Introduction
Malignant astrocytomas represent the most frequent
type of malignant brain tumors and are characterized by a strong
resistance to therapies and a dismal prognosis (1). Among the factors that determine this
resistance to treatment, anti-apoptotic signaling, for instance
through constitutive NF-κB pathway activation (2,3), and
altered DNA-damage response (4),
are believed to play major roles.
Casein kinase 2 (CK2), an ubiquitous serine
threonine kinase, consists of tetramers of 2 catalytic and 2
regulatory subunits. It has recently gained interest in the field
of cancer research as both a regulator of proliferation and
survival pathways and a modulator of the DNA-repair machinery
(5,6). CK2 was thus shown to regulate NF-κB
and STAT3 activation, P53 function (7), PTEN activity, Akt-dependant
signaling, mTOR stability and SIRT-dependent protein acetylation
(6,8–13).
CK2 also regulates the function of several enzymes of the
DNA-repair and DNA-damage sensing machinery, such as XRCC1 and 4,
Rad9 and DNA-PK (14–16). As a corollary, pre-clinical studies
have shown that CK2 inhibitors elicit anti-tumoral effects against
leukemias, prostate carcinomas, breast cancers, and some PTEN or
TP53 mutated malignant gliomas (17,18).
Based on these reports, CK2 inhibitors entered the field of
clinical trials (8,19–22).
Among them, apigenin is a naturally occurring plant flavonoid and a
specific inhibitor of the catalytic subunits of CK2 (8,23,24).
It was shown to reduce the proliferation and to induce apoptosis in
several carcinoma cells (25–27)
as well as in some human glioma cell lines (28), and has recently been used in a
phase II trial for the prevention of colorectal cancer recurrence
(NCT00609310).
Given this growing interest of clinicians and the
industry for CK2 inhibitors, and in view of the fundamental yet
disappointing role of radiation therapy for the treatment of
malignant gliomas (1), we
investigated whether CK2 inhibition would alter the
radiation-induced DNA repair response and whether these tumors
could be radiosensitized.
Materials and methods
Cell cultures, reagents and siRNA
Cell lines U87 and LN18 were obtained from the
American Type Culture Collection (ATCC) and grown in DMEM
(Dulbecco’s modified Eagle’s medium, Gibco, Gent, Belgium)
containing 10% of fetal bovine serum (FBS, Gibco) and penicillin.
Cultures were maintained at 37°C under a humidified atmosphere
containing 5% carbon dioxide.
Apigenin was purchased from Sigma (Bornem, Belgium),
dissolved in dimethylsulfoxide (DMSO) and used at final
concentration of 40 μM (stock solution, 100 mM). Control
cells were treated with a similar final concentration of DMSO as
the apigenin-treated cells. Irradiations of cell lines were
conducted with a research irradiator (Gammacell 40 Exactor,
Theratronics, Stockley Park, UK).
Subconfluent cultured cells were transfected with 50
nmol/l ON-TARGETplus non-targeting pool or SMARTpool human CSNK2A1
siRNA from Dharmacon (Fisher Scientific, Tournai, Belgium) using
oligofectamine (Invitrogen, Gent, Belgium) according to the
manufacturer’s instructions. Cells were harvested and assayed 48 h
after transfection. CK2 depletion was controlled using western blot
analysis of the expression of CK2-α.
CK2 and IKK-β kinase assays
Cells were lysed using RIPA buffer extraction kit
(Santa Cruz Biotechnology) and 300 μg of protein were taken
for immunoprecipitation. After a pre-cleared step, supernatant were
incubated with an anti-CK2 antibody (clone 1AD9, Millipore,
Overijse, Belgium) under rotary agitation for 4 h at 4°C. GammaBind
G Sephorase beads (25 μl/samples, GE Healthcare, Diegem,
Belgium) were then added to the sample and incubated on a rotating
system overnight at 4°C. After three washes, immunoprecipitated
proteins were processed with the CK2 assay kit (Upstate, Millipore)
or the IKK-β kinase assay kit (Cell Signaling, Bioke, Leiden, The
Netherlands), according to the manufacturer’s instructions.
NF-κB transcription assay
Cells were co-transfected by using TransIT-2020
transfection reagent (Mirus, Eke, Belgium) with: i) a
luciferase-coupled reporter gene for NF-κB and ii) a Renilla
luciferase reporter driven by a constitutive promoter. Radiation
(10 Gy) and apigenin treatment (40 μM) effects on NF-κB
transcriptional activity were assessed 24 h later. Briefly, cells
were lysed and luciferase activities were measured according to the
manufacturer’s instructions for the Dual Luciferase Assay System
(Promega, Leiden, The Netherlands) and using a Victor luminometer
(PerkinElmer, Zaventem, Belgium). The relative NF-κB luciferase
activity was normalized to the one of the Renilla.
