Introduction
Systemic lupus erythematosus (SLE) is a chronic, severe, multiorgan systemic autoimmune disease that predominantly affects women, with a complex genetic inheritance and strong clustering in families (1) It is characterized by the production of high titers of autoantibodies directed against native DNA, cell surface and other cellular constituents (2). SLE is associated with high morbidity rates (3). Genetic association and genome-wide association studies (GWAS) for susceptibility loci of SLE, performed in various ethnic populations, have provided novel insights into SLE and uncovered >100 common SLE risk loci, explaining disease up to 30% (4). Attempts to clarify the mechanisms underlying this disease may contribute to the development of disease-modifying therapeutic protocols. Of interest, accumulating evidence suggests that several genetic polymorphisms linked to SLE, are associated with other autoimmune diseases as well, such as rheumatoid arthritis, type 1 diabetes, psoriasis, Crohn's disease, ulcerative colitis, celiac disease, systemic sclerosis, multiple sclerosis and Behçet's disease (5).
The expansion of Genetics and Genomics in the 20th century has provided a basis for the development of novel techniques and applications. As a result of the rapid expansion in genomic technologies, genetics studies have become crucial in clinical practice and research (6). The molecular background and knowledge of genetics has become more understandable due to rapid technological advancements, including the whole-genome and whole-exome (WES) sequencing analyses (7). The massive accumulation and analysis of genomic data has resulted in the completion of The Human Genome Project and The 1000 Genome Project, which have contributed a great deal to the knowledge of genetic variants and their impact on human life and in harmful diseases (8).
At present, the focus of research is on personalized medicine, clinical genomics and the further involvement of computer science through data mining, semantic analyses and state of the art methods in bioinformatics (9,10). The discovery of the human genome was only the beginning, in the great effort to decipher it and associate it with the genetic variants and changes between populations, genes, diseases and mainly with the history of human existence. With the implementation of computer science and bioinformatics in the development of efficient applications of genetic and genomic analysis for clinical genomics and personalized medicine, we are at the beginning of an era that will provide novel discoveries in human health (10).
The importance of design and applying such methodical techniques and pipelines will grow as we continue to generate and integrate large quantities of genomics, proteomics, transcriptomics, lipidomics, metabolomics, secretomics and other -omics biological data (11). Examples of this type of specialized analyses include GWAS, gene classification per disease, single nucleotide polymorphism (SNP) classification per disease, correlation of human genomic data with a specific rare disease or a resistance in a well-known medication and various other applications (12). The Epione app webserver is an example that incorporates the application of bioinformatics and data mining technologies aiming to support the clinical genomic diagnosis process of SLE (Fig. 1).
 |
Figure 1
Epione application webserver pipeline. Left to right: Input parameters (FASTA or VCF file and a selected reference genome), Epione application pipeline, output files (SNP analysis results, candidate variants, patient profile and statistics charts, chromosome ideograms, relative publications with candidate variants and regulatory networks). VCF, Variant Call Format; SNPs, single nucleotide polymorphisms; SLE, systemic lupus erythematosus; dbSNP, Single Nucleotide Polymorphism Database.
|
Despite improvements in the identification of patients with SLE, the diagnosis of the disease is still a challenge for clinicians, particularly early in the course of the disease (13). The interval between the initial onset of symptoms and the actual diagnosis is still a number of years apart. The mean interval between the onset of symptoms and the diagnosis of SLE may be up to 2 years (14). Probably due to the lower suspicion, a longer time lag has been reported for children, males and late-onset disease (15). Importantly, increased healthcare utilization during the time preceding SLE diagnosis has been reported. The median number of GP consultations increased during the 5-year interval preceding SLE diagnosis, i.e., from median 1 in the 48-54 months before diagnosis to 38 in the 0-12 months before diagnosis (16). Notably, a study performed in 682 children and young patients (aged 10-24 years) with SLE also confirmed that they had significantly more health care visits than controls in the year before diagnosis (17). At 9-12 months prior to diagnosis, utilization of healthcare resources was increased by almost 2-fold. Of note, a number of young individuals with SLE carry psychiatric diagnoses prior to being diagnosed with SLE, which was also associated with increased pre-diagnosis healthcare use (17). SLE is no longer considered to be such a rare disease at the community level, thus there is likely a considerable number of patients who remain undiagnosed or experience significant diagnostic delays (18).
Patients with <6 months' delay may experience lower flare rates, less healthcare utilization and costs, as compared with those with at least 6 months' delay (19). Furthermore, for patients with major organ disease (nephritis, neurological), delay in prompt diagnosis and initiation of immunosuppressive therapy has been linked to adverse outcomes (20). Failure to achieve low disease activity in the first 6 months after diagnosis has been associated with early damage accrual (21). Finally, in patients at an early stage of the disease, all subscales of quality of life can be improved with proper therapy over a period of 2 years (22).
In the present study, the Epione application is presented, which is an online toolkit for clinical genomic and personalized medicine that is able to support the suspicion of physicians dealing with a possible case of SLE (10). The overall aim of the present study was to provide a reliable tool for the most effective study of SLE. The Epione application is able to analyze a patient's genetic or genomic data either as a FASTA or Variant Call Format (VCF) data file, and automatically scans input data against thousands of relevant recorded SNPs. The pipeline of the designed algorithm applies different filtering, processing and annotation techniques in several steps, towards identifying and visualizing the most probable prevalent variants related to SLE. Moreover, the application is capable of identifying and classifying the extracted SNPs using our SNP database and other genetic and clinical information from several online databases. At the same time, it recognizes individual SNPs with pathogenicity in SLE and other related disease, and it provides the user with additional information and direct links to several online databases, including The Single Nucleotide Polymorphism Database (dbSNP) and the LitVar database (23,24). Additionally, the Epione application analyzes and generates important information associated with the recognized SNP variants, including ideograms, statistic charts, a gene network based on the extracted SNPs and a number of related studies from the National Center for Biotechnology Information (NCBI) PubMed database.
