He S, Wang L, Miao L, Wang T, Du F, Zhao L
and Wang X: Receptor interacting protein kinase-3 determines
cellular necrotic response to TNF-alpha. Cell. 137:1100–1111. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ,
Chen X, Cai Q, Yang ZH, Huang D, Wu R and Han J: RIP1
autophosphorylation is promoted by mitochondrial ROS and is
essential for RIP3 recruitment into necrosome. Nat Commun.
8:143292017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Moquin DM, Mcquade T and Chan FK: CYLD
deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate
kinase activation and programmed necrosis. PLoS One. 8:pp.
e768412013, View Article : Google Scholar
|
|
43
|
Sun L, Wang H, Wang Z, He S, Chen S, Liao
D, Wang L, Yan J, Liu W, Lei X and Wang X: Mixed lineage kinase
domain-like protein mediates necrosis signaling downstream of RIP3
kinase. Cell. 148:213–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dondelinger Y, Declercq W, Montessuit S,
Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA
and Marquis RW: MLKL compromises plasma membrane integrity by
binding to phosphatidylinositol phosphates. Cell Rep. 7:971–981.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang H, Sun L, Su L, Rizo J, Liu L, Wang
LF, Wang FS and Wang X: Mixed lineage kinase domain-like protein
MLKL causes necrotic membrane disruption upon phosphorylation by
RIP3. Mol Cell. 54:133–146. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kaiser WJ, Sridharan H, Huang C, Mandal P,
Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J and Mocarski ES:
Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J
Biol Chem. 288:31268–31279. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
He S, Liang Y, Shao F and Wang X:
Toll-like receptors activate programmed necrosis in macrophages
through a receptor-interacting kinase-3-mediated pathway. Proc Natl
Acad Sci USA. 108:20054–20059. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lu J: Regulation of necroptosis and
autophagy in T cell homeostasis and function. University of
California; Irvine: 2014
|
|
49
|
Walsh CM: Grand challenges in cell death
and survival: Apoptosis vs. necroptosis. Front Cell Dev Biol.
2:32014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hoshino K, Takeuchi O, Kawai T, Sanjo H,
Ogawa T, Takeda Y, Takeda K and Akira S: Cutting edge: Toll-like
receptor 4 (TLR4)-deficient mice are hyporesponsive to
lipopolysaccharide: Evidence for TLR4 as the Lps gene product. J
Immunol. 162:3749–3752. 1999.PubMed/NCBI
|
|
51
|
Takaki H, Shime H, Matsumoto M and Seya T:
Tumor cell death by pattern-sensing of exogenous RNA: Tumor cell
TLR3 directly induces necroptosis by poly(I:C) in vivo, independent
of immune effector-mediated tumor shrinkage. Oncoimmunology. 6:pp.
e10789682015, View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yamamoto M, Sato S, Hemmi H, Hoshino K,
Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okab M, Takeda K and
Akira S: Role of adaptor TRIF in the MyD88-independent toll-like
receptor signaling pathway. Science. 301:640–643. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kawai T, Adachi O, Ogawa T, Takeda K and
Akira S: Unresponsiveness of MyD88-deficient mice to endotoxin.
Immunity. 11:115–122. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hoebe K, Du X, Georgel P, Janssen E,
Tabeta K, Kim SO, Goode J, Lin P, Mann N and Mudd S: Identification
of Lps2 as a key transducer of MyD88-independent TIR signalling.
Nature. 424:743–748. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Holler N, Zaru R, Micheau O, Thome M,
Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B and Tschopp
J: Fas triggers an alternative, caspase-8-independent cell death
pathway using the kinase RIP as effector molecule. Nature Immunol.
1:489–495. 2000. View
Article : Google Scholar
|
|
56
|
Geserick P, Hupe M, Moulin M, Wong WW,
Feoktistova M, Kellert B, Gollnick H, Silke J and Leverkus M:
Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting
RIP1 kinase recruitment. J Cell Biol. 187:1037–1054. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Robinson N, Mccomb S, Mulligan R, Dudani
R, Krishnan L and Sad S: Type I interferon induces necroptosis in
macrophages during infection with salmonella enterica serovar
typhimurium. Nature Immunol. 13:954–962. 2012. View Article : Google Scholar
|
|
58
|
Kaiser WJ, Upton JW and Mocarski ES: Viral
modulation of programmed necrosis. Curr Opin Virol. 3:296–306.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhang Y, Chen X, Gueydan C and Han J:
Plasma membrane changes during programmed cell deaths. Cell Res.