Western blot analysis
10% polyacrylamide precast gels (Mini Protean TGX,
Bio-Rad, Nazareth Eke, Belgium) were run for 30 min at 200 volts
with nuclear extract (20 μg/well) obtained from irradiated
and previously apigenin or DMSO treated cells. Protein extracts
were obtained using conventional RIPA buffer and phosphatase
inhibitors. After transfer to a PVDF membrane (Roche, Vilvoorde,
Belgium) for 2 h at 300 mA and blocking with Tris buffered saline
containing 0.2% Tween plus 5% dry milk powder, membranes were
incubated overnight at 4°C in the presence of primary antibody
directed against phospho(Thr68)-Chk2 (Cell Signaling, Bioké,
Leiden, The Netherlands). A horseradish peroxidase-coupled
secondary antibody was then incubated, and peroxidase activity was
evidenced with the Super Signal West Pico Chemiluminescent
substrate (Thermo Fisher Scientific, Aalst, Belgium) and the
ImageQuant LAS 4000 Mini Biomolecular Imager (GE Healthcare).
Cell survival assays
Cell survival in response to apigenin treatment and
radiation was assayed using clonogenic assays and MTS tests (One
Solution Cell Proliferation Assay, Promega). Clonogenic assays were
performed on cells plated at low-density as described previously
(29) and as recommended by the
manufacturer’s protocol for MTS assays.
DNA repair assays
Single-cell gel electrophoresis under alkaline
conditions and flow cytometry measurement of phosphorylated
γ-histone 2Ax (γ-H2Ax) foci were used to identify ds-DNA breaks and
associated repair mechanisms, respectively.
ds-DNA breaks following apigenin (or DMSO) treatment
and radiation were detected by using the CometAssay HT kit
(Trevigen, Sanbio, Uden, The Netherlands). Briefly, single cells
embedded in agarose were lysed to remove proteins and where then
submitted to electrophorese. Staining was performed with SYBR green
I (Trevigen) for 15 min. The slides were examined under a
fluorescent microscope (Zeiss Axiovert 10 microscope, Carl Zeiss)
and DNA tail lengths were quantified in a blinded manner by
counting a minimum of 50 cells per condition in independent
experiments.
Treated cells were harvested at different times and
were prepared for flow cytometer analysis. Approximately
2.5×106 cells/ml were resuspended in 250 μl of
