Introduction
Ulcerative colitis (UC) is a chronic disease
characterized by inflammation of the mucosa and submucosa of the
large intestine. The duration and extent to which a patient suffers
from UC are directly proportional to a propensity for colorectal
cancer (1,2). Ulcerative colitis patients have
elevated risk of colorectal cancer. This risk has been estimated to
be 2% after 10 years, 8% after 20 years and 18% after 30 years of
disease (3). Unlike sporadic
colorectal cancers that typically follow the adenoma-carcinoma
pathway, UC-associated colorectal tumors progress from areas of
dysplastic mucosa, following what it has been termed the
dysplasia-carcinoma pathway. These tumors typically develop as flat
lesions, which in many instances are difficult to detect,
especially in areas of active inflammation (4–8).
Owing to improvements in UC treatment this disease is rarely lethal
per se, and the associated mortality is largely determined
by the incidence of colorectal cancer. As of today, very little is
known about the molecular basis of the UC-associated colorectal
cancer. This highlights the urgent need to determine the early
genetic and epigenetic alterations germane to the UC-associated
carcinogenesis (9). Discerning the
mechanisms underlying UC-associated colorectal cancer will
facilitate the identification of markers for predisposition of the
UC-associated carcinogenesis, which in turn might improve the
prognosis and treatment of UC patients.
In several diseases characterized by chronic
inflammation, such as chronic hepatitis and Helicobacter
pylori infection chronic gastritis, aberrant DNA methylation of
promoter-associated CpG islands is associated with transcriptional
inactivation of tumor suppressor genes (10). In addition, aberrant DNA
methylation has been observed not only in cancer tissues but also
in tissues that appear to be histologically normal (11–13),
suggesting that some cancers arise from regions containing certain
epigenetic alterations that may be shared with an accompanying
cancer. This phenomenon is exemplified by the ‘field cancerization’
caused by carcinogen exposure (14). Molecular alterations such as p16
(15) and p14 hypermethylation
(16), were more frequently
observed in non-neoplastic epithelium of UC patients with neoplasia
than in those without neoplasia, suggesting that these molecular
alterations may be exploited as markers for identifying individuals
with UC at increased risk of neoplasia. Chronic inflammation has
been reported to associate with high levels of CpG island
hypermethylation, perhaps as a result of increased cell turnover,
and that age-related methylation marks (and may lead to) the field
defect that reflects acquired predisposition to colorectal
neoplasia (17). Fujii et
al, reported that ER gene methylation may be potentially useful
for identifying individuals at increased risk of neoplasia among
patients with long-standing and extensive UC (18). In addition, global DNA
hypomethylation in the rectal mucosa of patients with UC has been
proposed to be the result of the increased proliferative activity
triggered by the active inflammation, and that this epigenetic
change might predispose these individuals to develop colorectal
neoplasm (19). The genomic extent
of methylation alterations in UC mucosa, however, remains to be
accurately defined. In this regard, genome wide analyses of
methylation alterations provide an effective strategy to elucidate
the influence of epigenetic alterations on field cancerization.
Methylation sensitive amplified fragment length
polymorphism (MS-AFLP) is a fingerprinting technique developed by
Yamamoto et al as a tool to analyze DNA methylation in
hundreds of loci simultaneously (20,21).
MS-AFLP is based on the differential PCR amplification of
methylated vs. unmethylated NotI sites. We employed this
technique to demonstrate that the accumulation of DNA methylation
alterations in human colorectal and gastric cancers occurs in a
gradual fashion (22), and that
this accumulation associates with aging and precedes genomic damage
(23). More recently, we developed
a fluorescence-labeled MS-AFLP technique for the detection of
hyper- and hypomethylation alterations using a
fluorescence-detecting electrophoresis apparatus (24). One advantage of the MS-AFLP over
similar approaches is that approximately half (44%) of the
NotI recognition sites are located in or adjacent to CpG
islands, while the rest (56%) are located outside the CpG islands.
This distribution enables to detect both DNA hypermethylation,
generally occurring closer to CpG islands, and DNA hypomethylation,
generally affecting repetitive elements and single copy loci
outside CpG islands (25). Thus,
MS-AFLP provides unbiased insight into the complex picture of
hypermethylation and hypomethylation alterations, both of which are
known to be associated with cancer (26). In its original format, however,
MS-AFLP was limited by the number of bands than can be resolved in
a single electrophoretic gel, typically less than 100. This
limitation could be partly overcome by employing several primer
combinations that amplify different subsets of the
NotI-MseI fragment, but this solution is still
insufficient to analyze the nearly 10,000 NotI sites of the
human genome. A previous attempt to increase its throughput by
applying MS-AFLP to a DNA microarray hybridization demonstrated the
feasibility of the approach (27).
This methodology, however, required the cloning and sequencing of
individual MS-AFLP fragments before printing them onto the arrays,
two very labor-intensive steps that represented an important hurdle
for scaling up the throughput of the MS-AFLP microarrays.
In this study, we have designed a novel MS-AFLP
array-based platform containing probes consisting of
60-mer-oligonucleotides, which cover the sequences adjacent to all
the NotI sites identified in the human genome sequence.
Using this platform, we have identified methylation alterations in
non-neoplastic UC epithelia. Our results describe for the first
time the genome-wide methylation profile of UC mucosa samples, and
provides clues for the increased risk for developing colorectal
cancer.
Materials and methods
Cell culture, patients and tissues
The human colon cancer cell line CW-2 (RCB0778) was
provided by the RIKEN Cell Bank (RCB, Tsukuba, Japan). CW-2 cells
were cultured in Roswell Park Memorial Institute 1640 (RPMI-1640)
medium supplemented with 10% fetal bovine serum (FBS), 10,000 U/ml
penicillin G, 10 mg/ml streptomycin sulfate and 25 μg/ml
amphotericin B. The cells were maintained at 37°C in a humidified
incubator with 5% CO2.
Fourteen non-neoplastic UC epithelia biopsies (UCM)
were obtained from patients who had undergone surgery at Jichi
Medical University Saitama Medical Center and Jichi Medical
University hospital. In addition, 11 normal colon mucosal tissues
from healthy volunteers were collected and used as controls. The
study design was approved by the Institutional Review Board (IRB)
of Hamamatsu University School of Medicine and Jichi Medical
University.
Design of the methylation sensitive
amplified fragment array
To design the MS-AFLP array, we retrieved all the
80-bp upstream and downstream sequences adjacent to every one of
the 9,654 NotI sites previously mapped on the Build 36 of
the human genome sequence obtained from the NCBI. These sequences
were employed to design 19,308 60-mer microarray probes using
OligoArray 2.0, a program that designs specific oligonucleotides at
the genomic scale (28). This
process generated a set of 19,308 probes, two probes (one upstream
and one downstream) per every NotI site in the human genome
sequence. Using the same process, 2,200 additional
methylation-insensitive probes within MseI-MseI
fragments were designed to control for copy number alterations. All
these oligonucleotides were synthesized in duplicate onto array
slides by an ink-jet oligonucleotide synthesizer apparatus at
Agilent Technologies. The MS-AFLP array comprises a total number of
43,016 synthesized probes: 19,308×2 methylation sensitive probes,
plus 2,200×2 methylation-insensitive probes, and approximately two
thousand additional features required for the array scanning and
quality control. NotI sites are associated to DNA regions
with a high GC content, which are also rich in repetitive elements.
Mapped NotI sites (8.8%) occur within or adjacent to
repetitive sequences, in particular AluY elements. Hence, some
NotI-associated probes are unavoidably located inside
repetitive elements. To identify those probes complementary to
repetitive elements, every probe was re-mapped on the entire human
genome sequence allowing up to one mismatch. Of the 19,308 probes,
18,086 (93.7%) were unique in the genome while 1222 (6.3%) mapped
in two or more locations (896 probes mapping in 2–10 loci, 130
probes mapping in 11–100 loci, 110 probes mapping in 101–1,000
loci, 80 probes mapping in 1,001–10,000 loci, and 6 probes mapping
in >10,000 loci). Therefore, the MS-AFLP array design allows the
determination of methylation alterations in unique loci, moderately
repetitive DNA elements and highly repetitive DNA elements. The
MS-AFLP arrays employed in this study were printed at Agilent
Technologies facilities at Hachioji (Tokyo 192–8510, Japan), under
a collaborative arrangement.