28:9–21. 2018. View Article : Google Scholar :
|
|
60
|
Orozco S, Yatim N, Werner MR, Tran H,
Gunja SY, Tait SW, Albert ML, Green DR and Oberst A: RIPK1 both
positively and negatively regulates RIPK3 oligomerization and
necroptosis. Cell Death Differ. 21:1511–1521. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wajant H and Scheurich P: TNFR1-induced
activation of the classical NF-κB pathway. FEBS J. 278:862–876.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jaco I, Annibaldi A, Lalaoui N, Wilson R,
Tenev T, Laurien L, Kim C, Jamal K, Wicky John S, Liccardi G, et
al: MK2 phosphorylates RIPK1 to prevent TNF-induced cell death. Mol
Cell. 66:698–710.e5. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kearney CJ, Cullen SP, Danielle C and
Martin SJ: RIPK1 can function as an inhibitor rather than an
initiator of RIPK3-dependent necroptosis. FEBS J. 281:4921–4934.
2015. View Article : Google Scholar
|
|
64
|
Schenk B and Fulda S: Reactive oxygen
species regulate Smac mimetic/TNFα-induced necroptotic signaling
and cell death. Oncogene. 34:5796–5806. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ,
Lin SC, Dong MQ and Han J: RIP3, an energy metabolism regulator
that switches TNF-induced cell death from apoptosis to necrosis.
Science. 325:332–336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wen S, Wu X, Gao H, Yu J, Zhao W, Lu JJ,
Wang J, Du G and Chen X: Cytosolic calcium mediates RIP1/RIP3
complex-dependent necroptosis through JNK activation and
mitochondrial ROS production in human colon cancer cells. Free
Radic Biol Med. 108:433–444. 2017. View Article : Google Scholar
|
|
67
|
Kearney CJ and Martin SJ: An inflammatory
perspective on necroptosis. Mol Cell. 65:965–973. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Martin SJ: Cell death and inflammation:
The case for IL-1 family cytokines as the canonical DAMPs of the
immune system. FEBS J. 283:2599–2615. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kono H and Rock KL: How dying cells alert
the immune system to danger. Nat Rev Immunol. 8:279–289. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Fan H, Liu F, Dong G, Ren D, Xu Y, Dou J,
Wang T, Sun L and Hou Y: Activation-induced necroptosis contributes
to B-cell lymphopenia in active systemic lupus erythematosus. Cell
Death Dis. 5:pp. e14162014, View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Siegel RM: Caspases at the crossroads of
immune-cell life and death. Nat Rev Immunol. 6:308–317. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Bell BD, Leverrier S, Weist BM, Newton RH,
Arechiga AF, Luhrs KA, Morrissette NS and Walsh CM: FADD and
caspase-8 control the outcome of autophagic signaling in
proliferating T cells. Proc Natl Acad Sci USA. 105:16677–16682.
2009. View Article : Google Scholar
|
|
73
|
Lu JV and Walsh CM: Programmed necrosis
and autophagy in immune function. Immunol Rev. 249:205–217. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
O'Donnell JA, Lehman J, Roderick JE,
Martinez-Marin D, Zelic M, Doran C, Hermance N, Lyle S, Pasparakis
M, Fitzgerald KA, et al: Dendritic cell RIPK1 maintains immune
homeostasis by preventing inflammation and autoimmunity. J Immunol.
200:737–748. 2018. View Article : Google Scholar :
|
|
75
|
Giltiay NV, Chappell CP, Sun X, Kolhatkar
N, Teal TH, Wiedeman AE, Kim J, Tanaka L, Buechler MB, Hamerman JA,
et al: Overexpression of TLR7 promotes cell-intrinsic expansion and
autoantibody production by transitional T1B cells. J Exp Med.