Materials and methods
Epione Application Database (EAD) of SNPs and variants for SLE
All the genes, pseudogenes, promoters, enhancers, SNPs and variants associated with SLE, and reported in global available databases and studies were stored in the structured EAD. The PubMed database was initially used for detecting and extracting studies related to 'SLE'. The available studies were filtered to human-related studies only and were curated using data mining and semantic methods in order to identify those that refer to genes by using a dictionary from the Gene database of the NCBI (25) and those that contained SNP variants. A targeted query search was performed in the text using regular expressions by combining each gene or variant with their synonyms and the key word 'SLE' (26). The identified genes, SNPs and variants referred in the study datasets were stored in EAD. Additionally, appropriate studies from PubMed were mined for the provision of additional information, such as Medical Subject Headings (MeSH)/MEDLINE terms, genes, polymorphisms and mutations described and were examined for their role in SLE (26,27). Supplementary information was mined and included in the EAD from numerous available online databases, including Online Mendelian Inheritance in Man (OMIM) Database (28) and GWAS Catalog (29,30). The final dataset of SNPs and variants associated with SLE were annotated in the EAD using several external query searches in the dbSNP, ClinVar and LitVar databases of the NCBI (23,24,31). Moreover, for each entry a representative FASTA sequence was isolated using the human reference genome GRCh38. The main idea was to generate a representative FASTA sequence, using sliding windows of ~201 bases (100 before and 100 after the polymorphism), whether being a nucleotide change or deletion or insertion. After the collection, annotation and filtering processes, the information contained in the EAD was classified using a scoring function described below. Finally, the information contained in the EAD was classified according the scoring function described below and the final outcome was manually evaluated by medical experts in SLE using the annotated information, results and the sources of origin as follows (10): Score = (VNorFrePub ×0.1) + (VNorFreLitVar ×0.3) + (VClinVar ×0.2) + (VMedExpertsSNPs ×0.4). Where: i) VNorFrePub, the normalized frequency of the identified SNPs from the PubMed dataset (max, 1; min, 0); ii) VNorFreLitVar, the normalized frequency of the identified SNPs that were linked to SLE from the LitVar Database (Scalar value, max, 1 and min, 0); iii) VClinVar, Boolean Parameter (1, the SNP was identified in the ClinVar databases and was connected to SLE; 0, no connection to the ClinVar or no connection to SLE); and iv) VMedExperts, Boolean Parameter (1, if the given SNP was identified as being associated with endometriosis by the medical experts team; 0, no connection to the dataset). Scoring function was as follows: i) 'Strong-associated SNPs' Class, score ≥0.4; ii) 'High-associated SNPs' Class, score <0.4 and ≥0.2; and iii) 'Associated SNPs' Class, score <0.2.
VCF or FASTA file validation and filtering
The uploaded file in the Epione application pipeline was verified for compliance with the standardized genomic data formats, including FASTA/Pearson format or VCF 4 correspondingly (32). The FASTA file had to contain a header and sequence information, and each entry had to start with the symbol '>'. Minimum character count for the sequence information was set to 250 characters. No duplicated header string names were allowed. The VCF file at the beginning had to contain a header section with the preset column names as they were defined by the Global Alliance for Genomics and Health Data Working group file format team (https://www.ga4gh.org/) (32). The VCF file is a tab delimited array for storing variants and individual genotypes. It is able to include all variant calls from SNPs and variants to, small changes, and large-scale insertions and deletions. VCF file columns could not have any duplicated entries, and each entry must have only contained the appropriate information without gaps. The Epione application online toolkit provides the user with the ability to upload a single FASTA or VCF file of ≤ 1GB. After the file validation process, only nucleotides sequences or SNPs and gene variants that passed the quality and filtering controls were considered as an input in the main pipeline of the Epione application.
Identification of SNPs
The Epione app web-toolkit has two different SNP identification processes depending on the type of uploaded file (FASTA or VCF file). For each case, the webserver uses the EAD of SNPs associated with SLE to analyze and correlate the input curated dataset. In the case of a FASTA file, the application implements the process of the local alignments with the EAD. Input entries identified with 100% identity in a range of a window of 200 bases within a given nucleotide sequence from EAD were reported and marked to the system as a candidate polymorphism case SLE. In the second case of the VCF file, all the SLE-related SNPs were identified based on the EAD's directory with the reported positions of SNPs on each chromosome. Finally, all the identified cases in each case of the analysis were collected in a separated list with all the annotated information from the EAD.
Variant classification and interface representation
The Epione application classification procedure identified candidate and dominant deleterious SNPs in the list of exonic and non-coding polymorphisms. The graphic representation interface enables the user to see the patient SLE profile, which is presented through the three major classes of polymorphisms according to severity, namely 'Strong-associated SNPs', 'High-associated SNPs' and 'Associated SNPs'. All the identified SNPs were classified in these three major classes based on the annotated information contained in the EAD. An additional list of all identified variants with necessary information, such as 'snp_name', 'chromosome', 'position', 'reference genome', 'change', 'gene_name', 'variant_type', 'disease', 'litvar' and 'class' is also provided to the user. Moreover, for each identified variant, the application provides an external link to the dbSNP and the LitVar Database for reference to additional information.
A more specialized representation with bar charts and ideograms is presented based on the patient's identified polymorphism profile. This enables the user to better understand the general genetic profile for the patient and draw beneficial conclusions concerning the association of each chromosome with SLE development. With this more specialized analysis, conclusions could be drawn on how genes may be involved in SLE, not only as separate entities, but as part of specific chromosomal regions or as a cluster in a network or in a combination of both.
Data mining and semantic analysis
The MEDLINE and PubMed databases were searched for English-language publications that contained the key term 'Systemic lupus erythematosus,' with no date restriction (26). The MATLAB Bioinformatics toolbox functions for data mining and semantic analysis were used to extract gene names from the selected publications' abstracts using a dictionary of the gene, allele and pseudogene names for Homo sapiens (33,34). Furthermore, using the same techniques, all the polymorphisms reported by at least two studies from the dataset were extracted. A second-level analysis was performed in order to estimate the internal links between genes through selected publications. Internal links were created when genes, alleles, pseudogenes or transcription factors were mentioned in the same publication. Finally, all the mining knowledge was processed through semantic algorithms contained in the MATLAB 'Data Analysis for Computational Biology,' towards estimating correlations among genes and generating the regulator network in a graph representation for SLE (34-36).
Epione application web-toolkit security and availability
The Epione application web tool is run on a Secure XAMPP HTTP Apache webserver hosted on the computing facility of the School of Applied Biology and Biotechnology at the Agricultural University of Athens. All EADs and third-party software packages used are locally installed, so there is no additional information transferred to other web servers. The user genomic data uploaded in the webserver is used for the Epione application pipeline only, while the results are presented privately and securely for a period of 1 month and erased afterward. The pipeline for identifying the most probable SNPs causing SLE described above is executed in the webserver named Epione application web tool, using Windows, Apache, XAMPP, PHP, HTML, JavaScript, R and parallel computing architecture and is openly available online at http://geneticslab.aua.gr/epione/.
Epione application validation
The Epione application webserver validation was performed by a retrospective study on seven patients from a three-generation family with endometriosis and other autoimmune diseases (10,37). WES data of one female patient with SLE, from the first generation (F1), was reanalyzed using the Epione application webserver.