Preparation, labeling and hybridization
of DNA samples for MS-AFLP arrays
Genomic DNA was isolated by QIAamp DNA Mini Kit
(Qiagen, Hilden, Germany). The initial steps of the MS-AFLP were
performed as previously described (20). Briefly, 1 μg of genomic DNA was
digested overnight with 5 U of methylation-sensitive NotI
(Promega, Madison, WI, USA) and 2 U of methylation-insensitive
MseI (NE Biolabs, Beverly, MA, USA) at 37°C. Two pairs of
oligonucleotides were annealed overnight at 37°C to generate
NotI (5′-CTCGTAGACTGCGTAGG-3′ and 5′-GGCCCCTACGCAGTCTAC-3′)
and MseI (5′-GACGATG AGTCCTGAG-3′ and 5′-TACTCAGGACTCAT-3′)
specific adaptors. The digested DNA was ligated to 1.25 μl each of
5 pmol/μl NotI and 50 pmol/μl MseI adaptor using 1 U
of T4 DNA ligase (Promega) overnight at 16°C. The adaptor-ligated
template DNA was amplified by PCR using NotI primer
(5′-GACTGCGTAGGGGCCGCG-3′) and MseI primer
(5′-GATGAGTCCTGAGTAA-3′). The PCR mixture consisted of 6 ng of
NotI primer, 30 ng of MseI primer, 0.25 mM dNTP, and
1.5 U of AmpliTaq DNA polymerase (Applied Biosystems, Foster City,
CA, USA) in a final volume of 20 μl. The PCR started at 72°C for 30
sec and 94°C for 30 sec, followed by 35 cycles of 94°C for 30 sec,
52°C for 30 sec, and 72°C for 2 min. The final extension was
performed for 10 min at 72°C. The reactions were then kept at 10°C
until the amplified DNA fragments were isolated using a QIA PCR
Clean-up kit (Qiagen). DNA was eluted into 50 μl of elution
buffer.
Prior to hybridization on the MS-AFLP arrays, the
DNA samples were labeled as previously described (27). Briefly, fluorescently labeled
fragments were prepared using the Bioprime labeling system
(Invitrogen). Each sample of PCR-amplified DNA (50 ng/2.5 μl) was
mixed with 5 μl of water and 5 μl of random primer mix solution.
The mixtures were boiled at 100°C for 2 min, quickly placed on ice
for 1 min, and briefly centrifuged for 10 sec. Then 1 μl of either
CY5 mix solution (1.56 mM each of dGTP, dATP and dTTP, 0.22 mM
dCTP, and 0.11 mM Fluorolink CY5-dCTP) or CY3 mix solution (1.56 mM
each of dGTP, dATP, and dTTP, 0.22 mM dCTP, and 0.11 mM Fluorolink
CY3-dCTP) was added. Fluorolink CY5-dCTP and CY3-dCTP were
purchased from Amersham-Pharmacia. Klenow fragment of E.
coli DNA polymerase was then added to a final concentration of
0.8 U per μl. The mixtures were incubated at 37°C for 1 h before
adding 2 μl of stop solution (0.5 M EDTA) to terminate the
reaction. The CY5 and CY3 fluorescently labeled DNA fragments were
separated from the unincorporated dNTPs by filtration through
Microcon YM-30 columns (Millipore, Bedford, MA, USA). Each sample
was reconstituted with 1X TE (pH 8.0) to a final volume of 37 and 2
μl of each sample was taken to determine the yield of labeled
genomic DNA and the specific activity after labeling and clean-up.
Exposure of samples to light was minimized during all experimental
procedures.
The Cy3 and Cy5 labeled DNA samples were mixed in a
siliconized tube with 70 μl of Agilent 2X Hi-RPM buffer (Agilent,
Santa Clara, CA, USA). The mix was heated at 95°C for 3 min and
centrifuged at 6000 × g for 1 min to collect the sample at the
bottom of the tube. Hybridization sample mixture (110 μl) was
applied slowly to the gasket slide into the Agilent SureHyb chamber
base. Then, one microarray slide was placed onto the gasket slide,
with the active side facing down. The SureHyb chamber was covered
onto the slides, and the clamp assembly was slid onto both pieces.
The assembled slide chamber was placed in a rotator rack inside a
hybridization oven and rotated at 20 rpm and hybridized at 65°C for
40 h. After hybridization, array slides were washed with Oligo aCGH
wash buffer 1 at room temperature for 5 min and Oligo aCGH wash
buffer 2 at 37°C for 1 min. To prevent Cy5 degradation by ozone,
the slides were washed with acetonitrile for 30 sec and then with
stabilization and drying solution for 30 sec. The arrays were
scanned using an Agilent G2565BA DNA Microarray Scanner.
MS-AFLP array data analysis
After scanning, data extraction was conducted using
the Feature Extraction software version A.10.5.1.1 (Agilent
Technologies). GeneSpring version 10 (Agilent Technologies) was
used to export the array data. For the MS-AFLP array data analysis,
an ad hoc pipeline was developed in R (29). The background corrected signals
were used to calculate the log2 ratio of the CY5 vs CY3 channel for
every array feature. The log2 ratios were subsequently normalized
by LOWESS method (locally weighted scatterplot smoothing), using
the information from all the features in the array. Then, the
normalized log2 ratios from the 19,308 methylation-sensitive probes
were extracted from the dataset. Since these probes are synthesized
in duplicate in the array, two independent log2 ratio values were
obtained for each probe. These values were combined into a single
value by calculating their weighted mean, where the weights were
given by the total intensity of the replicates in both the CY3 and
CY5 channels. Finally, the probes within the 30% lowest intensity
in both the CY3 and CY5 channels in both replicates, likely
representing cross-hybridization noise, were excluded from the
subsequent analyses. The thresholds for hypermethylation and
hypomethylation were set at log2 ratio < -1 and log2 ratio >
1.
Bisulfite sequencing
Bisulfite sequencing was performed as previously
described (30). Genomic DNA was
modified with EpiTect Bisulfite Kit™ (Qiagen). Bisulfite-modified
DNA was analyzed by PCR with primers specifically designed to
amplify the NotI-containing products of interest. The
primers and annealing temperature for the bisulfite sequencing
were: spot a (NotI site no. 5391, primers 5′-TGTTATAGGGATGGA
TTTTGTTAT-3′ and 5′-CCACCCCTATATCATACCCT-3′, annealing temperature:
56°C); spot b (NotI site no. 2627, primers
5′-GGTGGTTTGTTTTTTTGAAGA-3′ and 5′-AACCTATAT CAAAACACCCCC-3′,
annealing temperature 50°C); spot c (NotI site no. 6105,
primers 5′-GAGTGATTGAATTAAGA AGAGTAAAA-3′ and
5′-AAACTCAAACCCAATTAATT AAA-3′, annealing temperature 46°C); spot d
(NotI site no. 374, primers 5′-TGTGTTAATATTTGGGGGTAGAG-3′
and 5′-TACTTCTACCCTAACCTCCCCT-3′, annealing temperature 53°C); spot
e (NotI site no. 563, primers 5′-GTG TTTTAGGAGGATTTGGG-3′
and 5′-AAAAAATACCCC TATCTCACCTC-3′, annealing temperature 51°C);
and spot f (NotI site no. 3511, primers
5′-GTTTTTATTTGAGGGGGA ATG-3′ and 5′-TACTCTTAACCTAACTCACCCCC-3′,
annealing temperature 51°C). PCR amplifications were carried out in
20 μl reaction mixtures containing 1X PCR buffer, 0.25 mM (each)
dNTP mix, 0.025 U/μl HotStarTaq Plus DNA Polymerase (Qiagen) and
0.5 μM each primer. The PCR conditions were 95°C for 5 min,
followed by 38 cycles of 95°C for 45 sec, then 45 sec at the
specific annealing temperature, 72°C for 1 min, and then 72°C for
10 min. PCR products were purified by EXOSAP-IT (USB Corp.,
Cleveland, OH, USA), and cloned into the pGEM-T Easy Vector
(Promega). Recombinant plasmids were sequenced with an ABI PRISM
3100 Genetic Analyzer (Applied Biosystems) using SP6
(5′-TATTTAGGTGACACTATAG-3′) and T7 (5′-TAA TACGACTCACTATAGGG-3′)
primers. The methylation status of each CpG site was determined by
sequencing, as unmethylated cytosines are converted into thymines
after the bisulfite treatment and PCR amplification, whereas
methylated cytosines remain unaltered.
