Introduction
A characteristic feature of epithelial cancer is
aberrant glycosylation of glycoproteins. The Tn antigen, an
O-linked N-acetylgalactosamine (GalNAc) epitope, which should be
covered by other distal sugars in normal cells, is a well
characterized glyco-epitope expressed by breast, colorectal and
ovarian cancer cells (1–8).
MUC1 is a 500–1000-kDa transmembrane glycoprotein
expressed by normal and cancer cells. In healthy human epithelial
cells MUC1 is expressed on the apical surface of the cell, i.e.,
the side of the cell membrane that faces the tubular interior of a
vessel. As a mucin, its functions are to lubricate, to keep the
cell hydrated, and to protect from pathogen invasion. The
extracellular domain of MUC1 contains a variable number (25–125) of
tandem repeats of 20 amino acids in length (1–8),
each of which has five potential O-glycosylation sites:
(-His-Gly-Val-Thr-Ser-Ala-Pro-Asp-Thr-Arg-Pro-Ala-Pro-Gly-Ser-Thr-Ala-Pro-Pro-Ala-)n.
In healthy cells, MUC1 is heavily glycosylated, and the O-glycans
are mostly of the core 2 type, which is a trisaccharide. It may be
elongated by several LacNAc units, whereas fucose and/or sialic
acid are terminal sugars of the completed oligosaccharide. Unlike
normal cells, most carcinomas overexpress MUC1, and MUC1 is
distributed over the entire cell surface. The glycans are
‘abnormal’ due to incomplete glycosylation and premature
sialylation. Two common tumor associated antigens found in
carcinomas are the Tn (αGalNAc, 2), and the STn
(αNeuAc-2,6-αGalNAc). It is believed that these glycans are
truncated because there are different expression levels of glycosyl
transferases in carcinomas when compared to healthy cells, which
may be caused by mutation or inactivation of glycosyltransfeases,
or by lack of functional chaperone proteins for
glycosyltransferases. The mutation of COSMC, an X chromosome
located gene encoding a chaperon protein required by core-1
β1,3-galactosyltransferase, is believed to be associated with
expression of the Tn antigen (9).
Due to the overexpression of MUC1 by almost all epithelial
carcinomas, glycopeptide partial sequences with abnormal O-glycans
contained in the MUC1’s tandem repeats are ideal potential antigens
and biomarkers that could be detected by monoclonal antibodies.
A fundamental problem is that the exact epitopes at
the molecular level remain unknown. If and how epitope expression
changes over time as the cancer progresses is also poorly
understood (10). One could
envision isolating MUC1 glycopeptides directly from cancer cells,
and analyzing them by mass spectrometry. However, a major obstacle
is the resistance of MUC1 toward proteolytic digestion. Two methods
that have been used for the discovery or detection of MUC1 epitopes
both take advantage of glycostructure recognition by antibodies.
The first method utilizes synthetic glycopeptides believed to exist
on cancer MUC1, conjugation of these glycopeptides to create an
immunogen, immunization of mice (11), collection of anti-sera, and
analysis of the binding of antibodies to MUC1 expressed on cancer
cells by flow cytometry (FACS) analysis (12). The advantage of this method is that
polyclonal or monoclonal antibodies can be obtained that recognize
an epitope of known chemical structure, and using these antibodies,
cancer tissue can be analyzed for the presence of a particular
epitope. However, the obtained antibodies can only detect the
epitope for which they were elicited, and the presence of other
epitopes may remain undetected. The second approach utilizes modern
glyco-microarrays (13), which are
utilized for the screening of patient serum for the presence of
auto-antibodies that recognize compounds in the microarray. While
this approach is capable of detecting multiple epitopes given that
the patient has produced auto-antibodies, large micro-arrays are
required, and not every patient produces sufficient levels of
auto-antibodies against MUC1. Thus, cancer may not be detected in
all patients due to an insufficient diversity of the microarray, or
due to no (or weak) immune responses to the cancer. While large
glyco-microarrays have been printed, including glycopeptides of
MUC1, undoubtedly even more diverse glyco-microarrays are needed
for the discovery of new biomarkers using the screening
methodologies already in place. There are currently no tools
available that allow for a comprehensive screening of cancer tissue
for the presence of a large number of specific biomarkers.