Results
Epione application SLE database
The Epione application SLE database is an integrated resource for genes, alleles, pseudogenes and SNPs associated with SLE. The Epione database currently holds information on 2,158 genes, alleles, pseudogenes and transcription factors, 1,274 SNPs, and 70,000 related publications (Fig. 2). Moreover, 100 SNPs were detected in the coding region sites of genes (Fig. 3). All the SNPs associated with SLE were manually curated and classified into three major classes, including 'Strong-associated SNPs' with 221 members, 'High associated SNPs' with 100 members, and 'Associated SNPs' with 953 members (Fig. 2). The database also includes information from the Gene Database, dbSNP, LitVar Database, ClinVar Database, OMIM Database and PubMed Database. The information within the database was structured in several fields, and the knowledge was organized in a specific way in order to serve the webserver application immediately and quickly (Fig. 3).
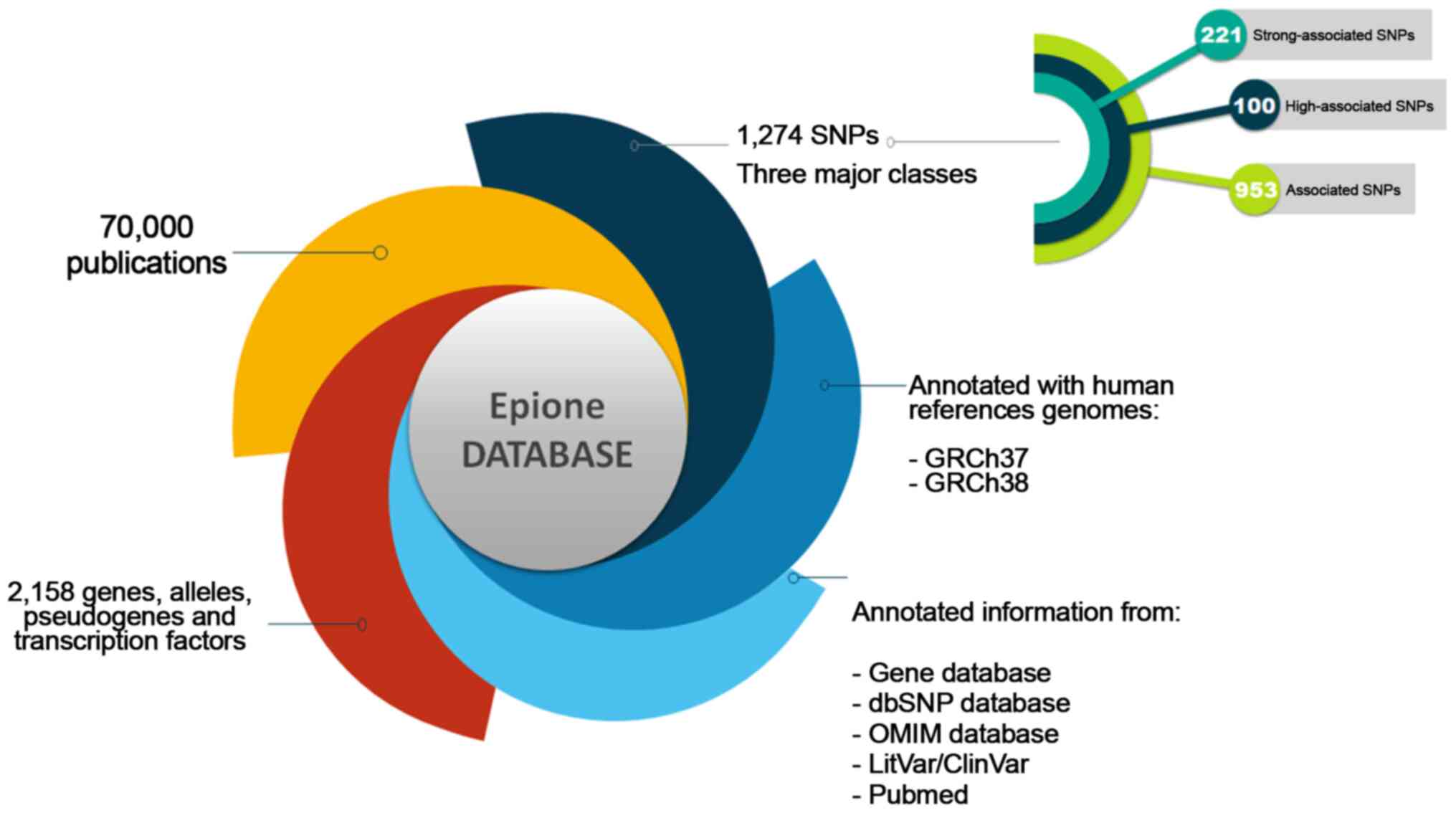 |
Figure 2
Epione application presenting the systemic lupus erythematosus database. SNP, single nucleotide polymorphism; dbSNP, Single Nucleotide Polymorphism Database; OMIM, Online Mendelian Inheritance in Man.
|
 |
Figure 3
Database analysis results. (A) 'X1', 'X2', 'X3' corresponds to the number of affected regions per SNP. (B) The five identified categories within the Epione database. (C) The identified types of SNPs within the Epione database. (D) The two major categories of the genomic regions within the Epione database. SNPs, single nucleotide polymorphisms; N/A, not applicable; LOC, locations; LINC, long intergenic non-coding; MIR, microRNA.
|
Data mining and semantic analysis for SLE
A systematic data mining and semantic analysis of the most frequently reported genes and polymorphisms was performed in order to identify those that are directly associated with SLE and thus may be of value in clinical genomics (10). A total of 70,000 publications were screened that contained the term 'SLE' in the title or abstract of the MEDLINE file. In the first level of the analysis, 2,158 genes, alleles, pseudogenes, and transcription factor names or synonyms were identified, and 230 key terms were found that described SLE, which were present in >10 publications within the dataset (Fig. 4). In Table I, the 30 most frequently identified key terms describing SLE are shown. Moreover, within the dataset, 420 different SNPs and 457 SLE-associated genes (Figs. 4 and 5) were reported and imported from online databases. Therefore, the analysis allowed us to identify polymorphisms that could potentially be included in the EAD, alongside the other SNPs that could predispose individuals to SLE. In the second level of analysis, 4,994 internal links among genes, alleles, pseudogenes and transcription factors were estimated through publications, and the regulatory network was calculated in a graph representation (Fig. 3). The major goal of this step of the analysis was to provide an exhaustive regulatory network in genes directly related to SLE (Fig. 5), apart from other SLE gene networks that have been presented previously (38).
 |
Figure 4
Selection of genes, alleles, pseudogenes and transcription factors for data mining and semantic analysis. SLE, systemic lupus erythematosus; MeSH, Medical Subject Headings.