Statistical analysis
Statistical analyses were performed using the R
statistical environment (29).
Fisher’s exact was used to examine associations between two
categorical variables. Continuous variable comparisons between two
groups were performed with the Student’s t-test for those variables
following a normal distribution, or with the non-parametric
Mann-Whitney-Wilcoxon test for those variables that do not follow a
normal distribution. Normality was assessed using the Shapiro-Wilk
test. The level of statistical significance was set at P<0.01,
unless otherwise specified. Holm’s multi-hypothesis testing
correction was applied when appropriate (31). Hierarchical analysis and clustering
analysis were performed using MeV (32). Gene category enrichment analyses
were performed using DAVID (33,34),
which employs a more robust variation of the Fisher’s exact test,
termed EASE score (35). To
account for the bias due to the partial gene representation in the
MS-AFLP array, all the gene enrichment analyses were performed
using the list of the genes present in the array as a background,
instead of the total number of genes in the human genome.
Results
Set-up and testing the MS-AFLP array
To estimate the false-positive rate of the MS-AFLP
array we performed a self-self hybridization experiment using a
mixture of DNAs obtained from the colonic mucosa of 11 healthy
volunteers (see Materials and methods). This mixture of DNAs was
used as reference sample in all the subsequent MS-AFLP arrays
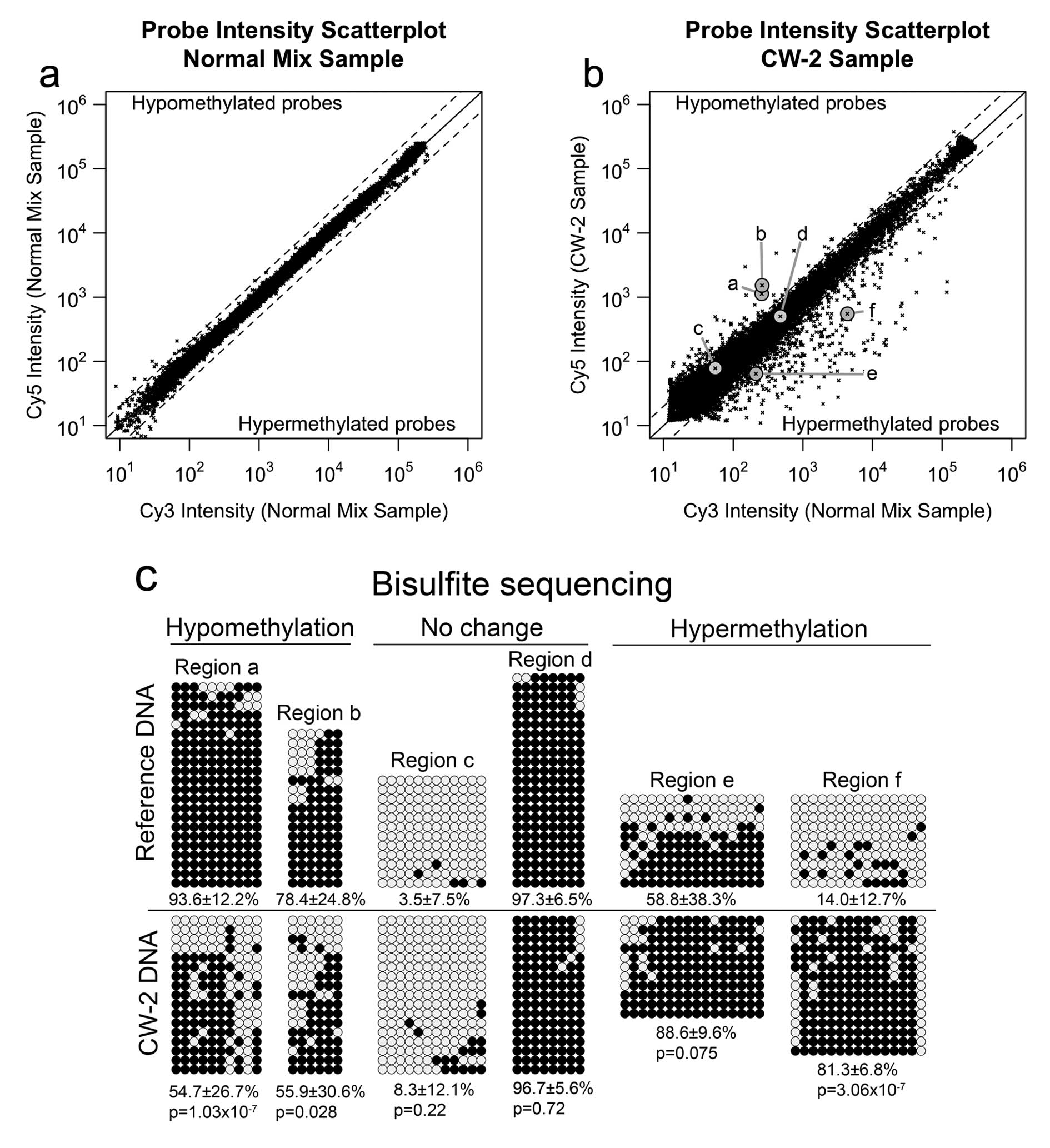
presented in this study. The results are shown in the scatter plot
of Fig. 1a. In this plot, the
logarithmic x-axis and y-axis represent the average signal in the
Cy3 and Cy5 channels, respectively. The no-change line (log2 ratio
= 0) is in solid black. The diagonal dashed lines indicate the
alteration calling thresholds, set at log2 ratio > 1 for
hypomethylation and log2 ratio < -1 for hypermethylation.
According to the self-self hybridization experiment, the MS-AFLP
array exhibited a 0.12% of false positives mostly in the
low-intensity subset of probes, more susceptible to spurious
fluctuations due to probe cross-hybridization. Applying a very
stringent floor filter excluding the 30% lowest-intensity probes,
the false positive rate was reduced to <0.015%. This filter was
applied by default in all subsequent MS-AFLP array analyses.
The colon cancer cell line CW-2 was selected to
determine the capabilities of the array-based MS-AFLP to detect DNA
methylation alterations. CW-2 is a colorectal cancer cell line
derived from a 60-year-old female patient with lymph node
metastases. The cells display microsatellite instability (MSI) due
to an inactivating frameshift mutation in hMLH1. This
alteration is fixed in homozygosis due to copy neutral LOH
affecting the 62-Mb distal region of the 3p chromosome, where
hMLH1 is located (data from The Cancer Genome Project of The
Wellcome Trust Sanger Institute). Except for the loss of a 13-Mb
region of chromosome X, CW-2 cells harbor very few copy number
alterations (CNAs), and they are mostly diploid with 84% of cells
harboring a normal content of 46 chromosomes (information from the
RIKEN BioResource Center).
The result of the MS-AFLP array analysis of CW-2 DNA
relative to a pooled sample of normal colon tissue DNAs is shown in
Fig. 1b. To validate the
methylation changes detected by the MS-AFLP array, we selected six
probes representing two hypomethylated NotI sites (a and b),
two non-altered NotI sites (c and d) and two hypermethylated
NotI sites (e and f). These loci were analyzed by bisulfite
genomic sequencing, the current gold standard technique to analyze
DNA methylation, using the primers indicated in Materials and
methods. The results obtained by bisulfite sequencing (Fig. 1c) confirmed the results from the
MS-AFLP array in all the analyzed loci, demonstrating the
capability of the MS-AFLP array to detect DNA methylation
alterations. In addition, no association between the chromosomal
location of the DNA methylation alterations and the copy number
variations reported in CW-2 was observed (data not shown), which
further supports the validity of this platform to accurately detect
somatic epigenetic changes even within chromosomal regions that had
undergone moderate copy number alterations.