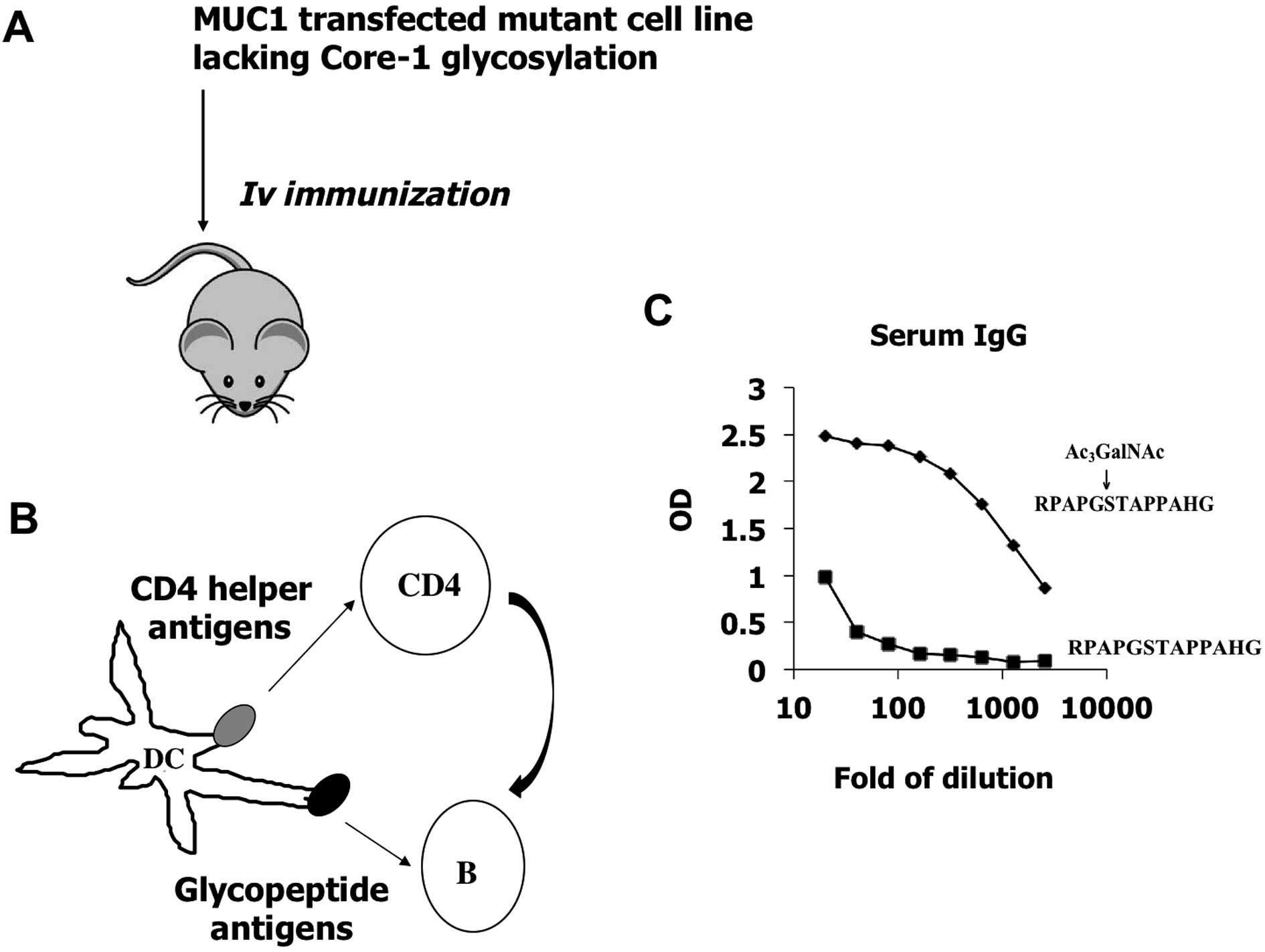
In this study, we generated glycopeptide-specific
antibodies which recognize authentic glycoforms expressed by tumor
cells (Fig. 1). The method will
allow us to generate a library of monoclonal antibodies that
recognize all existing forms of glycopeptides abnormally expressed
by tumor cells deficient in O-glycosylation, which may lead to
feasible methods to study the O-linked glycoproteome in cancer
cells.