|
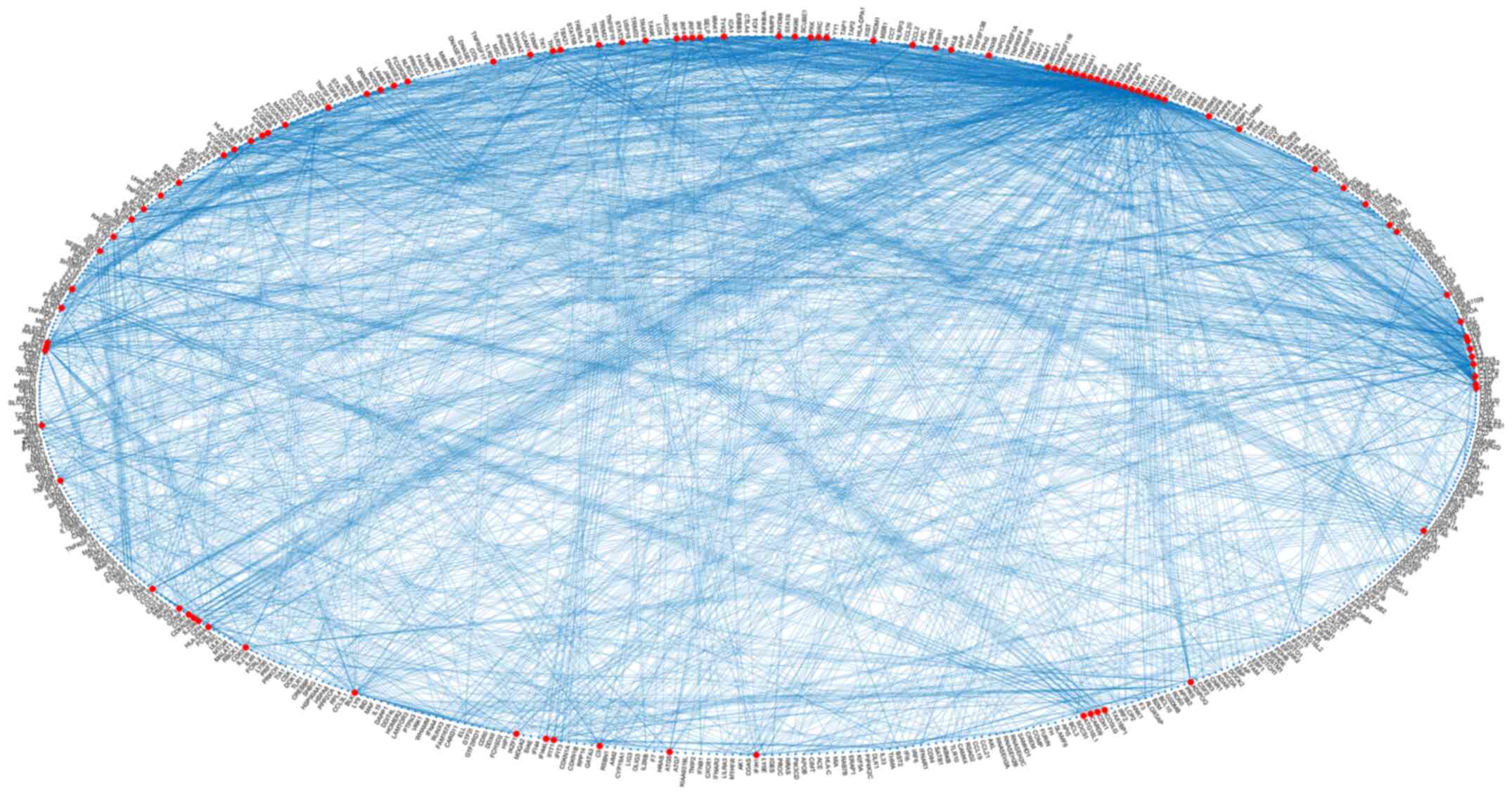 |
Figure 5
Systemic lupus erythematosus gene regulatory network of the class 'Strong-associated SNPs' in a graph representation. SNPs, single nucleotide polymorphisms.
|
 |
Table I
List of the 30 most frequently shown key terms describing SLE within the dataset.
|
Table I
List of the 30 most frequently shown key terms describing SLE within the dataset.
| A/A |
Key term |
Frequency |
| 1 |
'systemic lupus erythematosus' |
7,979 |
| 2 |
'lupus' |
1,151 |
| 3 |
'lupus erythematosus' |
1,028 |
| 4 |
'lupus nephritis' |
962 |
| 5 |
'autoimmune diseases' |
881 |
| 6 |
'rheumatoid arthritis' |
790 |
| 7 |
'autoimmunity' |
738 |
| 8 |
'antiphospholipid syndrome' |
460 |
| 9 |
'autoantibodies' |
456 |
| 10 |
'inflammation' |
445 |
| 11 |
'lupus nephritis'a |
293 |
| 12 |
'lupus erythematosus/therapy'a |
291 |
| 13 |
'disease activity' |
243 |
| 14 |
'lupus erythematosus, discoid'a |
232 |
| 15 |
'hydroxychloroquine' |
232 |
| 16 |
'pregnancy' |
218 |
| 17 |
'antiphospholipid antibodies' |
215 |
| 18 |
'biomarker' |
201 |
| 19 |
'epidemiology' |
195 |
| 20 |
'lupus anticoagulant' |
173 |
| 21 |
'lupus erythematosus, disseminated'a |
155 |
| 22 |
'lupus erythematosus/complications'a |
142 |
| 23 |
'cytokines' |
136 |
| 24 |
'nephritis' |
133 |
| 25 |
'lupus/therapy'a |
131 |
| 26 |
'meta-analysis' |
131 |
| 27 |
'cardiovascular disease' |
129 |
| 28 |
'atherosclerosis' |
129 |
| 29 |
'rituximab' |
129 |
| 30 |
'b cells' |
121 |
| 31 |
'dermatomyositis'a |
120 |
| 32 |
'quality of life' |
108 |
| 33 |
'le cells'a |
104 |
| 34 |
'lupus erythematosus/diagnosis'a |
102 |
| 35 |
'glomerulonephritis' |
102 |
| 36 |
'apoptosis' |
100 |
| 37 |
'cutaneous lupus erythematosus' |
100 |
| 38 |
'antiphospholipid syndrome'a |
98 |
| 39 |
'lupus eritematoso sistémico' |
96 |
| 40 |
'multiple sclerosis' |
91 |
| 41 |
'discoid lupus erythematosus' |
89 |
| 42 |
'cyclophosphamide' |
89 |
| 43 |
'glomerulonephritis'a |
86 |
| 44 |
'children' |
85 |
| 45 |
'drug therapy'a |
84 |
| 46 |
'autoimmune' |
84 |
| 47 |
'complement' |
84 |
| 48 |
'antibodies'a |
82 |
| 49 |
'collagen diseases'a |
82 |
| 50 |
'infection' |
82 |
| 51 |
'diagnosis'a |
81 |
| 52 |
'chloroquine'a |
80 |
| 53 |
'adolescence'a |
80 |
| 54 |
'autoantibody' |
79 |
| 55 |
'adrenal cortex hormones'a |
78 |
| 56 |
'mycophenolate mofetil' |
78 |
| 57 |
'arthritis' |
78 |
| 58 |
'belimumab' |
78 |
| 59 |
'diagnosis' |
77 |
Epione application webserver
The Epione application webserver assists health experts in supporting an SLE diagnosis for a patient using genetic information. This effective pipeline has been designed by geneticists able to benefit from bioinformatics support and by medical experts in SLE aiming to evaluate and classify all the determined gene variants related to SLE. Due to the large amounts of data required for analysis and the computational complexity of this pipeline, advanced bioinformatics techniques and parallel programming have been applied. It is estimated that using a parallel processing on the webserver requires 10× less time to analyze and extract the final results. Based on various tests executed on the performance of this application, it was estimated that this webserver has the ability to analyze a VCF file of 37,000 variants and create a personalized patient profile in <20 min. The Epione application has been designed to reduce complexity and minimize probable mistakes, allowing health experts to inset only a patient's genomic data from FASTA or VCF file towards estimating a clear and concise output HTML file with the patient profile (Fig. 6).