210:2773–2789. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Mina-Osorio P, LaStant J, Keirstead N,
Whittard T, Ayala J, Stefanova S, Garrido R, Dimaano N, Hilton H,
Giron M, et al: Suppression of glomerulonephritis in lupus-prone
NZB x NZW mice by RN486, a selective inhibitor of Bruton's tyrosine
kinase. Arthritis Rheum. 65:2380–2391. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Biton S and Ashkenazi A: NEMO and RIP1
control cell fate in response to extensive DNA damage via TNF-α
feedforward signaling. Cell. 145:92–103. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Xu C, Wu X, Zhang X, Xie Q, Fan C and
Zhang H: Embryonic lethality and host immunity of relA-deficient
mice are mediated by both apoptosis and necroptosis. J Immunol.
200:271–285. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Lu JV, Weist BM, van Raam BJ, Marro BS,
Nguyen LV, Srinivas P, Bell BD, Luhrs KA, Lane TE, Salvesen GS and
Walsh CM: Complementary roles of fas-associated death domain (FADD)
and receptor interacting protein kinase-3 (RIPK3) in T-cell
homeostasis and antiviral immunity. Proc Natl Acad Sci USA.
108:15312–15317. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zhang H, Zhou X, Mcquade T, Li J, Chan FK
and Zhang J: Functional complementation between FADD and RIP1 in
embryos and lymphocytes. Nature. 471:373–376. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Liu Y, Fan C, Zhang Y, Yu X, Wu X, Zhang
X, Zhao Q, Zhang H, Xie Q, Li M, et al: RIP1 kinase
activity-dependent roles in embryonic development of fadd-deficient
mice. Cell Death Differ. 24:1459–1469. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Dillon CP, Oberst A, Weinlich R, Janke LJ,
Kang TB, Ben-Moshe T, Mak TW, Wallach D and Green DR: Survival
function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep.
1:401–407. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Kaiser WJ, Daleybauer LP, Thapa RJ, Mandal
P, Berger SB, Huang C, Sundararajan A, Guo H, Roback L, Speck SH,
et al: RIP1 suppresses innate immune necrotic as well as apoptotic
cell death during mammalian parturition. Proc Natl Acad Sci USA.
111:7753–7758. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Dillon CP, Weinlich R, Rodriguez DA,
Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F,
Gong YN, et al: RIPK1 blocks early postnatal lethality mediated by
caspase-8 and RIPK3. Cell. 157:1189–1202. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Dowling JP, Nair A and Zhang J: A novel
function of RIP1 in postnatal development and immune homeostasis by
protecting against RIP3-dependent necroptosis and FADD-mediated
apoptosis. Front Cell Dev Biol. 3:122015. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Degterev A, Hitomi J, Germscheid M, Ch'en
IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al:
Identification of RIP1 kinase as a specific cellular target of
necrostatins. Nat Chem Biol. 4:313–321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Takahashi N, Duprez L, Grootjans S,
Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van
Hauwermeiren F, Libert C, et al: Necrostatin-1 analogues: Critical
issues on the specificity, activity andin vivouse in experimental
disease models. Cell Death Dis. 3:pp. e4372012, View Article : Google Scholar
|
|
88
|
Harris PA, Berger SB, Jeong JU, Nagilla R,
Bandyopadhyay D, Campobasso N, Capriotti CA, Cox JA, Dare L, Dong
X, et al: Discovery of a first-in-class receptor interacting
protein 1 (RIP1) kinase specific clinical candidate (GSK2982772)
for the treatment of inflammatory diseases. J Med Chem.
60:1247–1261. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Ren Y, Su Y, Sun L, He S, Meng L, Liao D,
Liu X, Ma Y, Liu C, Li S, et al: Discovery of a highly potent,
selective, and metabolically stable inhibitor of
receptor-interacting protein 1 (RIP1) for the treatment of systemic
inflammatory response syndrome. J Med Chem. 60:972–986. 2017.
View Article : Google Scholar
|
|
90
|
Martens S, Goossens V, Devisscher L,
Hofmans S, Claeys P, Vuylsteke M, Takahashi N, Augustyns K and
Vandenabeele P: RIPK1-dependent cell death: A novel target of the
Aurora kinase inhibitor Tozasertib (VX-680). Cell Death Dis.