The MS-AFLP array analysis of the genome of CW-2 DNA
revealed 500 probes (3.75%) with log2 ratio below the
hypermethylation threshold, and 129 probes (0.95%) with log2 ratio
above the hypomethylation threshold (Fig. 1b). Since this cell line is derived
from a female patient, the data from the Y chromosome probes was
removed from the analysis. Due to design strategy constrains, the
MS-AFLP array contains 94 probes with mononucleotide repeats of 9
or more nucleotides (polyN). Shortenings and expansions of
mononucleotide repeats are the archetypical feature of the MSI
phenotype resulting from defects in the mismatch repair mechanisms
(36). Since CW-2 is an MSI cell
line, we investigated whether the polyN-containing probes exhibited
a particular behavior. Out of the 94 probes with polyN sequences, 8
where excluded due to low intensity in both the Cy3 (reference
sample) and Cy5 (CW-2 sample) channels. Of the remaining 86 probes,
17 (19.8%) exhibited a log2 ratio below the hypermethylation
threshold (i.e., significantly hypermethylated), a much higher
frequency than that of probes without polyN (483/13015, 3.7%). None
of the polyN-containing probes exhibited log2 ratios above the
hypomethylation threshold (i.e., significantly hypomethylated).
Hence, there was a very strong association between the presence of
mononucleotide repeats in the probe sequences and the reduction in
their signal intensity in the CW-2 sample, resulting in a negative
log2 ratio (OR=6.6, 95%CI=3.6–11.5, P=1.3×10−8 Fisher’s
exact test). While the possibility that some of these probes
indicate actual methylation alterations can not be discarded, it
seems more plausible that the reduction of signal intensity is a
consequence of microsatellite shortenings and expansions in the
genome of CW-2 cells, resulting in loss of perfect complementarity
between the MS-AFLP amplicons and the MS-AFLP array probes.
Therefore, to avoid miss-interpretation errors, polyN-containing
probes were not considered for the scoring of alterations in the
subsequent analysis of CW-2 MS-AFLP array. Of the remaining 13,395
probes, 482 (3.6%) indicated hypermethylation and 128 (0.96%)
indicated hypomethylation.
The majority of the alterations were detected by
probes mapping in a single location (558/610, 91.5%) or in 2–10
locations (47/610, 7.7%). Only five of the altered probes mapped in
>10 locations in the genome, all of them within repetitive
sequences (data not shown). We also investigated whether DNA
methylation alterations were more frequently detected in
NotI sites located close to gene transcriptional start sites
(TSS), where presumably these alterations might be associated to
transcriptional variations. Since the TSS is not well defined for
all the annotated human genes, we considered a promoter-containing
region the 10-kb region surrounding the annotated 5’ end of the
genes (5 kb upstream and 5 kb downstream). In CW-2, both
hypermethylation and hypomethylation alterations were actually more
frequent in the NotI sites located outside of these
promoter-related regions. The negative association of
hypermethylation alterations with the 5′ regions exhibited an odds
ratio (OR) of 0.80 (95% CI 0.66–0.96, P=0.018 Fisher’s exact test).
The negative association of hypomethylation alterations with the 5′
regions exhibited an OR=0.62 (95% CI 0.43–0.89, P=0.008). This
observation is in agreement with recent findings showing that in
colon cancer most DNA methylation alterations occur not in
promoters or in CpG islands, but in ‘shore’ sequences up to 2 kb
distant (25). A list of the
NotI sites with methylation alterations in CW-2, their
genomic location, and the associated genes is available upon
request.
Using the database for annotation, visualization and
integrated discovery v6.7 (DAVID) (33,34)
we analyzed the enrichment of gene families among the
hypermethylated or hypomethylated genes versus the list of genes
present in the array. For hypermethylation, this analysis revealed
a highly significant overrepresentation of genes encoding
transcriptional factors (GOterm 0003700, n=49, fold enrichment,
1.86; EASE score, 1.14×10−5), in particular,
transcriptional factors with homeobox domains (Interpro IPR001356,
n=25, fold enrichment, 3.32; EASE score, 1.9×10−7). In
addition, forty-two of the hypermethylated genes are involved in
developmental processes (fold enrichment, 1.99; EASE score,
1.74×10−5). Among the hypomethylated genes, we found an
enrichment of genes involved in epithelial cell development (GO
term 0060429, n=6, fold enrichment, 4.37; EASE score, 0.01).
DNA methylation alterations in the
colonic mucosa of UC patients
Once the MS-AFLP array was validated in the CW-2
colorectal cancer cell line, we analyzed 14 colonic mucosa samples
obtained from UC patients (UCM). Table
I shows the clinicopathological features of the patients
recruited in this study and the frequency of DNA methylation
alterations detected by the MS-AFLP array analyses. No association
was found between patient age, duration of disease or inflammation
grade and the incidence of methylation alterations. The analysis of
the data from all the UCM samples together revealed a negative
association between methylation alterations and the 5′ region of
genes (OR=0.84, P=0.0013, 95%CI 0.75–0.93 for hypermethylation, and
OR=0.78, P=7.5×10−5, 95%CI=0.69–0.88 for
hypomethylation. Fisher’s exact test).
 | Table IPatient and tissue characteristics,
and epigenetic alterations. |
Table I
Patient and tissue characteristics,
and epigenetic alterations.
| | | | | Methylation
alterationsb |
|---|
| | | | |
|
|---|
| Patient | Gender | Age | Disease
duration | Matts’
scorea | Hyper | Hypo | Total |
|---|
| UC.1 | F | 46 | 14 | 3.3 | 0.64% | 0.64% | 1.27% |
| UC.2 | F | 67 | 13 | 3.3 | 0.41% | 0.14% | 0.55% |
| UC.3 | F | 23 | 2 | 3.3 | 0.24% | 0.37% | 0.61% |
| UC.4 | M | 37 | 12 | 3.7 | 0.66% | 0.31% | 0.97% |
| UC.5 | M | 56 | 5 | 2.3 | 1.07% | 0.64% | 1.71% |
| UC.6 | M | 28 | 10 | 2.7 | 0.48% | 0.46% | 0.94% |
| UC.7 | M | 36 | 16 | 1.3 | 0.23% | 0.61% | 0.84% |
| UC.8 | M | 19 | 1.5 | 2.7 | 0.97% | 1.02% | 1.99% |
| UC.9 | M | 42 | 10 | 2 | 0.82% | 0.68% | 1.50% |
| UC.10 | M | 26 | 6 | 1.7 | 1.12% | 0.72% | 1.83% |
| UC.11 | F | 43 | 3 | 2.3 | 0.98% | 0.50% | 1.48% |
| UC.12 | F | 23 | 9 | 2.3 | 1.04% | 0.34% | 1.38% |
| UC.13 | F | 32 | −9 | 3 | 0.87% | 0.80% | 1.67% |
| UC.14 | M | 35 | 10 | 2.7 | 0.42% | 0.38% | 0.81% |
| Mean ± SD | 57% M | 36.6±13.4 | 8.6±4.5 | 2.6±0.7 | 0.7±0.3% | 0.5±0.2% | 1.3±0.5% |
| 43% F | | | | | | |
Identification of frequently altered
loci
To identify the probes altered in the UCM samples
with a statistically significant frequency, we applied a binomial
test based on the frequency of alterations empirically obtained
from every individual sample, under the null hypothesis that every
probe had the same probability of being altered within a sample.
The level of statistical significance was set at P<0.01 after
correction for multi-hypothesis testing by the Holm method
(31). Results are shown in
Fig. 2. This analysis rendered 133
probes indicating hypermethylation in 3–12 of the 14 samples
(frequently hypermethylated set), and 118 probes indicating
hypomethylation in 3–14 of the 14 samples (frequently
hypomethylated set). Twelve probes were common to both
hypermethylated and hypomethylated sets, that is, hypermethylated
in three or more samples and hypomethylated in three or more
different samples.
The frequently hypermethylated probe set was
enriched in genes participating in transcriptional processes, i.e.,
encoding transcriptional factors and transcriptional modulators and
proteins with RNA-binding function, and genes involved in signal
transduction, such as protein kinases and SH2 domain-containing
proteins (data not shown). Interestingly, the second most
frequently hypermethylated probe is located in the promoter of MPG,
a gene that codes for the N-methylpurine-DNA glycosylase involved
in the repair of DNA alkylation lesions caused by reactive oxygen
and nitrogen species (RONS), which are typical products of the
chronic inflammation process. In addition, six of the frequently
hypermethylated genes, i.e., CCND1, COL4A2, HDAC2, AXIN2, ABL1 and
GLI2, are included in the pathways in cancer map from the KEGG
database (37).