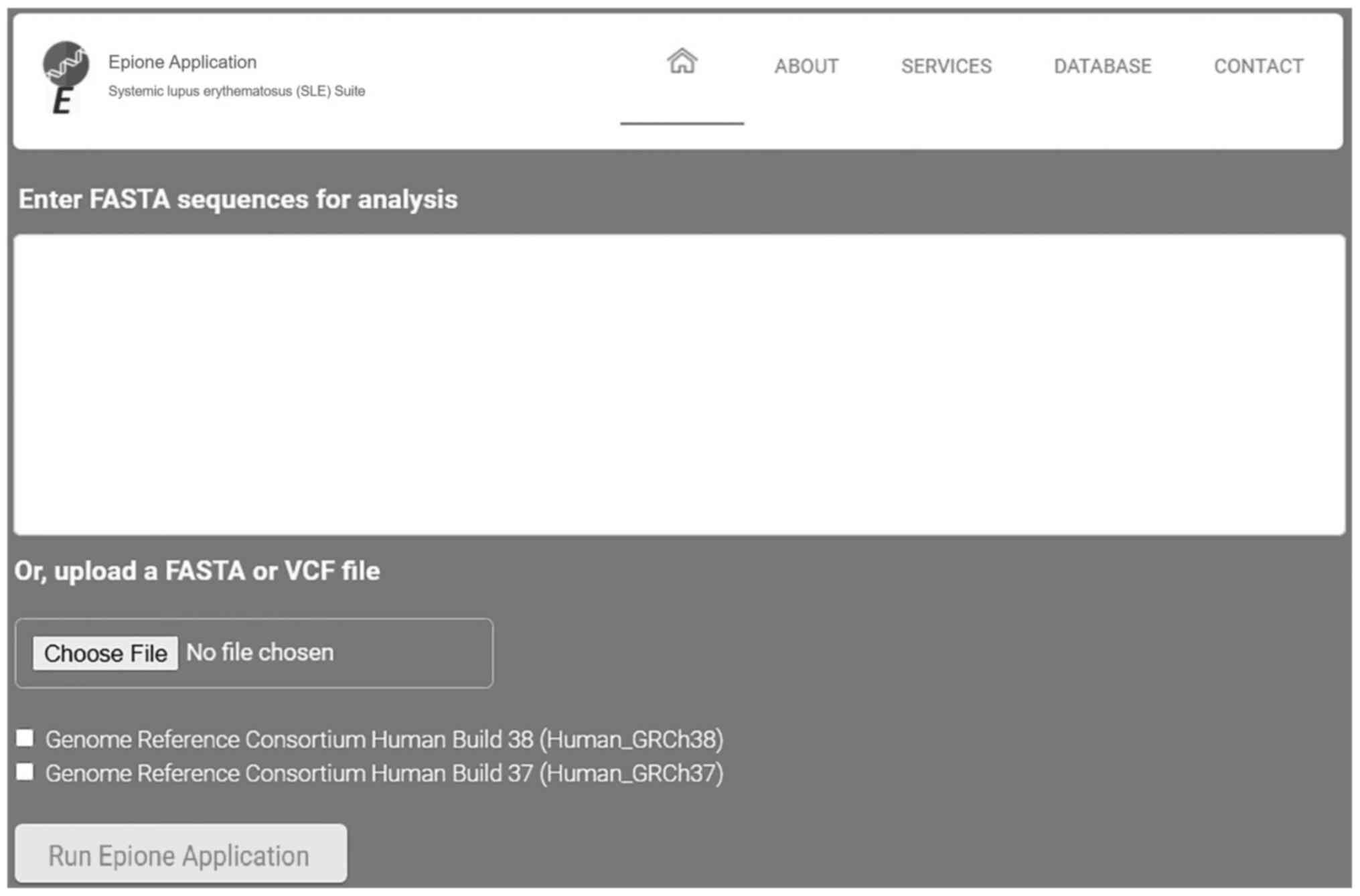 |
Figure 6
Epione application user interface. VCF, Variant Call Format.
|
The Epione application output is a HTML file that describes the patient profile through six major areas of results, including 'Server output details', 'SNPs Analysis Results for SLE', 'Statistic Charts', 'GWAS Analysis Results', 'Semantic and Data mining of identified Genes' and 'Downloads' (Figs. 7Figure 8-9). In the first results section, a summary of the analyzed information is presented, including the type of the data file analyzed, the number of identified SNPs and the date the analysis was performed. In the second section, the results of the SNP classification are shown in three separated charts and a list of all identified SNPs with extra information for each SNP as extracted from the Epione database. The third results section is concerned with various statistics charts regarding identified SNPs and the overall SNPs contained in the Epione database. The fourth section provides GWAS analysis results in a graphical representation of the chromosome ideogram, where all the identified SNPs in each genetic locus per chromosome have been marked. Moreover, a statistical chart that presents the identified SNPs per chromosome are shown. In the sixth section, the results from the data mining and semantic analysis are presented. A list of all identified genes is provided with all the information mined from the relative publications towards calculating and drawing the regulatory network in a graph representation. The user can filter the list in several ways and has the option to retrieve the relevant publications that describe each internal link within the network. Moreover, the beneficial knowledge of all connected genes with the identified genes is provided to the users. In the last results section, the user has the choice to download and save all the generated results from the Epione application webserver.
 |
Figure 7
Example of Epione application output part A. SLE, systemic lupus erythematosus; SNPs, single nucleotide polymorphisms.
|
 |
Figure 8
Example of Epione application output part B. SNPs, single nucleotide polymorphisms; GWAS, genome wide association studies.
|
 |
Figure 9
Example of Epione application output part C. SLE, systemic lupus erythematosus; SNPs, single nucleotide polymorphisms.