Materials and methods
Predicting glycopeptide sequences by
computational glycomics
We have analyzed the family of mucin glycoproteins,
focusing on tandem repeat sequences which are heavily
O-glycosylated, and express Tn antigens in cancer conditions. A
theoretical database covering possible O-glycopepetide sequences
has been constructed by MATLAB language. Fig. 2 shows the example of MUC1 tandem
repeat modified by Tn antigen (GalNAc) and sialyl Tn antigen
(NeuAcα6GalNAc).
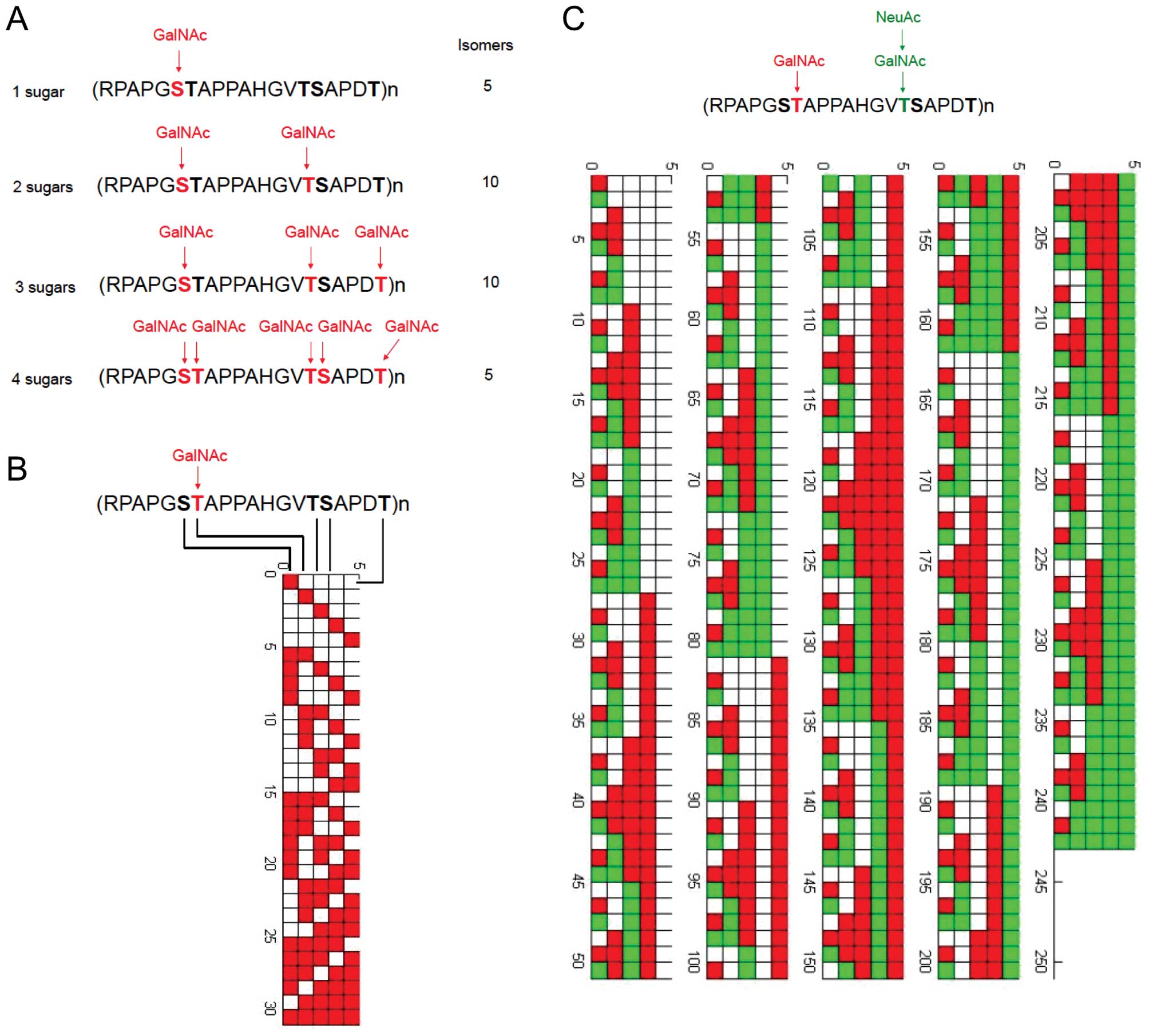 | Figure 2Theoretical glycopeptide epitopes
expressed by MUC1. (A) MUC1 protein is heavily glycosylated in the
tandem repeat domain of 20 amino acids. Each TR domain contains 5
potential O-glycosylation sites. We have generated a database of
glycopeptide sequences with 1, 2, 3, 4, and 5 GalNAc residues,
respectively. Of 31 possible MUC1 glycopeptides that vary in number
and location of Tn epitopes, four MUC1 sequences that bear one,
two, three, or four Tn moieties are illustrated. For TR domain
which may bear 1 GalNAc residue, there may exist 5 different
isomers for antibody recognition. For TR domain which bears 2
GalNAc residues, there may exist 10 different isomers for antibody
recognition. For TR domain which bears 3 GalNAc residues, there may
exist 10 different isomers for antibody recognition. For TR domain
which bears 4 GalNAc residues, there may exist 5 different isomers
for antibody recognition. In reality, cross-reactivity must be
considered for monoclonal antibody recognition, thus the exact
number of epitopes which may be uniquely recognized by monoclonal
antibodies must be determined by experiments. (B) All possible
modification results by GalNAc sugar (31 results in total) in a
single sequence of RPAPGSTAPPAHGVTSAPDT, as constructed by MATLAB
language. (C) All possible modification results (242 results in
total) by GalNAc and NeuAc sugars in a single sequence of
RPAPGSTAPPAHGVTSAPDT. |
In a MATLAB program specially designed to create
Fig. 2B, each of the five
modifiable loci was represented by binary digits. After simple
calculation, a 5x31 matrix with all zeros was created. Functions
existing in MATLAB were utilized to change zeros to ones in the
matrix sequentially. Each element of zero represents a locus