Hypomethylation associates with
repetitive elements
In comparison with the frequently hypermethylated
probe set, the gene enrichment analysis of the frequently
hypomethylated probe set revealed a lower number of
highly-significant gene categories. Of note, we found a borderline
significant enrichment of genes with protein kinase C-like domains
(RASGRP1, CDC42BPB and PRKCB; EASE score, 0.14; Fisher’s exact test
P=0.016). Two of these genes, RASGRP1 and PRKCB, function as
positive regulators of the Ras signaling pathway. The analysis also
revealed a significant enrichment (fold enrichment, 8.0; EASE
score, 0.05; Fisher’s exact test P=0.006) of genes associated with
cell differentiation (NOG, FOXC1 and HNRNPAB).
Among the 118 frequently hypomethylated probes,
seven were complementary to high copy number elements, revealing a
strong association between hypomethylation and DNA repetitive
elements (OR=6.3; 95%CI 2.4–13.7; P=2.1×10−4, Fisher’s
exact test). A more extensive analysis of the 196 MS-AFLP array
probes complementary to high-copy repetitive elements (>100 hits
in the genome) revealed that more than half of the samples (8 out
of 14) showed hypomethylation in at least one of the probes located
in Alu repetitive elements. In contrast, none of these 196 probes
indicated hypermethylation in any of the 14 analyzed cases.
Demethylation of repetitive elements is a generally accepted
surrogate of global DNA hypomethylation (26). Hence, this finding is in agreement
with previous reports that global genome-wide demethylation is a
common phenomenon in the inflamed colonic mucosa of UC patients
(19).
Clustering of altered loci in the colonic
mucosa of UC patients
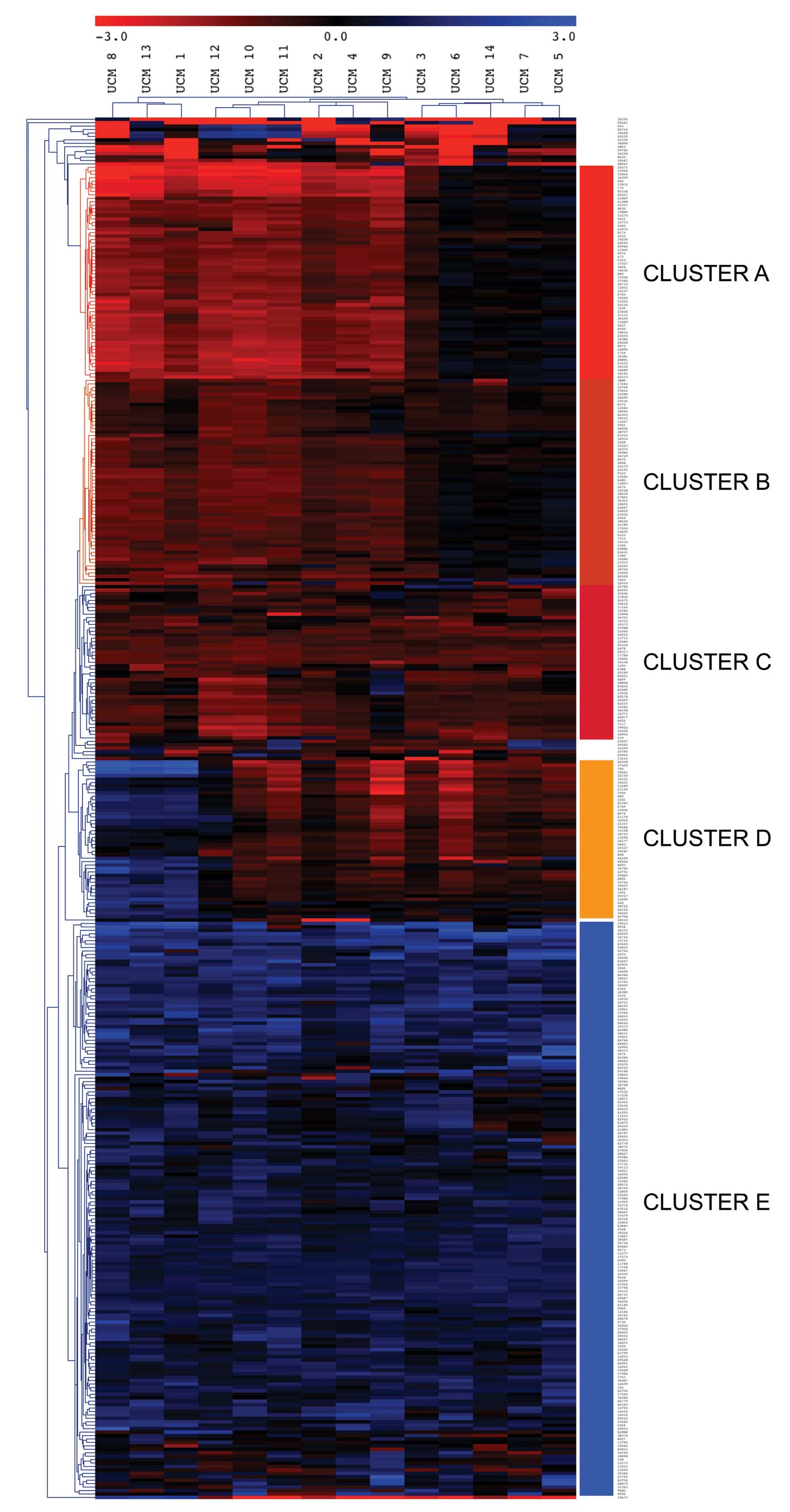
We performed a Euclidean distance unsupervised
cluster analysis with the data from all the probes hypermethylated
in 2 or more samples or hypomethylated in 2 or more samples
(Fig. 3). This analysis revealed 5
main gene clusters, named A–E, and three distinct groups of samples
that were primarily defined by differences in the methylation
status of three of these gene clusters (A, B and D). Clusters C and
E were mainly hypermethylated and hypomethylated, respectively, in
all the samples and exhibited lower classification power. The
clustering revealed three distinctive groups of patients. A group
of three cases (samples 1, 8 and 13, left of the heatmap) was
characterized by strong hypermethylation of cluster A, and
hypomethylation of cluster D. A second group of 6 patients (samples
2, 4, 9, 10, 11 and 12, center of the heatmap) showed
hypermethylation in all clusters, but cluster E. Finally, a group
of 5 patients (samples 3, 5, 6, 7 and 14, right of the heatmap)
exhibited a characteristic absence of hypermethylation in clusters
A and B. No obvious differences were found among the three groups
of patients regarding age, gender, degree of inflammation or
duration of the disease. A list of the NotI sites with DNA
methylation alterations in these UCM samples, their genomic
location, and the associated genes is available upon request.
Concordance of epigenetic alterations in
non-cancer colonic mucosa of UC patients and CW-2 colon cancer
cells
We compared the alterations in NotI sites
observed in the UCM samples with those identified in the previous
analysis of the CW-2 cancer cell line. There was a strong
concordance between the alterations found in the UCM samples and
those found in the CW-2 cell line, both for hypermethylation and
hypomethylation alterations (Table
II). The detailed gene-enrichment and gene ontology analysis of
these data will be published elsewhere.
| Table IIConcordance between hypermethylation
and hypomethylation alterations detected by MS-AFLP arrays in
cancer cell line CW-2 and non-cancer UCM samples. |
Table II
Concordance between hypermethylation
and hypomethylation alterations detected by MS-AFLP arrays in
cancer cell line CW-2 and non-cancer UCM samples.
| A, Hypermethylation
alterations |
|---|
|
|---|
| In UCM |
|---|
|
|
|---|
|
Hyper-methylateda |
Non-hyper-methylated |
|---|
| In CW-2 |
|
Hypermethylated | 62 | 421 |
|
Non-hypermethylated | 86 | 12946 |
| Odds ratio =
22.15 |
| 95% CI
15.48–31.56 |
| Fisher’s exact test
P<2.2×10−16 |
|
| B, Hypomethylation
alterations |
|
| In UCM |
|
|
|
Hyper-methylatedb | No
hyper-methylated |
|
| In CW-2 |
|
Hypomethylated | 27 | 102 |
|
Non-hypomethylated | 91 | 13295 |
| Odds ratio =
38.63 |
| 95% CI
23.13–62.91 |
| Fisher’s exact test
P<2.2×10−16 |
Discussion
We described a novel microarray-based platform,
termed MS-AFLP array, designed to analyze DNA methylation
alterations in the nearly 10,000 NotI sites of the human
genome. This platform is based on the robust MS-AFLP technique
(20) that we previously employed
to show that DNA methylation alterations are gradual and that
hypomethylation appears to be dominant over hypermethylation in
linking epigenotype with cancer genotype and clinical outcome in
sporadic gastro-intestinal cancers (22,23).