|
Epione application validation
A list with all known genes that were previously reported as 'SLE-associated' was properly identified in the final output HTML profile per patient, and by cross-comparison of the results, novel findings have emerged. The SNP analysis performed identified the common pathogenic variants that occurred within this family and were transmitted or imported from generation to generation (37). Moreover, a list of 'High-associated' and 'Strong-associated' polymorphisms that are directly related to SLE were identified and classified (Table II). The test was run with the Epione application using the default parameters on the human reference genome GRCh38. Further, the Epione application was also successfully evaluated with different well-confirmed SNPs located in genes, which may play a critical role in the development of SLE, as shown in Table II.
|
Table II
Major SNP cases identified in the seven patients with SLE.
|
Table II
Major SNP cases identified in the seven patients with SLE.
| SNP |
Chr |
Gene |
Class |
SNP type |
| rs3024866 |
Chr2 |
STAT4 |
A |
IV |
| rs17266594 |
Chr4 |
BANK1 |
A |
IV |
| rs10516487 |
Chr4 |
BANK1 |
A |
MV |
| rs280519 |
Chr19 |
TYK2 |
A |
IV |
| rs25487 |
Chr19 |
XRCC1 |
A |
MV |
| rs7530511 |
Chr1 |
IL23R |
A |
MV |
| rs549908 |
Chr11 |
IL18 |
A |
SV |
| rs3803800 |
Chr17 |
TNFSF13 |
A |
MV |
| rs344555 |
Chr19 |
C3 |
A |
IV |
| rs2476601 |
Chr1 |
PTPN22 |
A |
MV/IV |
| rs1061622 |
Chr1 |
TNFRSF1B |
A |
ICV |
| rs2230365 |
Chr6 |
NFKBIL1 |
A |
SV |
| rs419788 |
Chr6 |
SKIV2L |
A |
IV/UV |
| rs3813946 |
Chr1 |
CR2 |
A |
5′UTRV |
| rs1048971 |
Chr1 |
CR2 |
A |
SV |
| rs2246614 |
Chr11 |
CDHR5 |
A |
MV |
| rs2255336 |
Chr12 |
KLRC4-KLRK1 |
A |
MV/NCTV |
| rs17615 |
Chr1 |
CR2 |
A |
MV |
| rs945635 |
Chr1 |
FCRL3 |
A |
NCTV |
| rs3733197 |
Chr4 |
BANK1 |
A |
MV |
| rs2069763 |
Chr4 |
IL2 |
A |
SV |
| rs352140 |
Chr3 |
TLR9 |
A |
SV |
| rs315952 |
Chr2 |
IL1RN |
A |
MV |
| rs2326369 |
Chr20 |
MAVS |
A |
SV |
| rs315951 |
Chr2 |
IL1RN |
A |
3′UTRV |
| rs6133 |
Chr1 |
SELP |
A |
MV |
| rs763361 |
Chr18 |
CD226 |
A |
MV |
| rs2076530 |
Chr6 |
BTNL2 |
A |
MV/IV |
| rs4986938 |
Chr14 |
ESR2 |
A |
NCTV |
| rs2230201 |
Chr19 |
C3 |
A |
SV |
| rs3803665 |
Chr16 |
ZNF423 |
A |
SV |
| rs11552708 |
Chr17 |
TNFSF13 |
A |
MV/IV |
| rs6259 |
Chr17 |
SHBG |
A |
MV |
| rs3025000 |
Chr6 |
VEGFA |
A |
IV |
| rs513349 |
Chr6 |
BAK1 |
B |
IV |
| rs2229634 |
Chr6 |
ITPR3 |
B |
SV |
| rs7097397 |
Chr10 |
WDFY4 |
B |
MV |
| rs1061501 |
Chr11 |
IRF7 |
B |
SV |
| rs13181 |
Chr19 |
ERCC2 |
B |
StG/DV |
| rs20563 |
Chr1 |
LAMC1 |
B |
MV |
| rs4308977 |
Chr1 |
CR2 |
B |
MV |
| rs17616 |
Chr1 |
CR2 |
B |
MV |
| rs12150220 |
Chr17 |
NLRP1 |
B |
MV |
| rs396991 |
Chr1 |
FCGR3A |
B |
MV |
| rs1799793 |
Chr19 |
ERCC2 |
B |
MV |
| rs1801274 |
Chr1 |
FCGR2A |
B |
MV |
| rs3775291 |
Chr4 |
TLR3 |
B |
MV |
| rs3184504 |
Chr12 |
SH2B3 |
B |
MV |
| rs2279003 |
Chr19 |
MYO9B |
B |
SV |
| rs1782455 |
Chr1 |
MASP2 |
C |
SV/IV |
| rs6695096 |
Chr1 |
MASP2 |
C |
IV |
| rs11203366 |
Chr1 |
PADI4 |
C |
MV |
| rs11203367 |
Chr1 |
PADI4 |
C |
MV |
| rs874881 |
Chr1 |
PADI4 |
C |
MV |
| rs1748033 |
Chr1 |
PADI4 |
C |
MV |
| rs3790434 |
Chr1 |
LEPR |
C |
IV |
| rs6025 |
Chr1 |
F5 |
C |
MV |
| rs1137100 |
Chr1 |
LEPR |
C |
MV |
| rs2243188 |
Chr1 |
IL19 |
C |
IV/NCTV |
| rs3806268 |
Chr1 |
NLRP3 |
C |
SV |
| rs3747517 |
Chr2 |
IFIH1 |
C |
MV |
| rs2204640 |
Chr2 |
HECW2 |
C |
IV |
| rs708035 |
Chr3 |
IRAK2 |
C |
MV |
| rs818819 |
Chr3 |
SLC22A14 |
C |
MV |
| rs1137101 |
Chr1 |
LEPR |
C |
MV |
| rs1295686 |
Chr5 |
IL13 |
C |
IV |
| rs20541 |
Chr5 |
IL13 |
C |
MV |
| rs12522248 |
Chr5 |
HAVCR1 |
C |
MV |
| rs2075800 |
Chr6 |
HSPA1L |
C |
MV |
| rs1225944 |
Chr6 |
BLOC1S5-TXNDC5 |
C |
IV |
| rs1045642 |
Chr7 |
ABCB1 |
C |
MV |
Discussion
Epione application services can assist the diagnosis of SLE by filtering the individual's genetic profile through provided genomic SLE-related information that will eventually help to identify a patient's predisposition to SLE in the very early stages, even without any symptoms, similarly to a recently published article that used Epione to investigate endometriosis (10). In the case where medical experts lack a clear etiology for the patient's condition, Epione application results can provide useful information concerning the patient's profile and a list of the most critical genetic polymorphisms present in the patient's genome and their association with several biological pathways.