PBS and fixed by adding the same amount of 4% paraformaldehyde
(PFA, Merck, Overijse, Belgium). After permeabilization and
blocking with PBS containing 0.5% Triton X-100 (Acros Organics,
Geel, Belgium) and 5% donkey serum (Jackson Immunoresearch
Laboratories, Newmarket, UK) for 20 min, an anti-phosphorylated
Ser139 γ-H2Ax mouse monoclonal antibody (1:500, Millipore) was
incubated for 90 min at room temperature. Three PBS washes later,
we incubated cells with an FITC-conjugated secondary antibody
(1:500, Jackson Immunoresearch Laboratories). Indirect
immunofluorescence staining was immediately analyzed after three
more PBS washes (FACS Calibur, BD Biosciences, Erembodegem,
Belgium).
Statistical analysis
Statistical analyses were performed using the Prism
5.0c for Mac software (Graphpad Inc., La Jolla, CA). One-way ANOVA
and Mann-Whitney U tests were performed when appropriate and as
described in the results section.
Results
Irradiation-induced CK2 kinase activity
in malignant glioma cells
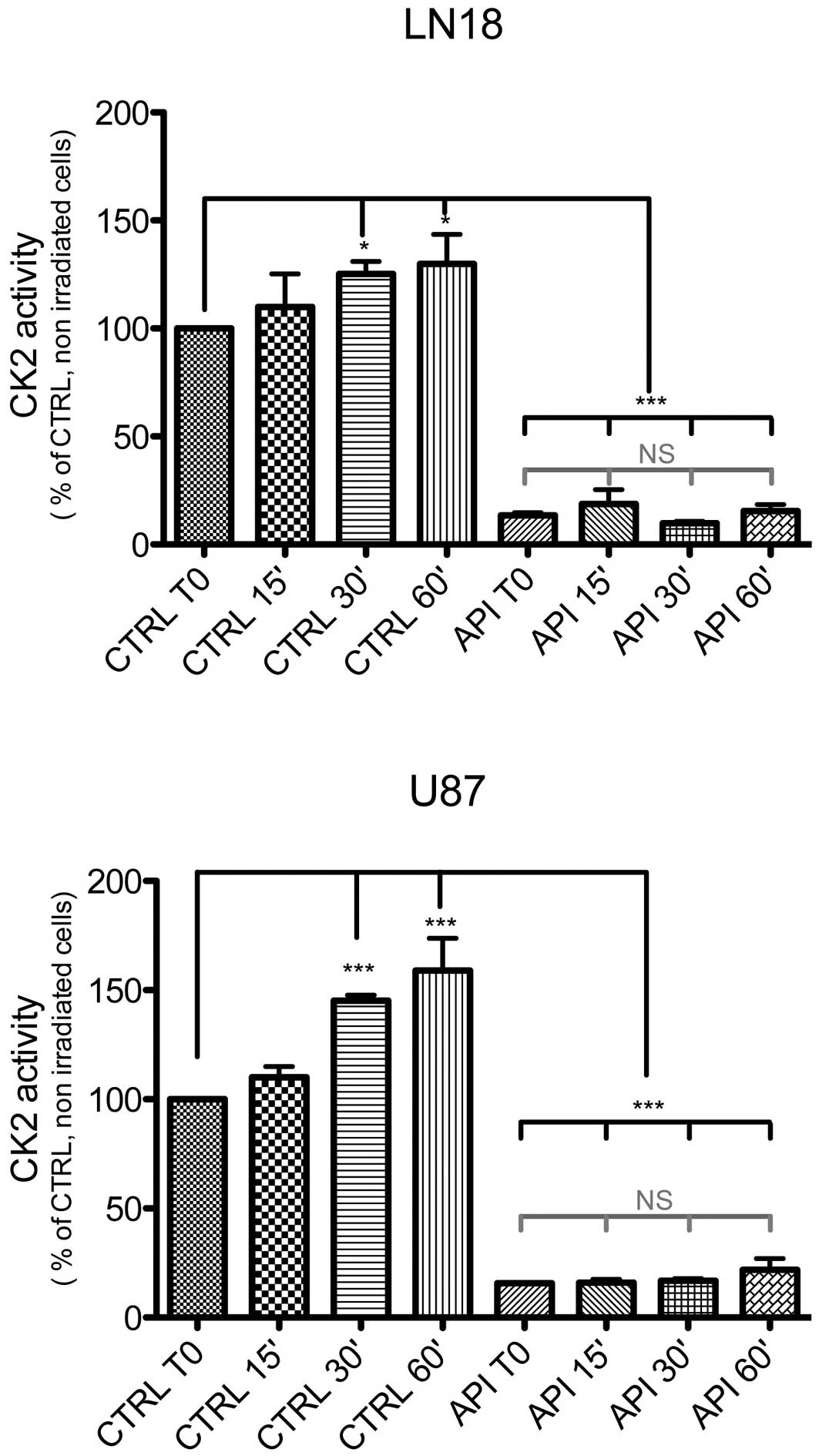
Exposure of LN18 and U87 cells to ionizing
radiations (γ rays, 4 Gy) increased the catalytic activity of CK2
within 30 min, by respectively 25±5% and 45±2.5%. Both the basal
and radiation-induced CK2 activities were significantly abolished
by pretreatment of the cell cultures with 40 μM Apigenin for
1 h (mean ± SD, n=3, P<0.05 for both, ANOVA with Tukey’s post
tests; Fig. 1).
Irradiation-induced NF-κB activation in
malignant glioma cells
Ionizing radiation activates NF-κB in tumors and
glioblastomas via an ATM-NEMO-IKK-kinase dependent pathway
(30). UV-induced DNA damage,
however, also activates CK2 (31),
leading to an IKK-kinase-independent C-terminal phosphorylation and
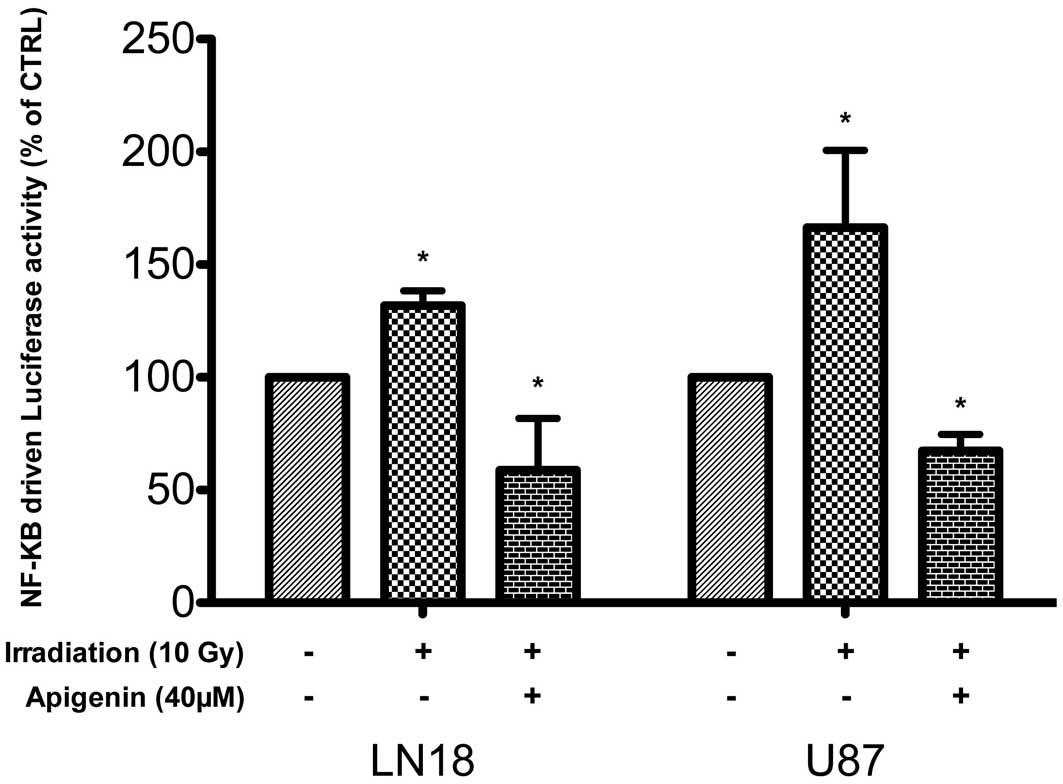
degradation of I-κBα, and NF-κB activation (32). In LN18 and in U87 cells, ionizing
irradiation (10 Gy) increased within 1 h the activity of an
NF-κB-driven luciferase reporter gene by 31±6.6% and 66±34%,
respectively, (mean ± SD, n=3, P<0.05, one-way ANOVA with
Tukey’s post tests). The baseline activity of the reporter gene was
inhibited following apigenin treatment and remained significantly
reduced in these cells despite irradiation (P<0.05, 40
μM, Fig. 2).