without modification, while one represents a locus with
modification of GalNAc. Finally, every element in the matrix was
inputed into a loop, creating a figure where X-axis is five
rectangles representing five loci in (RPAPGSTAPPAHGVTSAPDT)n and
Y-axis is different modification results (each modified locus was
stained by red color).
Based on the same principle, Fig. 2C was created, ternary digits were
utilized to represent different modification. Loci with
modification of GalNAc only was stained by red color. The
combination of GalNAc and NeuAc was stained by green color.
Biotinylated (glyco)peptides
The biotinylated glycopeptide
RPAPGS(Ac3GalNAc)TAPPAHG-dPEG™11-Biotin (Fig. 3), and non-glycosylated peptide,
RPAPGTAPPAHG-dPEG™11-Biotin were custom synthesized by Peptide
International Inc. (Louisville, KY). They were synthesized on an
automated peptide synthesizer from Protein Technologies, Inc.
(Tucson, AZ), model ‘Prelude’, using fluorenylmethyloxycarbonyl
(Fmoc)-protected amino acids as the building blocks,
6-chloro-benzotriazole-1-yl-oxy-tris-pyrrolidino-phosphonium
hexafluorophosphate (PyClock) as the coupling reagent, and
2-chlorotrityl resin preloaded with glycine, loading capacity 0.59
mmol/g, as the solid support. In each coupling cycle PyClock and
the Fmoc amino acid were used in 2.5-fold excess, and
N-methylmorpholine (NMM) in 4.25-fold excess in
N,N-dimethylformamide (DMF). Removal of the Fmoc group after each
coupling was performed with 20% piperidine in DMF. For the
glycopeptide preparation, the glycosylamino acid
Fmoc-Thr(Ac3GalNAc)-OH was coupled in only 1.2-fold
excess with the coupling reagent
O-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium
hexafluorophosphate (HATU) and NMM. The peptide and glycopeptide
were released from the resin with simultaneous deprotection by
treatment with cocktail R (TFA/thioanisole/EDT/anisole, 90:5:3:2)
and precipitated by cold ether. The crude peptides were purified by
preparative reversed-phase HPLC. The peptides were biotinylated via
the heterobifunctional cross linker
mono-N-t-Boc-amido-dPEG®11 amine. Coupling was performed
with diphenylphosphoryl azide and NMP in solution. Peptides were
characterized by mass spectrometry.