9:2112018. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Fauster A, Rebsamen M, Huber KVM,
Bigenzahn JW, Stukalov A, Lardeau CH, Scorzoni S, Bruckner M,
Gridling M, Parapatics K, et al: A cellular screen identifies
ponatinib and pazopanib as inhibitors of necroptosis. Cell Death
Dis. 6:pp. e17672015, View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Alcalá AM and Flaherty KT: BRAF inhibitors
for the treatment of metastatic melanoma: Clinical trials and
mechanisms of resistance. Clin Cancer Res. 18:33–39. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Li JX, Feng JM, Wang Y, Li XH, Chen XX, Su
Y, Shen YY, Chen Y, Xiong B, Yang CH, et al: The B-RafV600E
inhibitor dabrafenib selectivelyinhibits RIP3 and alleviates
acetaminophen-induced liver injury. Cell Death Dis. 5:pp.
e12782014, View Article : Google Scholar
|
|
94
|
Cruz SA, Qin Z, Afr S and Chen HH:
Dabrafenib, an inhibitor of RIP3 kinase-dependent necroptosis,
reduces ischemic brain injury. Neural Regen Res. 13:252–256. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Omoto S, Guo H, Talekar GR, Roback L,
Kaiser WJ and Mocarski ES: Suppression of RIP3-dependent
necroptosis by human cytomegalovirus. J Biol Chem. 290:11635–11648.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Liu J, Mil AV, Vrijsen K, Zhao J, Gao L,
Metz CH, Goumans MJ, Doevendans PA and Sluijter JP: MicroRNA-155
prevents necrotic cell death in human cardiomyocyte progenitor
cells via targeting RIP1. J Cell Mol Med. 15:1474–1482. 2011.
View Article : Google Scholar
|
|
97
|
Dhingra R, Lin J and Kirshenbaum LA:
Disruption of RIP1-FADD complexes by microRNA-103/107 provokes
necrotic cardiac cell death. Circ Res. 117:314–316. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Wang JX, Zhang XJ, Li Q, Wang K, Wang Y,
Jiao JQ, Feng C, Teng S, Zhou LY, Gong Y, et al: MicroRNA-103/107
regulate programmed necrosis and myocardial ischemia/reperfusion
injury through targeting FADD. Circ Res. 117:352–363. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Wo L, Lu D and Gu X: Knockdown of miR-182
promotes apoptosis via regulating RIP1 deubiquitination in
TNF-α-treated triple-negative breast cancer cells. Tumour Biol.
37:13733–13742. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Zheng M, Wu Z, Wu A, Huang Z, He N and Xie
X: MiR-145 promotes TNF-α-induced apoptosis by facilitating the
formation of RIP1-FADDcaspase-8 complex in triple-negative breast
cancer. Tumor Biol. 37:8599–8607. 2016. View Article : Google Scholar
|
|
101
|
Li D, Xu T, Cao Y, Wang H, Li L, Chen S,
Wang X and Shen Z: A cytosolic heat shock protein 90 and
cochaperone CDC37 complex is required for RIP3 activation during
necroptosis. Proc Natl Acad Sci USA. 112:5017–5022. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Li D, Li C, Li L, Chen S, Wang L, Li Q,
Wang X, Lei X and Shen Z: Natural product kongensin a is a
non-canonical HSP90 inhibitor that blocks RIP3-dependent
necroptosis. Cell Chem Biol. 23:257–266. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Jacobsen AV, Lowes KN, Tanzer MC, Lucet
IS, Hildebrand JM, Petrie EJ, van Delft MF, Liu Z, Conos SA, Zhang
JG, et al: HSP90 activity is required for MLKL oligomerisation and
membrane translocation and the induction of necroptotic cell death.
Cell Death Dis. 7:pp. e20512016, View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhao XM, Chen Z, Zhao JB, Zhang PP, Pu YF,
Jiang SH, Hou JJ, Cui YM, Jia XL and Zhang SQ: Hsp90 modulates the
stability of MLKL and is required for TNF-induced necroptosis. Cell
Death Dis. 7:pp. e20892016, View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Park SY, Shim JH, Chae JI and Cho YS: Heat
shock protein 90 inhibitor regulates necroptotic cell death via
down-regulation of receptor interacting proteins. Pharmazie.