CK2 inhibition and DNA-repair in
malignant glioma cells
CK2 has recently emerged as a regulator of the DNA
damage response machinery (33).
We thus performed COMET assays to measure the influence of CK2
inhibition on ds-DNA break formation in U87 and LN18 cells
following γ irradiation (10 Gy). As shown in Fig. 3A, si-mediated CK2 depletion
slightly decreased the peak amplitude of COMET tails in LN18 cells
3 h following a 10 Gy irradiation (P<0.05, Mann-Witney U test).
It however had the opposite effect in U87 cells (P<0.05,
Mann-Witney U test). The mean tail amplitude returned to baseline
in mock-treated and siCK2-treated LN18 cells after 24 h. Tail size
also returned to baseline in siCK2-treated U87 cells, in sharp
contrast to mock-transfected cells where tails still remained
significantly longer than at baseline at this time point
(P<0.0001, Mann-Witney U test).
We also assessed the kinetics of γ-H2Ax foci
formation in LN18 and U87 cells treated with apigenin (40
μM) using FACS cytometry. In both cell types, radiation
treatment (10 Gy) increased the amount of γ-H2Ax immunoreactivity
with respect to baseline conditions, with a peak within 1 to 3 h.
γ-H2Ax signal returned towards baseline in control and
apigenin-treated in both cell types within 24 h. Apigenin treatment
did not alter these post-irradiation kinetics of γ-H2Ax
immunoreactivity (data not shown).
CK2 is also known to inhibit the DNA-repair kinase
DNA-PK (15), and the Chk2
checkpoint kinase is phosphorylated on tyrosine 68 by DNA-PK
following irradiation (34). Tyr68
phosphorylation of Chk2 was induced in LN18 and U87 within 15 min
after irradiation (4 Gy). This event was potentiated and more
durable in both cell types by a pre-treatment with 40 μM
apigenin (Fig. 3B).
CK2 inhibition and cell survival
following γ irradiation
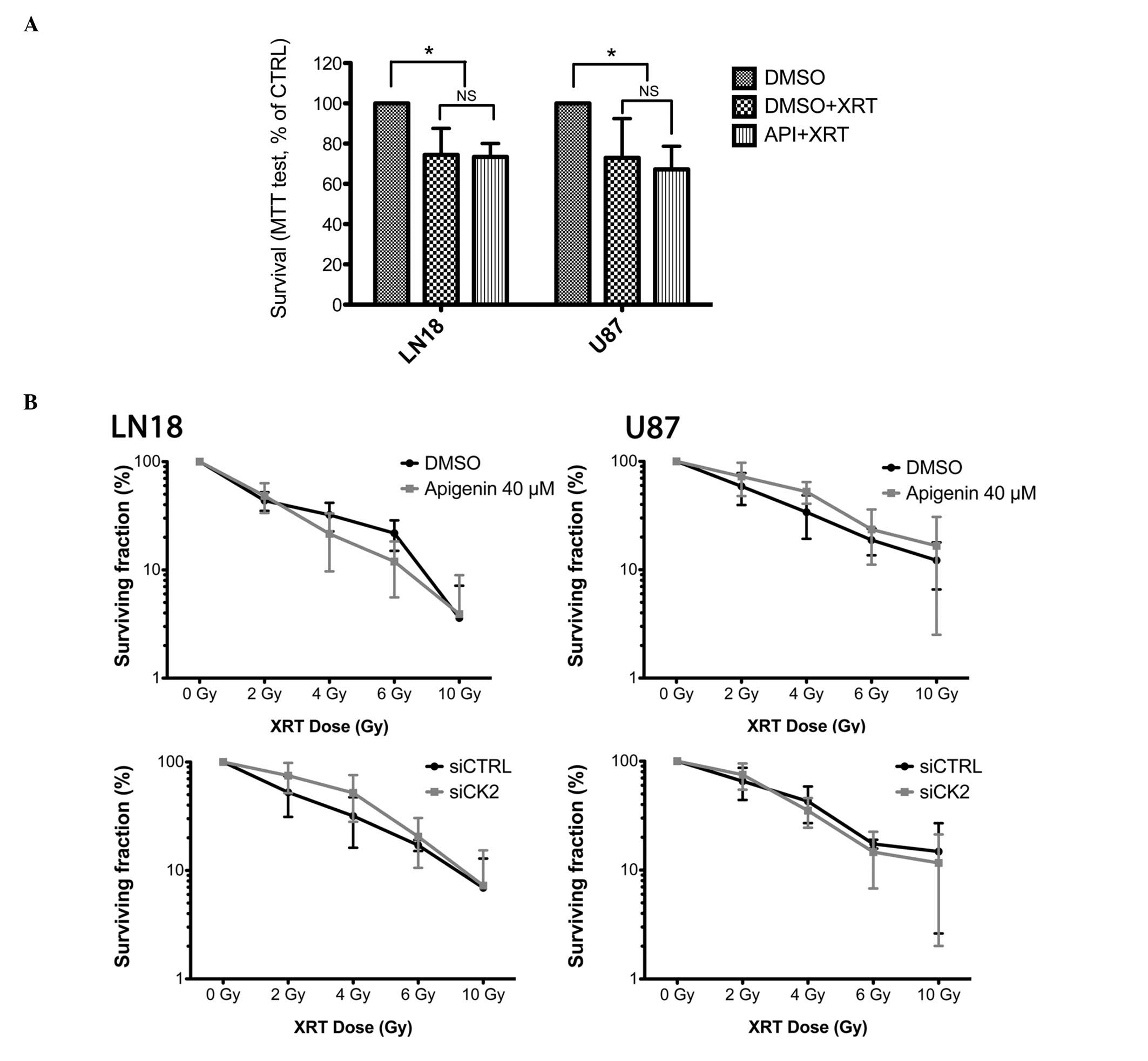
Both U87 and LN18 cells displayed a moderate but
significant reduction in viability following 4 G-rays of γ
irradiation (25.5±13.2% and 27±19.5%, respectively, P<0.05,
one-way ANOVA) as assessed using an MTS test (Fig. 4A). This viability was not further
reduced following co-treatment with 40 μM apigenin. At this
concentration, apigenin treatment also failed to radiosensitize U87
and LN18 cells in clonogenic assays (Fig. 4B, upper pannel).
As CK2-independent effects of apigenin have been
reported (35), we also assessed
the effect of siRNA-mediated CK2 kinase depletion on the
radiosensitization of malignant glioma cells. The clonogenic
survival of U87 and LN18 cells treated with CK2-targetting siRNA
prior to irradiation did not differ from scramble siRNA treated
controls (Fig. 4B, lower
pannel).
Discussion
CK2 has recently appeared as a regulator of ds-DNA
break (DSB)-triggered signaling cascades in normal, carcinoma and