The glycopeptide
RPAPGS(GalNAc)TAPPAHG-dPEG™11-Biotin was prepared by deacetylation
of RPAPGS(Ac3GalNAc) TAPPAHG-dPEG™11-Biotin. Briefly,
RPAPGS(Ac3GalNAc) TAPPAHG-dPEG™11-Biotin (600 μg
in 100 μl DMSO solution) was mixed with 5 μl
hydrazine. The mixture was shaken at room temperature for 30 min.
The deacetylation reaction was monitored by analytical HPLC on a
Waters 626 HPLC instrument with a Symmetry300™ C18 column (5.0
μm, 4.6x250 mm) at 40°C, eluted with a linear gradient of
0–60% MeCN containing 0.1% TFA within 20 min at a flow rate of 1
ml/min. Preparative HPLC was performed on a Waters 600 HPLC
instrument with a preparative Symmetry300 C18 column (particle size
7.0 μm, dimensions 19x250 mm), which were eluted with a
suitable gradient of aqueous acetonitrile containing 0.1% TFA at a
flow rate of 12 ml/min. The residue was neutralized with AcOH until
pH 4.0, and subject to the preparative HPLC purification to give
the de-acetylated product (420 μg as quantified by
analytical HPLC).
The glycopeptide containing the tri-O-acetylated
GalNAc residue (synthetic precursor of the de-O-acetylated
glycopeptide, RPAPGS(Ac3GalNAc)TAPPAHG-dPEG™11-Biotin)
was included in the binding studies in order to answer the question
whether OH-3, OH-4, and OH-6 of the GalNAc residue were important
for the binding specificity of the mAbs. ELISA experiments with the
non-glycosylated peptide allows for clues of the extent of peptide
involvement in binding.
Mass spectrometry analysis of
(glyco)peptides
Glycosylated and non-glycosylated peptides were
diluted in 80% ACN/0.1% FA to a final concentration of 0.1
μg/μl and 1 μl was directly injected at 300
nl/min into an ESI-linear ion trap-mass spectrometer (LTQ XL,
Thermo-Fisher Scientific), equipped at the front end with a
nano-electrospray ionization source (Thermo-Fisher Scientific). MS
spectra were collected in positive-ion mode for 120 sec, at the
400–2,000 m/z range, and the ions of interest were subjected to
collision-induced dissociation (35% normalized collision
energy).
Generation of a monoclonal antibody
specific for RPAPGS-(GalNAc)TAPPAHG
cDNA containing MUC1 tandem repeat sequences was
generated from the breast cancer cell line T47D by a RT-PCR kit
from Invitrogen (Carlsbad, CA), with PCR primers
5′-atgacaccgggcacccagtctcct-3′ and 5′-tcaggggagcatggggaaggaaaag-3′.
The amplified cDNA sequence was compared to published literature
(8, GenBank: X52228.1), and cloned into vector pcDNA-IRES-eGFP
(Invitrogen, Carlsbad, CA). The Jurkat cell line, which synthesizes
abnormal glycans (O-GalNAc residue) on all glycoproteins (14), was transfected by
pcDNA-IRES-eGFP-MUC1 and the mock vector pcDNA-IRES-eGFP,
respectively. Stable cell lines were generated by selection with
G418, and fluorescence-activated cell sorting for eGFP expressing
cells. C57B6 mice were immunized by Jurkat pcDNA-IRES-eGFP-MUC1.
Monoclonal antibodies were generated by screening against each
synthesized glycopeptide using ELISA methods as described below.
One monoclonal antibody, 16A, has been generated, which showed
stronger binding to RPAPGS(Ac3GalNAc)TAPPAHG in ELISA
experiments than the non-glycosylated RPAPGSTAPPAHG. Another
monoclonal antibody, 14A, which showed similar binding to
RPAPGS(Ac3GalNAc)TAPPAHG and RPAPGSTAPPAHG, was also
generated.
ELISA to determine antibody binding to
glycopeptides
The biotinylated (glyco)-peptides (1 μg/ml)
were bound to streptavidin-coated plates (2 μg/ml), and
incubated with serially diluted serum for 2 h. Binding of
glycopeptide-specific IgG was visualized by a secondary antibody
(goat anti-mouse IgG) followed by colorimetric detection. One
percent bovine serum albumin was used as blank for determining the
cutoff value.
Immunohistochemistry
The study subjects were female patients selected
from a clinical database at the University of Texas M.D. Anderson
Cancer Center. The institutional review boards (IRB) of the M.D.