The extracted knowledge from the data mining and semantic analysis for SLE is included in the Epione application in a seamless way, where for each patient profile the pre-analyzed information can be used to determine the corresponding gene regulatory network based on the identified genes from the SNP database. The Epione application webserver contains all the pre-analyzed data in order to calculate and draw the regulatory gene network of each patient. The application generates a personalized regulatory network graph based on the patient's profile using all the identified SNPs related to genes, alleles, pseudogenes and transcription factors from the previous steps of the described pipeline. Thus, in addition to the detected polymorphisms, the Epione application has the ability to provide a list of the genes directly involved in several biological processes as regards with the genes harboring these polymorphisms. Furthermore, beyond the generated graph, all the internal links are provided in a list along with genes and relative publications.
The quality of the data for variants identified in the VCF file uploaded by the user numerous times may provide low reliability and cause several limitations. To deal with such problems, the Epione application validates the VCF file and removes variants that do not pass the quality control thresholds. On the other hand, it can also enable the user to upload the raw sequences or genotype data and provides a pre-processed analysis through which a generated VCF file is passed into the main pipeline of the webserver. Thus, the end user has the option to analyze both VCF and FASTA files without any restrictions.
EAD contains all the identified SNPs related to SLE, classified into three major classes. The quality of the information in the individual databases has possible limitations, and clinical databases may include non-verified annotations, as clinical research is being produced at ever faster rates. In order to ensure the predictive performance and the reliability of the system, so far, we opted for the manual update of the SNP Epione database after validation and classification of the candidate SNPs by a team of medical experts.
The detection and identification of genetic and epigenetic targets that play an important role in the manifestation of a disease is the 'key' in understanding and interpreting the various pathological conditions that may be present (39). Since a disease can be manifested by a different combination of harmful genetic polymorphisms, their collection and classification is very important for the different interpretations of the findings in a patient every time (40). In the present study, a novel pipeline to the collection and evaluation of genetic targets for a given disease were described. The Epione application for SLE, is a principal example in understanding that the outcoming data of such a genomic study can readily be used in the development of efficient applications for other genetic polymorphism-related diseases. To apply this application to other diseases an indexed list of confirmed linked genetic polymorphisms is required together with an analysis of the literature information linking the polymorphisms to the specific disease.
A comprehensive application analyzing genetic data against multiple available genetic targets for several autoimmune diseases is currently under testing. It also includes further expansion in techniques on data mining, semantic and machine learning together with links to Gene Ontology and Kyoto Encyclopedia of Genes and Genomes disease and pathway analyses.
To conclude, SLE is an inherited multifactorial disease that is usually detected at a fairly advanced stage, thus preventing doctors from applying treatment at an early stage. The Epione application was designed to assist healthcare experts in the diagnosis of SLE, even from the onset, by using the genomic data of patients. The comprehensive interface of the Epione application was designed to be used by the clinical genomics scientists and numerous other healthcare experts (10). Its diagnosis-oriented output presents the patient profile through which the user is provided with a structured set of results in various categories, generated based on the list of the most prominent candidate gene variants related to SLE. The majority of the current clinical genomics tools, web tools and applications are scientifically oriented for geneticists and bioinformaticians and are not developed to be easily handled by medical doctors or other scientists. In this sense, the Epione application is an easy-to-use integrated public webserver for SLE, designed with the aim of bringing personalized medicine and personal genomics tools to the medical community.
Availability of data and materials
The data that support the findings of this study have been published before (29) and are available from GNG and IM but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of GNG and IM.
Authors' contributions
LP, HA, DV, GNG, GB, IM, MIZ, DAS and EE substantially contributed to the conception and design of the work, including acquisition, analysis and interpretation of data. LP, DV, GNG, GB, IM, MIZ, DAS and EE contributed towards drafting the work and revising it critically for important intellectual content and approved the version to be published. All authors agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors have read and approved the final manuscript. GNG and IM confirm the authenticity of all the raw data.
Ethics approval and consent to participate
The test WES data used were from a previous study (29), and thus no ethics approval was required for the present study, as this was previously obtained (Ethics Committee of Venizeleio General Hospital of Heraklion, Heraklion, Greece; approval no. 46/6686).
Patient consent for publication
Not applicable.
Competing interests
DAS is the Editor-in-Chief for the journal, but had no personal involvement in the reviewing process, or any influence in terms of adjudicating on the final decision, for this article. The other authors declare that they have no competing interests.
Acknowledgments
Not applicable.
Funding
Funding was received by ‘INSPIRED‑The National Research Infrastructures on Integrated Structural Biology, Drug Screening Efforts and Drug Target Functional Characterization’ (grant no. 5002550) and ‘OPENSCREENGR An Open‑Access Research Infrastructure of Chemical Biology and Target‑Based Screening Technologies for Human and Animal Health, Agriculture and the Environment’ (grant no. 5002691) projects, which are implemented under the Action ‘Reinforcement of the Research and Innovation Infrastructure’, funded by the Operational Program ‘Competitiveness, Entrepreneurship and Innovation’ (National Strategic Reference Framework; grant no. 2014‑2020) and co‑financed by Greece and the European Union (European Regional Development Fund).