even in some malignant glioma cells (36).
Accordingly, CK2-α, the active kinase subunit of
CK2, was activated within minutes of radiation treatment in our
malignant glioma cells. siRNA-mediated CK2 depletion significantly
increased the maximal peak of DSB in LN18 cells, but not in U87
cells. However, CK2 knock-down did not inhibit the repair of ds-DNA
breaks in our cell lines, but even slightly improved it, as
evidenced by the normalization of COMET assays within 24 h after
irradiation in both cell lines and the faster return of γ-H2Ax
immunoreactivity towards the baseline in U87 cells following
apigenin treatment.
In standard conditions, homologous recombination
(HR) only plays an accessory role in ds-DNA repair following
ionizing radiation in gliomas, and these tumors rather proceed via
non-homologous end joining (NHEJ) (37). During NHEJ, CK2 phosphorylates
XRCC4 and helps recruit repair enzymes like PNKP and APLF to this
scaffold protein (13,15,36,38,39).
According to this, and in contrast to our COMET and γ-H2Ax
findings, CK2 inhibition should thus inhibit DNA repair. In better
agreement with our results however, CK2 inhibition did not impair
ds-DNA rejoining in fibroblasts or colon carcinoma cells (40). As a tentative explanation, CK2 also
inhibits the DNA-dependent protein kinase subunit DNA-PKcs in
glioblastomas (15). DNA-PK is
itself a key inhibitor of HR (41), and gliomas might thus escape CK2
inhibition-induced NHEJ inhibition via an increase in homologous
recombination. In agreement with this hypothesis, apigenin
treatment of glioma cells increased the radiation-induced
phosphorylation of the DNA-PK target Chk2 (34,42)
in our experiments.
In colon carcinoma cells and fibroblasts, although
CK2 inhibition does not alter the rejoining of DSB, it does slow
down the dephosphorylation of γ-H2Ax and its dissociation from the
DNA after repair (40). Such a
lengthened γ-H2Ax decay is believed to amplify checkpoint signaling
in the presence of minimal residual DNA damage and lead to cell
death (43). We did not observe
this phenomenon in our experiments.
In our experiments and in agreement with previous
reports (44,45), CK2 inhibition also reduced the
constitutive level of NF-κB reporter activity in both cell lines.
The post-radiation transcriptional activity of this factor also
remained significantly lower in apigenin-treated irradiated cells
than in the control, non-irradiated cells. Apigenin-treated cells,
however, still responded to irradiation with a minimal induction of
NF-κB (data not shown), and our results thus do not contradict the
paradigm that CK2 triggers NF-κB activation in response to
UV-induced DNA damage but not following exposure to ionizing
radiation (46–49).