Our platform possesses some unique characteristics compared to
other methylation microarrays platforms. Most notably, it does not
rely on the treatment of DNA with bisulfite (30). Instead, the DNA samples are
digested with a combination of one methylation-sensitive
(NotI) and one methylation-insensitive (MseI)
restriction enzymes, then adaptors are ligated and the fragments
are amplified by PCR. Thus, the genomic distribution of probes is
dictated by the distribution of NotI sites, which are
located not only in promoter regions but also in intra- and
intergenic regions.
We tested the platform in the colorectal cancer cell
line CW-2, and showed the ability and accuracy of the MS-AFLP array
to detect DNA methylation alterations (Fig. 1). Hypermethylated probes (482) were
almost 5 times more frequent than the hypomethylated (128) probes.
This finding might seem counterintuitive, since DNA
hypomethylation, the almost constant companion to DNA
hypermethylation in cancer, preferentially occurs in repetitive
elements such as LINE-1 and Alu sequences that account for 34% of
the human genome, while hypermethylation generally occurs in unique
sequences, so the combined effect of these two types of alterations
is an overall loss of DNA methylation. In the CW-2 MS-AFLP array
context, however, it must be noted that: a) the vast majority of
NotI sites lay not in LINE-1 or Alu elements but in unique
loci, and therefore the detection is biased towards alterations
occurring in single-copy elements, and b) CW-2 cells exhibit a
particularly high level of global DNA methylation. In fact, this
cell line ranks third in terms of LINE-1 methylation levels when
compared to a panel of 13 colorectal cancer cell lines, only
surpassed by the heavily methylated cell lines HCT116 and SW48
(38). The high level of
hypermethylation of the CW-2 cells, however, does not seem to be
attributed to the reported elevated level of hypermethylation of
sporadic MSI colon cancer (39)
because the cells contain a double mutation of MLH1 (a nonsense
mutation fixed in homozygosis by copy neutral LOH) rather than its
epigenetic silencing by hypermethylation.
Hypermethylation alterations in the tumor cell line
were strongly associated to genes encoding transcriptional factors
(fold enrichment, 1.86; EASE score, 1.14×10−5). The
epigenetic silencing of transcriptional factors likely reflects a
dramatic epigenetic reprogramming that globally alters the gene
expression profile of the cancer cell. Many of these changes are
probably associated to the malignant transformation of the primary
tumor from which this cell line is derived rather than to the
subsequent in vitro culturing conditions of CW-2 cells
(40). This conclusion is based on
the similarity of the findings between this tumor cell line and
results with primary colon cancers (data not shown). For instance,
the subgroup of transcription factors exhibiting the highest
statistical significant over-representation comprised the
homeobox-containing genes (IPR: IPR001356, fold enrichment, 3.32;
EASE score, 2.95×10−8). This result confirms our
previous finding, back in 1999, by MS-AFLP fingerprinting, that the
homeobox-containing genes are preferential targets of methylation
alterations in colon cancer (41),
later confirmed by other groups (42–44).
A possible mechanism underlying the association
between homeobox-containing genes and aberrant hypermethylation has
been proposed recently. The two main cell memory systems involved
in the maintenance of a stem cell state, i.e., the Trithorax (Trx)
and Polycomb repressor complexes (PRCs), have been suggested to
participate in the aberrant DNA hypermethylation found in
colorectal cancers (45–47). It has been found that PRC-target
genes, including the Hox gene clusters, are up to 12-fold more
likely to have cancer-specific promoter DNA hypermethylation than
non-targets (47) and that
methylation on Lys27 of histone H3, a modification mediated by the
Polycomb repressor complex 2 (PRC2), pre-marks genes for de
novo methylation in cancer (46). This proposed mechanism, however, is
insufficient to explain all the hypermethylation alterations
observed in colorectal cancer cells, because many of the frequently
altered genes are not polycomb targets. For instance, hMLH1,
hypermethylated in the majority of sporadic MSI colorectal cancers
and classified as one of the targets of the so-called CIMP (CpG
island methylator phenotype) (39). Hence, the mechanisms that drive
aberrant hypermethylation in cancer remains to date to be
explained.
Among the hypomethylated loci, the most significant
also were transcription factors involved in epithelium cell
differentiation or proliferation, secreted proteins and proteins
involved in acetylation. Of note, several homeobox-containing genes
were also identified as hypomethylated (HOXB5, PHOX2A and HLX),
indicating that these genes are under epigenetic regulation other
than DNA hypermethylation.
In the second phase of this study, we employed the
MS-AFLP array to study methylation alterations in the non-cancer
colonic mucosa of UC patients. Overall, hypermethylated probes were
also slightly more abundant that hypomethylated probes (difference,
0.17±0.29%; P=0.047, exact Wilcoxon signed rank test), but whereas
hypermethylation occurred exclusively in unique or very low copy
number sequences, hypomethylation exhibited a very strong positive
association with highly repeated DNA elements, in particular with
Alu repetitive elements which have been shown to undergo
demethylation in colorectal cancer (48). Hence, this result supports the
previous observation that global DNA hypomethylation is a common
phenomenon in UC colonic mucosa (19), and shows a link between chronic
inflammation and cancer in UC patients and accumulative
demethylation of Alu repetitive elements (26,48).
We ranked the alterations based on their incidence,
and then applied a strict threshold (P<0.01) to identify those
exhibiting an incidence above the random chance. This analysis
rendered 133 probes hypermethylated in 3–12 samples, associated to
111 genes, and 118 probes hypomethylated in 3–14 samples and
associated to 93 genes (Fig.
2).
A significant number of the hypermethylated genes
encode transcriptional factors and chromatin remodelers (KLF7,
SLC2A4RG, TBX2, IRX2, GLI2, SOX8, YBX1, GABPB1, HDAC2, SIX1, RFX1,
MKX, FOXD2, RUNX2, TLX2, TLX1), some already reported to be
associated to colon cancer (49–51).
Five of these transcriptional factors (IRX2, SIX1, MKX, TLX2, TLX1)
contain a homeobox domain. In addition, there were alterations in
genes that might contribute to the deregulation of
cancer-associated signaling pathways. For instance, we found
frequent hypermethylation of two probes corresponding to one
NotI site located in the gene body of AXIN2. AXIN2 acts as
negative regulator of canonical Wnt signaling pathway by enhancing
the recruitment of the β-catenin degradation complex. Inactivating
mutations and epigenetic silencing of AXIN2 have been previously
reported to be involved in MSI colorectal cancer (52,53),
mainly by deregulating the Wnt signaling pathway. The MS-AFLP array
analysis also revealed epigenetic alterations in other members of
the Wnt (WNT2, WNT3, WNT3A, WNT6 and WNT10B) and MAPK (CACNG8,
EGFR, HSPA8, IKBKB, MAP2K3, PPP3CC, PRKCB RASGRP1, RRAS2 and TAB1)
signaling pathways. In total, twenty-two of the frequently altered
genes (ABL1, AXIN2, CCND1, COL4A2, DAPK3, EGFR, EPAS1, FLT3, GLI2,
HDAC2, IKBKB, JUP, LAMA1, MSH6, PRKCB, PTEN, RUNX1T1, WNT10B, WNT2,
WNT3, WNT3A, WNT6) belong to the pathways in cancer KEGG map
(hsa05200). In addition, we found very frequent (78.6%)
hypermethylation of the promoter region of MPG, a gene that encodes
an alkylpurine DNA glycosylase responsible for the reparation of
alkylated adenines and guanines. This type of DNA lesion can be
caused from inflammation-associated reactive oxygen and nitrogen
species (RONS). A recent study demonstrated that mice deficient for
the murine MPG-homologous gene, i.e., Aag, accumulate more DNA
lesions after dextran sulfate-induced colonic inflammation and are
more susceptible to colon tumorigenesis (54). Hence, the finding of epigenetic
alterations in MPG in UC patients suggests that the colonic mucosa
of these patients are not only more exposed to the deleterious
effect of the RONS generated by the inflammatory response, but also
that they might suffer the impairment of the alkylated-DNA
reparation mechanism, thus exacerbating the effect of these
mutagens. We also found hypermethylation of the probe associated to
the gene BOP1 in 36% of the UCM samples. This gene has been
suggested to contribute to colorectal tumorigenesis by deregulating
chromosomal segregation (55).