References
|
1
|
Crispín JC, Liossis SN, Kis-Toth K, Lieberman LA, Kyttaris VC, Juang YT and Tsokos GC: Pathogenesis of human systemic lupus erythematosus: Recent advances. Trends Mol Med. 16:47–57. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rahman A and Isenberg DA: Systemic lupus erythematosus. N Engl J Med. 358:929–939. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Harley JB, Kelly JA and Kaufman KM: Unraveling the genetics of systemic lupus erythematosus. Springer Semin Immunopathol. 28:119–130. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kwon YC, Chun S, Kim K and Mak A: Update on the Genetics of Systemic Lupus Erythematosus: Genome-Wide Association Studies and Beyond. Cells. 8:E11802019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ramos PS, Criswell LA, Moser KL, Comeau ME, Williams AH, Pajewski NM, Chung SA, Graham RR, Zidovetzki R, Kelly JA, et al: International Consortium on the Genetics of Systemic Erythematosus: A comprehensive analysis of shared loci between systemic lupus erythematosus (SLE) and sixteen autoimmune diseases reveals limited genetic overlap. PLoS Genet. 7:e10024062011. View Article : Google Scholar
|
|
6
|
Roberts J and Middleton A: Genetics in the 21st Century: Implications for patients, consumers and citizens. F1000 Res. 6:20202017. View Article : Google Scholar
|
|
7
|
Koboldt DC, Steinberg KM, Larson DE, Wilson RK and Mardis ER: The next-generation sequencing revolution and its impact on genomics. Cell. 155:27–38. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tam V, Patel N, Turcotte M, Bossé Y, Paré G and Meyre D: Benefits and limitations of genome-wide association studies. Nat Rev Genet. 20:467–484. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lightbody G, Haberland V, Browne F, Taggart L, Zheng H, Parkes E and Blayney JK: Review of applications of high-throughput sequencing in personalized medicine: Barriers and facilitators of future progress in research and clinical application. Brief Bioinform. 20:1795–1811. 2019. View Article : Google Scholar :
|
|
10
|
Papageorgiou L, Zervou MI, Vlachakis D, Matalliotakis M, Matalliotakis I, Spandidos DA, Goulielmos GN and Eliopoulos E: Demetra Application: An integrated genotype analysis web server for clinical genomics in endometriosis. Int J Mol Med. 47:1152021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Perakakis N, Yazdani A, Karniadakis GE and Mantzoros C: Omics, big data and machine learning as tools to propel understanding of biological mechanisms and to discover novel diagnostics and therapeutics. Metabolism. 87:A1–A9. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hirschhorn JN and Daly MJ: Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 6:95–108. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ugarte-Gil MF, González LA and Alarcón GS: Lupus: The new epidemic. Lupus. 28:1031–1050. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ozbek S, Sert M, Paydas S and Soy M: Delay in the diagnosis of SLE: The importance of arthritis/arthralgia as the initial symptom. Acta Med Okayama. 57:187–190. 2003.PubMed/NCBI
|
|
15
|
Feng X, Zou Y, Pan W, Wang X, Wu M, Zhang M, Tao J, Zhang Y, Tan K, Li J, et al: Associations of clinical features and prognosis with age at disease onset in patients with systemic lupus erythematosus. Lupus. 23:327–334. 2014. View Article : Google Scholar
|
|
16
|
Nightingale AL, Davidson JE, Molta CT, Kan HJ and McHugh NJ: Presentation of SLE in UK primary care using the Clinical Practice Research Datalink. Lupus Sci Med. 4:e0001722017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chang JC, Mandell DS and Knight AM: High health care utilization preceding diagnosis of systemic lupus erythematosus in youth. Arthritis Care Res (Hoboken). 70:1303–1311. 2018. View Article : Google Scholar
|
|
18
|
Gergianaki I and Bertsias G: Systemic lupus erythematosus in primary care: an update and practical messages for the general practitioner. Front Med (Lausanne). 5:1612018. View Article : Google Scholar
|
|
19
|
Oglesby A, Korves C, Laliberté F, Dennis G, Rao S, Suthoff ED, Wei R and Duh MS: Impact of early versus late systemic lupus erythematosus diagnosis on clinical and economic outcomes. Appl Health Econ Health Policy. 12:179–190. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Esdaile JM, Mackenzie T, Barré P, Danoff D, Osterland CK, Somerville P, Quintal H, Kashgarian M and Suissa S: Can experienced clinicians predict the outcome of lupus nephritis? Lupus. 1:205–214. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Piga M, Floris A, Cappellazzo G, Chessa E, Congia M, Mathieu A and Cauli A: Failure to achieve lupus low disease activity state (LLDAS) six months after diagnosis is associated with early damage accrual in Caucasian patients with systemic lupus erythematosus. Arthritis Res Ther. 19:2472017. View Article : Google Scholar
|
|
22
|
Urowitz M, Gladman DD, Ibañez D, Sanchez-Guerrero J, Bae SC, Gordon C, Fortin PR, Clarke A, Bernatsky S, Hanly JG, et al: Changes in quality of life in the first 5 years of disease in a multi-center cohort of patients with systemic lupus erythematosus. Arthritis Care Res (Hoboken). 66:1374–1379. 2014. View Article : Google Scholar
|
|
23
|
Allot A, Peng Y, Wei CH, Lee K, Phan L and Lu Z: LitVar: A semantic search engine for linking genomic variant data in PubMed and PMC. Nucleic Acids Res. 46(W1): W530–W536. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM and Sirotkin K: dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 29:308–311. 2001. View Article : Google Scholar :
|
|
25
|
Brown GR, Hem V, Katz KS, Ovetsky M, Wallin C, Ermolaeva O, Tolstoy I, Tatusova T, Pruitt KD, Maglott DR, et al: Gene: A gene-centered information resource at NCBI. Nucleic Acids Res. 43(D1): D36–D42. 2015. View Article : Google Scholar :
|
|
26
|
Kim S, Yeganova L, Comeau DC, Wilbur WJ and Lu Z: PubMed Phrases, an open set of coherent phrases for searching biomedical literature. Sci Data. 5:1801042018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lipscomb CE: Medical Subject Headings (MeSH). Bull Med Libr Assoc. 88:265–266. 2000.PubMed/NCBI
|
|
28
|
Hamosh A, Scott AF, Amberger JS, Bocchini CA and McKusick VA: Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 33:D514–D517. 2005. View Article : Google Scholar :
|
|
29
|
Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, et al: The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47(D1): D1005–D1012. 2019. View Article : Google Scholar :
|
|
30
|
Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, et al: The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 42(D1): D1001–D1006. 2014. View Article : Google Scholar :
|
|
31
|
Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al: ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46(D1): D1062–D1067. 2018. View Article : Google Scholar :
|
|
32
|
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al: 1000 Genomes Project Analysis Group: The variant call format and VCFtools. Bioinformatics. 27:2156–2158. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu JL and Zhao M: A PubMed-wide study of endometriosis. Genomics. 108:151–157. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Banchs RE: Text Mining With MATLAB®. Springer; New York, NY: 2013
|
|
35
|
Xiao H, Yang L, Liu J, Jiao Y, Lu L and Zhao H: Protein-protein interaction analysis to identify biomarker networks for endometriosis. Exp Ther Med. 14:4647–4654. 2017.PubMed/NCBI
|
|
36
|
Jurca G, Addam O, Aksac A, Gao S, Özyer T, Demetrick D and Alhajj R: Integrating text mining, data mining, and network analysis for identifying genetic breast cancer trends. BMC Res Notes. 9:2362016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Albertsen HM, Matalliotaki C, Matalliotakis M, Zervou MI, Matalliotakis I, Spandidos DA, Chettier R, Ward K and Goulielmos GN: Whole exome sequencing identifies hemizygous deletions in the UGT2B28 and USP17L2 genes in a three-generation family with endometriosis. Mol Med Rep. 19:1716–1720. 2019.PubMed/NCBI
|
|
38
|
Frangou EA, Bertsias GK and Boumpas DT: Gene expression and regulation in systemic lupus erythematosus. Eur J Clin Invest. 43:1084–1096. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gallagher MD and Chen-Plotkin AS: The Post-GWAS Era: From Association to Function. Am J Hum Genet. 102:717–730. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Suzuki A, Guerrini MM and Yamamoto K: Functional genomics of autoimmune diseases. Ann Rheum Dis. Jan 6–2021.Epub ahead of print. View Article : Google Scholar
|