Pharmacological NF-κB inhibitors are nonetheless
known to modulate the fate of tumor cells following irradiation.
They were reported to radiosensitize glioblastomas (50,51),
but NF-κB was also, on the contrary, recently reported to mediate
apoptosis following the irradiation of primary cultures and
progenitor cells of gliomas lines (52). In line with these contrasting
reports and its favorable effect on DSB-repair in our experiments,
CK2 inhibition did not radiosensitize our glioma cells. This
neutrality seems to be independent of TP53 mutational status, as we
confirmed by exon sequencing that LN18 and U87 cells express,
respectively mutant and wild-type variants of this CK2 target (data
not shown) (53–55). Aspecific effects of apigenin were
also ruled out by repeating clonogenic assays following siRNA
mediated depletion of CK2-α.
Although we cannot rule out that CK2 inhibition
could radio-sensitize glioblastoma cells with defective DNA-PK, the
lack of radiosensitization of gliomas that we have observed
contrasts with that of non-small cell lung carcinomas, fibroblasts
and colon carcinomas cells following CK2 inhibition (6,40).
Since DNA-PK mutations occur in only 3% of glioblastomas (TCGA data
portal, accessed January 16th, 2011; the TCGA research network)
(56) we believe that glioma
patients should not be included in clinical trials that assess the
radiosensitizing role of CK2 inhibitors. Further studies of DNA
repair mechanisms in primary brain tumors and a preclinical
evaluation of therapies combining CK2 inhibitor with other
DNA-damaging agents with DNA-PK inhibitors are required to improve
the therapeutic options for these tumors.
In spite of its modulation of DNA-damage signaling
cascades, CK2 inhibition fails to inhibit DNA repair following
ionizing radiation and to radiosensitize glioma cells,
independently of their TP53 status. This contrasts with other tumor
types, urging caution regarding the inclusion of malignant glioma
patients in clinical studies that will assess the radiosensitizing
role of CK2 inhibitors in solid cancers.
Acknowledgements
We wish to thank Dr Sandra Ormenese
for her help with the flow cytometry, as well as Mr. Olivier
Pierard and Ms. Catherine Waltener for their expert technical
assistance. P.A.R. is a research associate at the Belgian Fund for
Scientific Research. This study was supported by grant PNC 29-006
of the Belgian Ministry of Health to V.B. and P.A.R., grant
1.5.162.10 of the National Research Fund of Belgium to P.A.R., a
grant from the Belgian Foundation against Cancer to V.B. and
P.A.R., and a grant from the Centre Anticancéreux of the University
of Liège to V.B.
References
|
1
|
Stupp R, Mason WP, van den Bent MJ, et al:
Radiotherapy plus concomitant and adjuvant temozolomide for
glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Robe PA, Bentires-Alj M, Bonif M, et al:
In vitro and in vivo activity of the nuclear factor-kappaB
inhibitor sulfasalazine in human glioblastomas. Clin Cancer Res.
10:5595–5603. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bredel M, Scholtens DM, Yadav AK, et al:
NFKBIA deletion in glioblastomas. N Engl J Med. 364:627–637. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Squatrito M and Holland EC: DNA damage
response and growth factor signaling pathways in gliomagenesis and
therapeutic resistance. Cancer Res. 71:5945–5949. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Becherel OJ, Jakob B, Cherry AL, et al:
CK2 phosphorylation-dependent interaction between aprataxin and
MDC1 in the DNA damage response. Nucleic Acids Res. 38:1489–1503.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin YC, Hung MS, Lin CK, et al: CK2
inhibitors enhance the radiosensitivity of human non-small cell
lung cancer cells through inhibition of stat3 activation. Cancer
Biother Radiopharm. 26:381–388. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brown MS, Diallo OT, Hu M, et al: CK2
modulation of NF-κB, TP53, and the malignant phenotype in head and
neck cancer by anti-CK2 oligonucleotides in vitro or in vivo via
Sub-50-nm nanocapsules. Clin Cancer Res. 16:2295–2307. 2010.
|
|
8
|
Eddy SF, Guo S, Demicco EG, et al:
Inducible IkappaB kinase/IkappaB kinase epsilon expression is
induced by CK2 and promotes aberrant nuclear factor-kappaB
activation in breast cancer cells. Cancer Res. 65:11375–11383.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yu M, Yeh J and Van Waes C: Protein kinase
casein kinase 2 mediates inhibitor-kappaB kinase and aberrant
nuclear factor-kappaB activation by serum factor(s) in head and
neck squamous carcinoma cells. Cancer Res. 66:6722–6731. 2006.