The concordance between many loci altered in the UCM
samples and the cancer cells from the CW-2 cell line (Table II) indicates that the epithelial
cells of the colonic mucosa of UC patients have undergone a number
of epigenetic alterations shared with colon cancer cells.
Therefore, some of these epigenetic changes may be useful as
surrogate biomarkers for prediction of colon cancer development in
normal colonic mucosa.
In conclusion, to the best of our knowledge, this
work presents the first genomic DNA methylation profile underlying
ulcerative colitis. Many of these alterations occur in genes that
participate in well-established oncogenic pathways and are shared
with fully malignant cells. Thus, our data support the concept of
an epigenetic field for cancerization associated to the long-term
risk for carcinogenesis in UC patients, by the accumulation of
epigenetic alterations, followed by genetic alterations, that
persist after the inflammation is treated and predispose the
epithelial cells to progress into malignancy.
Acknowledgements
This work was supported in part by a grant-in-aid
for post-graduate students from Jichi Medical University and by
grant PI09/02444 from the Fondo de Investigación Sanitarias (FIS)
from the Instituto de Salud Carlos III, Ministry of Science and
Innovation, Spain. Toshiki Taya is an employee of Agilent
Technologies.
Abbreviations:
|
UC
|
ulcerative colitis
|
|
UCM
|
non-neoplastic UC mucosa
|
|
MS-AFLP
|
methylation sensitive amplified
fragment length polymorphism
|
|
OR
|
odds ratio
|
|
CI
|
confidence interval
|
References
|
1
|
Lashner BA, Silverstein MD and Hanauer SB:
Hazard rates for dysplasia and cancer in ulcerative colitis.
Results from a surveillance program. Dig Dis Sci. 34:1536–1541.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ekbom A, Helmick C, Zack M and Adami HO:
Ulcerative colitis and colorectal cancer. A population-based study.
N Engl J Med. 323:1228–1233. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Eaden JA, Abrams KR and Mayberry JF: The
risk of colorectal cancer in ulcerative colitis: a meta-analysis.
Gut. 48:526–535. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Itzkowitz SH and Present DH: Consensus
conference: colorectal cancer screening and surveillance in
inflammatory bowel disease. Inflamm Bowel Dis. 11:314–321. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pohl C, Hombach A and Kruis W: Chronic
inflammatory bowel disease and cancer. Hepatogastroenterology.
47:57–70. 2000.PubMed/NCBI
|
|
6
|
Geboes K: Ulcerative colitis and
malignancy. Acta Gastroenterol Belg. 63:279–283. 2000.PubMed/NCBI
|
|
7
|
Provenzale D and Onken J: Surveillance
issues in inflammatory bowel disease: ulcerative colitis. J Clin
Gastroenterol. 32:99–105. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Itzkowitz SH and Yio X: Inflammation and
cancer IV. Colorectal cancer in inflammatory bowel disease: the
role of inflammation. Am J Physiol Gastrointest Liver Physiol.
287:G7–G17. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Garrity-Park MM, Loftus EV Jr, Sandborn
WJ, Bryant SC and Smyrk TC: Methylation status of genes in
non-neoplastic mucosa from patients with ulcerative
colitis-associated colorectal cancer. Am J Gastroenterol.
105:1610–1619. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Issa JP: CpG-island methylation in aging
and cancer. Curr Top Microbiol Immunol. 249:101–118.
2000.PubMed/NCBI
|
|
11
|
Maekita T, Nakazawa K, Mihara M, Nakajima
T, Yanaoka K, Iguchi M, Arii K, Kaneda A, Tsukamoto T, Tatematsu M,
Tamura G, Saito D, Sugimura T, Ichinose M and Ushijima T: High
levels of aberrant DNA methylation in Helicobacter pylori-infected
gastric mucosae and its possible association with gastric cancer
risk. Clin Cancer Res. 12:989–995. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nakajima T, Maekita T, Oda I, Gotoda T,
Yamamoto S, Umemura S, Ichinose M, Sugimura T, Ushijima T and Saito
D: Higher methylation levels in gastric mucosae significantly
correlate with higher risk of gastric cancers. Cancer Epidemiol
Biomarkers Prev. 15:2317–2321. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kondo Y, Kanai Y, Sakamoto M, Mizokami M,
Ueda R and Hirohashi S: Genetic instability and aberrant DNA
methylation in chronic hepatitis and cirrhosis: a comprehensive
study of loss of heterozygosity and microsatellite instability at
39 loci and DNA hypermethylation on 8 CpG islands in microdissected
specimens from patients with hepatocellular carcinoma. Hepatology.
32:970–979. 2000.
|
|
14
|
Ushijima T: Epigenetic field for
cancerization. J Biochem Mol Biol. 40:142–150. 2007. View Article : Google Scholar
|
|
15
|
Hsieh CJ, Klump B, Holzmann K, Borchard F,
Gregor M and Porschen R: Hypermethylation of the
p16INK4a promoter in colectomy specimens of patients
with long-standing and extensive ulcerative colitis. Cancer Res.
58:3942–3945. 1998.PubMed/NCBI
|
|
16
|
Sato F, Harpaz N, Shibata D, Xu Y, Yin J,
Mori Y, Zou TT, Wang S, Desai K, Leytin A, Selaru FM, Abraham JM
and Meltzer SJ: Hypermethylation of the p14(ARF) gene in ulcerative
colitis-associated colorectal carcinogenesis. Cancer Res.
62:1148–1151. 2002.PubMed/NCBI
|
|
17
|
Issa JP, Ahuja N, Toyota M, Bronner MP and
Brentnall TA: Accelerated age-related CpG island methylation in
ulcerative colitis. Cancer Res. 61:3573–3577. 2001.PubMed/NCBI
|
|
18
|
Fujii S, Tominaga K, Kitajima K, Takeda J,
Kusaka T, Fujita M, Ichikawa K, Tomita S, Ohkura Y, Ono Y, Imura J,
Chiba T and Fujimori T: Methylation of the oestrogen receptor gene
in non-neoplastic epithelium as a marker of colorectal neoplasia
risk in long-standing and extensive ulcerative colitis. Gut.
54:1287–1292. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gloria L, Cravo M, Pinto A, De Sousa LS,
Chaves P, Leitao CN, Quina M, Mira FC and Soares J: DNA
hypomethylation and proliferative activity are increased in the
rectal mucosa of patients with long-standing ulcerative colitis.
Cancer. 78:2300–2306. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yamamoto F, Yamamoto M, Soto JL, Kojima E,
Wang EN, Perucho M, Sekiya T and Yamanaka H: Notl-Msell
methylation-sensitive amplied fragment length polymorhism for DNA
methylation analysis of human cancers. Electrophoresis.
22:1946–1956. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Samuelsson JK, Alonso S, Yamamoto F and
Perucho M: DNA fingerprinting techniques for the analysis of
genetic and epigenetic alterations in colorectal cancer. Mutat Res.