70:193–198. 2015.PubMed/NCBI
|
|
106
|
Declercq W, Vanden BT and Vandenabeele P:
RIP kinases at the crossroads of cell death and survival. Cell.
138:229–232. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Chan FK, Luz NF and Moriwaki K: Programmed
necrosis in the cross talk of cell death and inflammation. Annu Rev
Immunol. 33:79–106. 2015. View Article : Google Scholar :
|
|
108
|
Ariana A: Dissection of TLR4-Induced
Necroptosis Using Specific Inhibitors of Endocytosis and P38 MAPK.
Department of Biochemistry, Microbiology and Immunology University
of Ottawa; Ottawa, Canada: 2017
|
|
109
|
Upton JW, Kaiser WJ and Mocarski ES: DAI
complexes with RIP3 to mediate virus-induced programmed necrosis
that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe.
11:290–297. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Gupta K, Phan N, Wang Q and Liu B:
Necroptosis in cardiovascular disease-a new therapeutic target. J
Mol Cell Cardiol. 118:26–35. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Lin J, Li H, Yang M, Ren J, Huang Z, Han
F, Huang J, Ma J, Zhang D, Zhang Z, et al: A role of RIP3-mediated
macrophage necrosis in atherosclerosis development. Cell Rep.
3:200–210. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Meng L, Jin W and Wang X: RIP3-mediated
necrotic cell death accelerates systematic inflammation and
mortality. Proc Natl Acad Sci USA. 112:11007–11012. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Zhang Y, Cheng J, Zhang J, Wu X, Chen F,
Ren X, Wang Y, Li Q and Li Y: Proteasome inhibitor PS-341 limits
macrophage necroptosis by promoting cIAPs-mediated inhibition of
RIP1 and RIP3 activation. Biochem Biophys Res Commun. 477:761–767.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Oerlemans MI, Liu J, Arslan F, den Ouden
K, van Middelaar BJ, Doevendans PA and Sluijter JP: Inhibition of
RIP1-dependent necrosis prevents adverse cardiac remodeling after
myocardial ischemia-reperfusion in vivo. Basic Res Cardiol.
107:2702012. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Zhu P, Hu S, Jin Q, Li D, Tian F, Toan S,
Li Y, Zhou H and Chen Y: Ripk3 promotes ER stress-induced
necroptosis in cardiac IR injury: A mechanism involving calcium
overload/XO/ROS/mPTP pathway. Redox Biol. 16:157–168. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Zhao M, Qin Y, Lu L, Tang X, Wu W, Fu H
and Liu X: Preliminary study of necroptosis in cardiac hypertrophy
induced by pressure overload. Sheng Wu Yi Xue Gong Cheng Xue Za
Zhi. 32:618–623. 2015.In Chinese. PubMed/NCBI
|
|
117
|
Zhang L, Feng Q and Wang T: Necrostatin-1
protects against paraquat-induced cardiac contractile dysfunction
via RIP1-RIP3-MLKL-dependent necroptosis pathway. Cardiovasc
Toxicol. 18:346–355. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Meng MB, Wang HH, Cui YL, Wu ZQ, Shi YY,
Zaorsky NG, Deng L, Yuan ZY, Lu Y and Wang P: Necroptosis in
tumori-genesis, activation of anti-tumor immunity, and cancer
therapy. Oncotarget. 7:57391–57413. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Chen D, Yu J and Zhang L: Necroptosis: An
alternative cell death program defending against cancer. Biochim
Biophys Acta. 1865:228–236. 2016.PubMed/NCBI
|
|
120
|
Yang Y, Hu W, Feng S, Ma J and Wu M: RIP3
beta and RIP3 gamma, two novel splice variants of
receptor-interacting protein 3 (RIP3), downregulate RIP3-induced
apoptosis. Biochem Biophys Res Commun. 332:181–187. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Kasof GM, Prosser JC, Liu D, Lorenzi MV
and Gomes BC: The RIP-like kinase, RIP3, induces apoptosis and
NF-kappaB nuclear translocation and localizes to mitochondria. FEBS
Lett. 473:285–291. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Wu W, Liu P and Li J: Necroptosis: An
emerging form of programmed cell death. Crit Rev Oncol Hematol.