Anderson Cancer Center approved the retrospective review of the
medical records and identification and analysis of tumor blocks for
the purposes of the present study. Breast cancer diagnosis was made
by core needle or excisional biopsy of the breast tumor. All
pathologic specimens were reviewed by dedicated breast
pathologists. The histological type of the tumor specimens was
defined according to the World Health Organization Classification
System (15).
Immunohistochemistry was performed as previously
described (16). Briefly,
5-μm paraffin-fixed tissue sections were deparaffinized in
xylene and rehydrated through using a gradient of alcohol (100, 95
to 80%, Sigma, St. Louis, MO). Antigen retrieval was carried out
for 30 min using PT Module (Lab Vision Corp., USA) in Tris-EDTA
buffer (pH 9.0). After cooling down, the slides were thoroughly
washed in distilled water and washed three times in 1X
phosphate-buffered saline (PBS), 2 min each. Endogenous peroxidase
activity was quenched by immersion in 3% hydrogen peroxide (Sigma),
then in methanol for 10 min at room temperature followed by rinsing
for 2 min in 1X PBS three times. Nonspecific binding of the primary
antibody was blocked by incubating the sections with 10% normal
horse serum for 30 min at room temperature. Sections were then
incubated with primary anti-16A, 14A, or C595 (17, Abcam,
Cambridge, MA) 4°C overnight, at 1 μg/ml concentration.
The second day, after washing three times in 1X PBS
(2 min each), the slides were incubated with secondary anti-mouse
IgG-biotin antibody (1:200, Vectastain Elite ABC kit; Vector
laboratories, CA, USA) at room temperature for 1 h and rinsed in 1X
PBS three times (2 min each). After another 1-h incubation with the
avidin-biotin peroxidase complex (1:100, Vectastain Elite ABC Kit;
Vector Laboratories, CA, USA) and repeated washing steps with 1X
PBS, visualization was performed with the chromagen
3,3′-diaminobenzidine (DAB, Dako, Carpinteria, CA, USA). The slides
were counterstained with hematoxylin and coverslipped with
PerMount. Sections of Jurkat-pcDNA-IRES-eGFP-MUC1 and
Jurkat-pcDNA-IRES-eGFP were used as positive and negative controls,
respectively. Isotype IgG and omission of the primary antibody were
used as negative controls for staining.
Surface plasmon resonance (SPR)
measurement of antibody affinity toward glycopeptides
Interactions of RPAPGSTAPPAHG,
RPAPGS(Ac3GalNAc)TAPPAHG, and RPAPGS(GalNAc)TAPPAHG with
immobilized antibody 14A and 16A were determined by surface plasmon
resonance on a Biacore T100 (GE Healthcare) instrument (18). Antibody 14A and 16A were
immobilized on a CM5 chip until reaching 5000 response units. A
reference channel was immobilized with ethanolamine, respectively.
Immobilizations were carried out at protein concentrations of 25
μg/ml in 10 mM acetate pH 5.0 by using an amine coupling kit
supplied by the manufacturer. Measurements were carried out at 25°C
in 10 mM HEPES, pH 7.4 containing 150 mM NaCl and 0.005% surfactant
P20 at a flow rate of of 30 μl/min. The association time was
120 sec and dissociation time was 200 sec. The surface was
regenerated by 3 M MgCl2 solution. Data were analyzed
with BIA evaluation software (GE Healthcare).
Results and Discussion
Generation of a monoclonal antibody, 16A,
toward glycopeptide RPAPGS(GalNAc)TAPPAHG
Immunizing mice by MUC-1 transfected mutant cancer
cells induced IgG antibody responses against authentic
glyco-epitopes expressed on tumor cell surface, which could be
measured by ELISA experiments using chemically synthesized
glycopeptide RPAPGS(Ac3GalNAc)TAPPAHG. We observed
individual variation of IgG titers, and selected one mouse with
titer above 5000 for hybridoma fusion. Supernatants of hybridoma
cultures were screened toward both glycopeptide
RPAPGS(Ac3GalNAc) TAPPAHG and non-glycosylated peptide
RPAPGSTAPPAHG.
A hybridoma clone 16A, which produces a monoclonal
antibody belonging to the IgG1 subclass, showed stronger binding to
RPAPGS(Ac3GalNAc)TAPPAHG (EC50=6.509±0.8019
ng/ml), but approx. 40-fold weaker binding to non-glycosylated
peptide, RPAPGSTAPPAHG (EC50=247.3±16.29 ng/ml), as measured by
ELISA experiments (Fig. 4A).