View Article : Google Scholar
|
|
10
|
Siepmann M, Kumar S, Mayer G and Walter J:
Casein kinase 2 dependent phosphorylation of neprilysin regulates
receptor tyrosine kinase signaling to Akt. PLoS One. 5:E131342010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Horejsí Z, Takai H, Adelman CA, et al: CK2
phospho-dependent binding of R2TP complex to TEL2 is essential for
mTOR and SMG1 stability. Mol Cell. 39:839–850. 2010.PubMed/NCBI
|
|
12
|
Maccario H, Perera NM, Davidson L, Downes
CP and Leslie NR: PTEN is destabilized by phosphorylation on
Thr366. Biochem J. 405:439–444. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kang H, Jung JW, Kim MK and Chung JH: CK2
is the regulator of SIRT1 substrate-binding affinity, deacetylase
activity and cellular response to DNA-damage. PLoS One.
4:E66112009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Takeishi Y, Ohashi E, Ogawa K, Masai H,
Obuse C and Tsurimoto T: Casein kinase 2-dependent phosphorylation
of human Rad9 mediates the interaction between human Rad9-Hus1-Rad1
complex and TopBP1. Genes Cells. 15:761–771. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Olsen BB, Issinger OG and Guerra B:
Regulation of DNA-dependent protein kinase by protein kinase CK2 in
human glioblastoma cells. Oncogene. 29:6016–6026. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ström CE, Mortusewicz O, Finch D, et al:
CK2 phosphorylation of XRCC1 facilitates dissociation from DNA and
single-strand break formation during base excision repair. DNA
Repair. 10:961–969. 2011.PubMed/NCBI
|
|
17
|
Kaminska B, Ellert-Miklaszewska A, Oberbek
A, et al: Efficacy and mechanism of anti-tumor action of new
potential CK2 inhibitors toward glioblastoma cells. Int J Oncol.
35:1091–1100. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Prudent R, Moucadel V, Nguyen CH, et al:
Antitumor activity of pyridocarbazole and benzopyridoindole
derivatives that inhibit protein kinase CK2. Cancer Res.
70:9865–9874. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pierre F, Chua PC, O’Brien SE, et al:
Pre-clinical characterization of CX-4945, a potent and selective
small molecule inhibitor of CK2 for the treatment of cancer. Mol
Cell Biochem. 356:37–43. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Battistutta R, Cozza G, Pierre F, et al:
Unprecedented selectivity and structural determinants of a new
class of protein kinase CK2 inhibitors in clinical trials for the
treatment of cancer. Biochemistry. 50:8478–8488. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schneider CC, Hessenauer A, Montenarh M
and Götz C: p53 is dispensable for the induction of apoptosis after
inhibition of protein kinase CK2. Prostate. 70:126–134.
2010.PubMed/NCBI
|
|
22
|
Shehata M, Schnabl S, Demirtas D, et al:
Reconstitution of PTEN activity by CK2 inhibitors and interference
with the PI3-K/Akt cascade counteract the antiapoptotic effect of
human stromal cells in chronic lymphocytic leukemia. Blood.
116:2513–2521. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li C, Liu X, Lin X and Chen X:
Structure-activity relationship of 7 flavonoids on recombinant
human protein kinase CK2 holoenzyme. Zhong Nan Da Xue Xue Bao Yi
Xue Ban. 34:20–26. 2009.PubMed/NCBI
|
|
24
|
Sarno S, de Moliner E, Ruzzene M, et al:
Biochemical and three-dimensional-structural study of the specific
inhibition of protein kinase CK2 by
[5-oxo-5,6-dihydroindolo-(1,2-a)quinazolin-7-yl] acetic acid (IQA).
Biochem J. 374:639–646. 2003.PubMed/NCBI
|
|
25
|
Zhao M, Ma J, Zhu HY, et al: Apigenin
inhibits proliferation and induces apoptosis in human multiple
myeloma cells through targeting the trinity of CK2, Cdc37 and
Hsp90. Mol Cancer. 10:1042011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhong Y, Krisanapun C, Lee SH, et al:
Molecular targets of apigenin in colorectal cancer cells:
involvement of p21, NAG-1 and p53. Eur J Cancer. 46:3365–3374.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Slusarz A, Shenouda NS, Sakla MS, et al:
Common botanical compounds inhibit the hedgehog signaling pathway
in prostate cancer. Cancer Res. 70:3382–3390. 2010. View Article : Google Scholar
|
|
28
|
Das A, Banik NL and Ray SK: Flavonoids
activated caspases for apoptosis in human glioblastoma T98G and
U87MG cells but not in human normal astrocytes. Cancer.
116:164–176. 2010.PubMed/NCBI
|
|
29
|
Chakravarti A, Zhai GG, Zhang M, et al:
Survivin enhances radiation resistance in primary human
glioblastoma cells via caspase-independent mechanisms. Oncogene.
23:7494–7506. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Miyamoto S: Nuclear initiated NF-κB
signaling: NEMO and ATM take center stage. Cell Res. 21:116–130.