82:249–258. 2012. View Article : Google Scholar
|
|
123
|
Cabal-Hierro L and O'Dwyer PJ: TNF
signaling through RIP1 kinase enhances SN38-induced death in colon
adenocarcinoma. Mol Cancer Res. 15:395–404. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Xin J, You D, Breslin P, Li J, Zhang J,
Wei W, Cannova J, Volk A, Gutierrez R, Xiao Y, et al: Sensitizing
acute myeloid leukemia cells to induced differentiation by
inhibiting the RIP1/RIP3 pathway. Leukemia. 124:1154–1165. 2017.
View Article : Google Scholar
|
|
125
|
Li Y, Liu X, Gong P and Tian X: Bufalin
inhibits human breast cancer tumorigenesis by inducing cell death
through the ROS-Mediated RIP1/RIP3/PARP-1 pathways. Carcinogenesis.
39:700–707. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Larocca TJ, Sosunov SA, Shakerley NL, Ten
VS and Ratner AJ: Hyperglycemic conditions prime cells for
RIP1-dependent necroptosis. J Biol Chem. 291:13753–13761. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Mccaig WD, Patel PS, Sosunov SA, Shakerley
NL, Smiraglia TA, Craft MM, Walker KM, Deragon MA, Ten VS and
LaRocca TJ: Hyperglycemia potentiates a shift from apoptosis to
RIP1-dependent necroptosis. Cell Death Discov. 4:552018. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Wang K, Hu L and Chen JK: RIP3-deficience
attenuates potassium oxonate-induced hyperuricemia and kidney
injury. Biomed Pharmacother. 101:617–626. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Dara L, Liu ZX and Kaplowitz N: Questions
and controversies: The role of necroptosis in liver disease. Cell
Death Discov. 2:160892016. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Deutsch M, Graffeo CS, Rokosh R, Pansari
M, Ochi A, Levie EM, Van Heerden E, Tippens DM, Tippens DM, Greco
S, et al: Divergent effects of RIP1 or RIP3 blockade in murine
models of acute liver injury. Cell Death Dis. 6:pp. e17592015,
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Zhang YF, He W, Zhang C, Liu XJ, Lu Y,
Wang H, Zhang ZH, Chen X and Xu DX: Role of receptor interacting
protein (RIP)1 on apoptosis-inducing factor-mediated necroptosis
during acetaminophen-evoked acute liver failure in mice. Toxicol
Lett. 225:445–453. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Ramachandran A, McGill MR, Xie Y, Ni HM,
Ding WX and Jaeschke H: Receptor interacting protein kinase 3 is a
critical early mediator of acetaminophen-induced hepatocyte
necrosis in mice. Hepatology. 58:2099–2108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Roychowdhury S, McMullen MR, Pisano SG,
Liu X and Nagy LE: Absence of receptor interacting protein kinase 3
prevents ethanol-induced liver injury. Hepatology. 57:1773–1783.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Wang S, Ni HM, Dorko K, Kumer SC, Schmitt
TM, Nawabi A, Komatsu M, Huang H and Ding WX: Increased hepatic
receptor interacting protein kinase 3 expression due to impaired
protea-somal functions contributes to alcohol-induced steatosis and
liver injury. Oncotarget. 7:17681–17698. 2016.PubMed/NCBI
|
|
135
|
Afonso MB, Rodrigues PM, Carvalho T,
Caridade M, Borralho P, Cortez-Pinto H, Castro RE and Rodrigues CM:
Necroptosis is a key pathogenic event in human and experimental
murine models of non-alcoholic steatohepatitis. Clin Sci (Lond).
129:pp. 721–739. 2015, View Article : Google Scholar
|
|
136
|
Choi HS, Kang JW and Lee SM: Melatonin
attenuates carbon tetrachloride-induced liver fibrosis via
inhibition of necroptosis. Transl Res. 166:292–303. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Linkermann A, Brasen JH, Himmerkus N, Liu
S, Huber TB, Kunzendorf U and Krautwald S: Rip1
(receptor-interacting protein kinase 1) mediates necroptosis and
contributes to renal ischemia/reperfusion injury. Kidney Int.