EC50 of binding to RPAPGS(GalNAc)TAPPAHG by 16A was
9.278±1.059 ng/ml, indicating at least 25-fold higher affinity as
compared to RPAPGSTAPPAHG.
 | Figure 4A monoclonal antibody, 16A, binds to
glycopeptide RPAPGS(GalNAc) TAPPAHG with high affinity. (A)
Monoclonal antibody 16A was prepared as described in the text. Its
binding to glycopeptides RPAPGS(GalNAc) TAPPAHG (▪),
RPAPGS(Ac3GalNAc)TAPPAHG (♦) was compared to peptide
control RPAPGSTAPPAHG (▴), 2 μg/ml of biotinylated peptides
were bound to streptavidin coated ELISA plates. Monoclonal antibody
was added at indicated concentration, and binding was detected by
secondary goat anti-mouse IgG antibody, which was conjugated to
HRP. At a working concentration of 10 ng/ml, 16A antibody showed
strong binding to glycopeptide, but much weaker binding to peptide
alone. (B) A control monoclonal antibody, 14A, was generated by
screening the supernatant of hybridomas against nonglycosylated
control peptide RPAPGSTAPPAHG. 14A antibody showed same binding to
glycosylated and non-glycosylated peptides. Both 16A and 14A
antibodies showed no reactivity with 2 irrelevant glycopeptides
modified by GalNAc, PAHGVT(GalNAc)SAPD and PAHGVTS(GalNAc)APD. |
As a control, we also selected another hybridoma,
14A, by ELISA using RPAPGSTAPPAHG peptide. Not surprisingly, 14A
monoclonal antibody showed same binding specificity toward
RPAPGS(Ac3GalNAc)TAPPAHG and non-glycosylated RPAP
GSTAPPAHG. EC50 for RPAPGS(Ac3GalNAc)TAPPAHG,
RPAPGS(GalNAc)TAPPAHG, and RPAPGSTAPPAHG were 189.3±52.55 ng/ml,
245.7±54.80 ng/ml, and 348.3±79.25 ng/ml respectively (Fig. 4).
The 16A and 14A antibodies do not bind to two other
GalNAc modified glycopeptides synthesized by our group,
PAHGVT(GalNAc)SAPD, or PAHGVTS(GalNAc)APD, as measured by ELISA
experiments, indicating that antibody binding is not only toward
the GalNAc sugar residue, but also to the specific peptide backbone
involved.
However, surface plasmon resonance measurement of
the antibody affinity to glyopeptides showed similar binding of 16A
antibody toward both glycosylated and non-glycosylated peptides
(Table II). This suggests that the
25-fold higher affinity of 16A antibody binding to glycosylated
peptides observed in ELISA experiments (Fig. 4) might be due to the conformational
changes of glycopeptides when the 2-fragment antigen-binding sites
of IgG1 molecules bind to glycopeptides in a bivalent fashion.
 | Table IISPR measurement of dissociation
constants for the binding of 14A and 16A monoclonal antibodies to
glycopeptides. |
Table II
SPR measurement of dissociation
constants for the binding of 14A and 16A monoclonal antibodies to
glycopeptides.
| (Glyco)peptide | Ka (1/Ms) | Kd (1/s) | KD (nM) | Chi2
(RU2) |
|---|
| 16A | | | | |
|
RPAPGS(Ac3GalNAc)TAPPAHG | 5.721E+4 | 0.02794 | 488.5 | 18.6 |
|
RPAPGS(GalNAc)TAPPAHG | 3.353E+4 | 0.03120 | 930.4 | 23.1 |
| RPAPGSTAPPAHG | 7208 | 0.003898 | 540.8 | 4.95 |
| 14A | | | | |
|
RPAPGS(Ac3GalNAc)TAPPAHG | 1.600E+5 | 0.07362 | 460.2 | 10.3 |
|
RPAPGS(GalNAc)TAPPAHG | 1.172E+5 | 0.05141 | 438.6 | 19.4 |
| RPAPGSTAPPAHG | 1.218E+4 | 0.002851 | 234.0 | 3.35 |
Binding of 16A antibody to patient
samples
We further examined whether 16A antibody binds to
breast cancer tissue sections. Among 10 patients studied, we
observed strong positive staining in four patients (Table I). Fig. 5 shows a representative staining of
an estrogen receptor, progesterone receptor, and HER2-positive
breast tumor. In contrast, 14A antibody showed very weak binding in
immunohistochemistry experiments (data not shown).