2010.
|
|
31
|
Keller DM, Zeng X, Wang Y, et al: A DNA
damage-induced p53 serine 392 kinase complex contains CK2, hSpt16,
and SSRP1. Mol Cell. 7:283–292. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tsuchiya Y, Asano T, Nakayama K, Kato T
Jr, Karin M and Kamata H: Nuclear IKKbeta is an adaptor protein for
IkappaBalpha ubiquitination and degradation in UV-induced NF-kappaB
activation. Mol Cell. 39:570–582. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cheung WL, Turner FB, Krishnamoorthy T, et
al: Phosphorylation of histone H4 serine 1 during DNA damage
requires casein kinase II in S. cerevisiae. Curr Biol. 15:656–660.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li J and Stern DF: DNA damage regulates
Chk2 association with chromatin. J Biol Chem. 280:37948–37956.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Agullo G, Gamet-Payrastre L, Manenti S, et
al: Relationship between flavonoid structure and inhibition of
phosphatidylinositol 3-kinase: a comparison with tyrosine kinase
and protein kinase C inhibition. Biochem Pharmacol. 53:1649–1657.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Koch CA, Agyei R, Galicia S, et al: Xrcc4
physically links DNA end processing by polynucleotide kinase to DNA
ligation by DNA ligase IV. EMBO J. 23:3874–3885. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Quiros S, Roos WP and Kaina B: Rad51 and
BRCA2 - new molecular targets for sensitizing glioma cells to
alkylating anti-cancer drugs. PLoS One. 6:E271832011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Macrae CJ, McCulloch RD, Ylanko J,
Durocher D and Koch CA: APLF (C2orf13) facilitates nonhomologous
end-joining and undergoes ATM-dependent hyperphosphorylation
following ionizing radiation. DNA Repair. 7:292–302. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Iles N, Rulten S, El-Khamisy SF and
Caldecott KW: APLF (C2orf13) is a novel human protein involved in
the cellular response to chromosomal DNA strand breaks. Mol Cell
Biol. 27:3793–3803. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zwicker F, Ebert M, Huber PE, Debus J and
Weber KJ: A specific inhibitor of protein kinase CK2 delays
gamma-H2Ax foci removal and reduces clonogenic survival of
irradiated mammalian cells. Radiat Oncol. 6:152011. View Article : Google Scholar
|
|
41
|
Neal JA and Meek K: Choosing the right
path: does DNA-PK help make the decision? Mutat Res. 711:73–86.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hill R and Lee PW: The DNA-dependent
protein kinase (DNA-PK): more than just a case of making ends meet?
Cell Cycle. 9:3460–3469. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kinner A, Wu W, Staudt C and Iliakis G:
Gamma-H2AX in recognition and signaling of DNA double-strand breaks
in the context of chromatin. Nucleic Acids Res. 36:5678–5694. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shen J, Channavajhala P, Seldin DC and
Sonenshein GE: Phosphorylation by the protein kinase CK2 promotes
calpain-mediated degradation of IkappaBalpha. J Immunol.
167:4919–4925. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Viatour P, Merville MP, Bours V and
Chariot A: Phosphorylation of NF-κB and IκB proteins: implications
in cancer and inflammation. Trends Biochem Sci. 30:43–52. 2005.
|
|
46
|
Janssens S and Tschopp J: Signals from
within: the DNA-damage-induced NF-κB response. Cell Death Differ.
13:773–784. 2006.
|
|
47
|
Kato T, Delhase M, Hoffmann A and Karin M:
CK2 is a C-terminal IkappaB kinase responsible for NF-kappaB
activation during the UV response. Mol Cell. 12:829–839. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li N: ATM is required for Ikappa B kinase
(IKK) activation in response to DNA double strand breaks. J Biol
Chem. 276:8898–8903. 2000. View Article : Google Scholar
|
|
49
|
Veuger SJ, Hunter JE and Durkacz BW:
Ionizing radiation-induced NF-κB activation requires PARP-1
function to confer radioresistance. Oncogene. 28:832–842. 2008.
|
|
50
|
Hunter JE, Willmore E, Irving JAE,
Hostomsky Z, Veuger SJ and Durkacz BW: NF-κB mediates
radio-sensitization by the PARP-1 inhibitor, AG-014699. Oncogene.
31:251–264. 2012.
|
|
51
|
Tsuboi Y, Kurimoto M, Nagai S, et al:
Induction of autophagic cell death and radiosensitization by the
pharmacological inhibition of nuclear factor-kappa B activation in
human glioma cell lines. J Neurosurg. 110:594–604. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Berger R, Jennewein C, Marschall V, et al:
NF-κB is required for Smac mimetic-mediated sensitization of
glioblastoma cells for γ-irradiation-induced apoptosis. Mol Cancer
Ther. 10:1867–1875. 2011.
|
|
53
|
Asai A, Miyagi Y, Sugiyama A, et al:
Negative effects of wild-type p53 and s-Myc on cellular growth and
tumorigenicity of glioma cells. Implication of the tumor suppressor
genes for gene therapy. J Neurooncol. 19:259–268. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wischhusen J, Naumann U, Ohgaki H,
Rastinejad F and Weller M: CP-31398, a novel p53-stabilizing agent,
induces p53-dependent and p53-independent glioma cell death.
Oncogene. 22:8233–8245. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Meek DW and Cox M: Induction and
activation of the p53 pathway: a role for the protein kinase CK2?
Mol Cell Biochem. 356:133–138. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Cancer Genome Atlas Research Network:
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|