81:751–761. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Linkermann A, Brasen JH, Darding M, Jin
MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H,
et al: Two independent pathways of regulated necrosis mediate
ischemia-reperfusion injury. Proc Natl Acad Sci USA.
110:12024–12029. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z,
Wang Y, Huang Z, Ren J, Liu S, et al: A role for tubular
necroptosis in cisplatin-induced AKI. J Am Soc Nephrol.
26:2647–2658. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Xiao X, Du C, Yan Z, Shi Y, Duan H and Ren
Y: Inhibition of necroptosis attenuates kidney inflammation and
interstitial fibrosis induced by unilateral ureteral obstruction.
Am J Nephrol. 46:131–138. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Murakami Y, Trichonas G, Thanos A,
Mantopulos D, Morizane Y, Kayama M, Hisatomi T, Miller J and Vavvas
D: The role of RIP-mediated necrosis and autophagy in photoreceptor
death after retinal detachment. Invest Ophthalmol Vis Sci.
52:65882011.
|
|
142
|
Trichonas G, Murakami Y, Thanos A,
Morizane Y, Kayama M, Debouck CM, Hisatomi T, Miller JW and Vavvas
DG: Receptor interacting protein kinases mediate retinal
detachment-induced photoreceptor necrosis and compensate for
inhibition of apoptosis. Proc Natl Acad Sci USA. 107:21695–21700.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Murakami Y, Ikeda Y, Nakatake S, Miller
JW, Vavvas DG, Sonoda KH and Ishibashi T: Necrotic cone
photoreceptor cell death in retinitis pigmentosa. Cell Death Dis.
6:pp. e20382015, View Article : Google Scholar
|
|
144
|
Murakami Y, Matsumoto H, Roh M, Suzuki J,
Hisatomi T, Ikeda Y, Miller JW and Vavvas DG: Receptor interacting
protein kinase mediates necrotic cone but not rod cell death in a
mouse model of inherited degeneration. Proc Natl Acad Sci USA.
109:14598–14603. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Sato K, Li S, Gordon WC, He J, Liou GI,
Hill JM, Travis GH, Bazan NG and Jin M: Receptor interacting
protein kinase-mediated necrosis contributes to cone and rod
photoreceptor degeneration in the retina lacking interphotoreceptor
retinoid-binding protein. J Neurosci. 33:17458–17468. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Kataoka K, Matsumoto H, Kaneko H, Notomi
S, Takeuchi K, Sweigard JH, Atik A, Murakami Y, Connor KM, Terasaki
H, et al: Macrophage- and RIP3-dependent inflammasome activation
exacerbates retinal detachment-induced photoreceptor cell death.
Cell Death Dis. 6:pp. e17312015, View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Ito Y, Ofengeim D, Najafov A, Das S,
Saberi S, Li Y, Hitomi J, Zhu H, Chen H, Mayo L, et al: RIPK1
mediates axonal degeneration by promoting inflammation and
necroptosis in ALS. Science. 353:603–608. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Hébert MJ and Jevnikar AM: The impact of
regulated cell death pathways on alloimmune responses and graft
injury. Curr Transpl Rep. 2:242–258. 2015. View Article : Google Scholar
|
|
149
|
Lau A, Wang S, Jiang J, Haig A, Pavlosky
A, Linkermann A, Zhang ZX and Jevnikar AM: RIPK3-mediated
necroptosis promotes donor kidney inflammatory injury and reduces
allograft survival. Am J Transplant. 13:2805–2818. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Becker DS: Toxic epidermal necrolysis.
Lancet. 351:1417–1420. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Kim SK, Kim WJ, Yoon JH, Ji JH, Morgan MJ,
Cho H, Kim YC and Kim YS: Upregulated RIP3 expression potentiates
MLKL phosphorylation-mediated programmed necrosis in toxic
epidermal necrolysis. J Investi Dermatol. 135:2021–2030. 2015.
View Article : Google Scholar
|