693:61–76. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yamashita K, Dai T, Dai Y, Yamamoto F and
Perucho M: Genetics supersedes epigenetics in colon cancer
phenotype. Cancer Cell. 4:121–131. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Suzuki K, Suzuki I, Leodolter A, Alonso S,
Horiuchi S, Yamashita K and Perucho M: Global DNA demethylation in
gastrointestinal cancer is age dependent and precedes genomic
damage. Cancer Cell. 9:199–207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kageyama S, Shinmura K, Yamamoto H, Goto
M, Suzuki K, Tanioka F, Tsuneyoshi T and Sugimura H:
Fluorescence-labeled methylation-sensitive amplified fragment
length polymorphism (FL-MS-AFLP) analysis for quantitative
determination of DNA methylation and demethylation status. Jpn J
Clin Oncol. 38:317–322. 2008. View Article : Google Scholar
|
|
25
|
Irizarry RA, Ladd-Acosta C, Wen B, Wu Z,
Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H,
Potash JB, Sabunciyan S and Feinberg AP: The human colon cancer
methylome shows similar hypo- and hypermethylation at conserved
tissue-specific CpG island shores. Nat Genet. 41:178–186. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ehrlich M: DNA methylation in cancer: too
much, but also too little. Oncogene. 21:5400–5413. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yamamoto F and Yamamoto M: A DNA
microarray-based methylation-sensitive (MS)-AFLP hybridization
method for genetic and epigenetic analyses. Mol Genet Genomics.
271:678–686. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rouillard JM, Zuker M and Gulari E:
OligoArray 2.0: design of oligonucleotide probes for DNA
microarrays using a thermodynamic approach. Nucleic Acids Res.
31:3057–3062. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Team RDC: R: A language and environment
for statistical computing. R Foundation for Statistical Computing;
Vienna: 2009
|
|
30
|
Frommer M, McDonald LE, Millar DS, Collis
CM, Watt F, Grigg GW, Molloy PL and Paul CL: A genomic sequencing
protocol that yields a positive display of 5-methylcytosine
residues in individual DNA strands. Proc Natl Acad Sci USA.
89:1827–1831. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Holm S: A simple sequentially rejective
multiple test procedure. Scand J Statist. 6:65–70. 1979.
|
|
32
|
Saeed AI, Sharov V, White J, Li J, Liang
W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M,
Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E,
Borisovsky I, Liu Z, Vinsavich A, Trush V and Quackenbush J: TM4: a
free, open-source system for microarray data management and
analysis. Biotechniques. 34:374–378. 2003.PubMed/NCBI
|
|
33
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009.PubMed/NCBI
|
|
34
|
Huang da W, Sherman BT, Tan Q, Kir J, Liu
D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC and Lempicki
RA: DAVID Bioinformatics Resources: expanded annotation database
and novel algorithms to better extract biology from large gene
lists. Nucleic Acids Res. 35:W169–W175. 2007.PubMed/NCBI
|
|
35
|
Hosack DA, Dennis G Jr, Sherman BT, Lane
HC and Lempicki RA: Identifying biological themes within lists of
genes with EASE. Genome Biol. 4:R702003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ionov Y, Peinado MA, Malkhosyan S, Shibata
D and Perucho M: Ubiquitous somatic mutations in simple repeated
sequences reveal a new mechanism for colonic carcinogenesis.
Nature. 363:558–561. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kanehisa M and Goto S: KEGG: kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kawakami K, Matsunoki A, Kaneko M, Saito
K, Watanabe G and Minamoto T: Long interspersed nuclear element-1
hypomethylation is a potential biomarker for the prediction of
response to oral fluoropyrimidines in microsatellite stable and CpG
island methylator phenotype-negative colorectal cancer. Cancer Sci.
102:166–174. 2011. View Article : Google Scholar
|
|
39
|
Toyota M, Ahuja N, Ohe-Toyota M, Herman
JG, Baylin SB and Issa JP: CpG island methylator phenotype in
colorectal cancer. Proc Natl Acad Sci USA. 96:8681–8686. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Smiraglia DJ, Rush LJ, Fruhwald MC, Dai Z,
Held WA, Costello JF, Lang JC, Eng C, Li B, Wright FA, Caligiuri MA
and Plass C: Excessive CpG island hypermethylation in cancer cell
lines versus primary human malignancies. Hum Mol Genet.
10:1413–1419. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Perucho M, Tokino T and Nakamura Y: Cancer
genomics and molecular diagnosis - The Nineteenth International
Symposium of Sapporo Cancer Seminar. Jpn J Cancer Res.
90:1273–1276. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Raman V, Martensen SA, Reisman D, Evron E,
Odenwald WF, Jaffee E, Marks J and Sukumar S: Compromised HOXA5
function can limit p53 expression in human breast tumours. Nature.
405:974–978. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shiraishi M, Sekiguchi A, Oates AJ, Terry
MJ and Miyamoto Y: HOX gene clusters are hotspots of de novo
methylation in CpG islands of human lung adenocarcinomas. Oncogene.
21:3659–3662. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shah N and Sukumar S: The Hox genes and
their roles in oncogenesis. Nat Rev Cancer. 10:361–371. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Vire E, Brenner C, Deplus R, Blanchon L,
Fraga M, Didelot C, Morey L, van Eynde A, Bernard D, Vanderwinden
JM, Bollen M, Esteller M, Di Croce L, De Launoit Y and Fuks F: The
Polycomb group protein EZH2 directly controls DNA methylation.
Nature. 439:871–874. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Schlesinger Y, Straussman R, Keshet I,
Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E,
Reubinoff BE, Bergman Y, Simon I and Cedar H: Polycomb-mediated
methylation on Lys27 of histone H3 pre-marks genes for de novo
methylation in cancer. Nat Genet. 39:232–236. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Widschwendter M, Fiegl H, Egle D,
Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M,
Young J, Jacobs I and Laird PW: Epigenetic stem cell signature in
cancer. Nat Genet. 39:157–158. 2007. View
Article : Google Scholar
|
|
48
|
Rodriguez J, Vives L, Jorda M, Morales C,
Munoz M, Vendrell E and Peinado MA: Genome-wide tracking of
unmethylated DNA Alu repeats in normal and cancer cells. Nucleic
Acids Res. 36:770–784. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mazumdar T, De Vecchio J, Shi T, Jones J,
Agyeman A and Houghton JA: Hedgehog signaling drives cellular
survival in human colon carcinoma cells. Cancer Res. 71:1092–1102.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hanigan CL, Van Engeland M, De Bruine AP,
Wouters KA, Weijenberg MP, Eshleman JR and Herman JG: An
inactivating mutation in HDAC2 leads to dysregulation of apoptosis
mediated by APAF1. Gastroenterology. 135:1652–1664. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Slattery ML, Lundgreen A, Herrick JS, Caan
BJ, Potter JD and Wolff RK: Associations between genetic variation
in RUNX1, RUNX2, RUNX3, MAPK1 and eIF4E and riskof colon and rectal
cancer: additional support for a TGF-beta-signaling pathway.
Carcinogenesis. 32:318–326. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Koinuma K, Yamashita Y, Liu W, Hatanaka H,
Kurashina K, Wada T, Takada S, Kaneda R, Choi YL, Fujiwara SI,
Miyakura Y, Nagai H and Mano H: Epigenetic silencing of AXIN2 in
colorectal carcinoma with microsatellite instability. Oncogene.
25:139–146. 2006.PubMed/NCBI
|
|
53
|
Liu W, Dong X, Mai M, Seelan RS, Taniguchi
K, Krishnadath KK, Halling KC, Cunningham JM, Boardman LA, Qian C,
Christensen E, Schmidt SS, Roche PC, Smith DI and Thibodeau SN:
Mutations in AXIN2 cause colorectal cancer with defective mismatch
repair by activating beta-catenin/TCF signalling. Nat Genet.
26:146–147. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
54
|
Meira LB, Bugni JM, Green SL, Lee CW, Pang
B, Borenshtein D, Rickman BH, Rogers AB, Moroski-Erkul CA, McFaline
JL, Schauer DB, Dedon PC, Fox JG and Samson LD: DNA damage induced
by chronic inflammation contributes to colon carcinogenesis in
mice. J Clin Invest. 118:2516–2525. 2008.PubMed/NCBI
|
|
55
|
Killian A, Sarafan-Vasseur N, Sesboue R,
Le Pessot F, Blanchard F, Lamy A, Laurent M, Flaman JM and Frebourg
T: Contribution of the BOP1 gene, located on 8q24, to colorectal
tumorigenesis. Genes Chromosomes Cancer. 45:874–881. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Matts SG: The value of rectal biopsy in
the diagnosis of ulcerative colitis. Q J Med. 30:393–407.
1961.PubMed/NCBI
|