| Table IExpression of MUC1 in breast cancer
patients as measured by 16A monoclonal antibody. |
Table I
Expression of MUC1 in breast cancer
patients as measured by 16A monoclonal antibody.
| Patient ID | Histology | ER | PR | HER2 | MUC1 (16A) |
|---|
| 1 | IDC+ILC | + | + | − | + |
| 2 | IDC | + | − | + | − |
| 3 | IDC | + | − | − | − |
| 4 | IDC | + | − | + | + |
| 5 | IDC | + | + | + | + |
| 6 | IDC | − | − | − | − |
| 7 | IDC | + | − | − | + |
| 8 | IDC | + | − | − | − |
| 9 | IDC | + | − | + | − |
| 10 | IDC | − | − | − | − |
Problems with current approaches to the
discovery of MUC1 biomarkers
In the past, most approaches focused on targeting
nonglycosylated MUC1, or MUC1 peptides with undefined glycosylation
profiles (19). While the
importance of the glycosylation pattern for the immunogenic
properties of a glycoprotein has been recognized (20), the peptide portion should not be
neglected. Recently, Schietinger and coworkers found that in a
mouse fibrosarcoma, a mutant chaperone abolished function of a
glycosyltransferase, which disrupted O-glycan core 1 synthesis, and
created a transmembrane protein as a tumor-specific antigen. This
antigen was recognized by a monoclonal antibody with exquisite
specificity. X-ray-crystallographic analysis showed that the
cognate epitope consisted of both the Tn antigen and an octapeptide
portion of the underlying protein backbone (21). Furthermore, the
glycopeptide-specific antibody showed high affinity toward cancer
antigen (Kd=10−7 M) and cured cancer in mouse models,
which is in contrast to the low affinities often observed for
antibodies toward Tn antigen (GalNAc). This result suggests that
glycopeptide epitopes may be highly immunogenic, most likely more
immunogenic than the saccharide portion alone.
Since biochemical characterization of glycan
epitopes are often challenging, we have developed a complementary
approach, that is to predict the glycolipid or glycopeptide
structures based on the central dogma of glycobiology proposed by
Kornfeld and Kornfeld (22). Since
the glycoconjugates are assembled stepwise by glycosyltransferases,
we have written computer programs that predict the glycan
structures by sequential additions of sugar units. Here we take a
novel approach to the potential discovery of new MUC1 biomarkers
via monoclonal antibodies. This approach is based on the
immunization of mice with authentic tumor cell surface MUC1. The
MUC1 epitopes expressed on the cell surface trigger B cell
responses, while the CD4 helper signals are provided by tumor
proteins (Fig. 1), when tumor
cells are lysed by xenogenicity-induced cell lysis. From these
experiments, an entire library of monoclonal antibodies can
potentially be obtained. As a first proof-of-principle experiment,
we have generated monoclonal antibodies which show higher affinity
to glycopeptide RPAPGS(GalNAc) TAPPAHG. This approach may be
complemented with conventional monoclonal antibody production via
immunization of mice with certain MUC1 glycopeptides conjugated to
adjuvants (23). Our approach
could potentially lead to a massive toolbox of monoclonal
antibodies that could be used for the discovery of authentic
biomarkers on cancer cell surfaces, and for the development of new
tools for diagnostic imaging using radioisotope-labeled monoclonal
antibodies.
Cross reactivity is an issue of all monoclonal
antibodies. In this study, we generated 16A monoclonal antibody
which preferentially binds to glycosylated peptide. The 16A
antibody showed strong binding to glycopeptide RPAPGS(GalNAc)
TAPPAHG, but much weaker binding to peptide RPAPGSTAPPAHG alone. It
has no cross reactivity with 2 other glycopeptides modified by
GalNAc, PAHGVT(GalNAc)SAPD and PAHGVTS(GalNAc)APD (Fig. 4). Identifying individual
glycopeptide epitopes will need not one, but an entire set of
monoclonal antibodies. Thus, multiple monoclonal antibodies with
preferential binding specificities will provide specific
information for an individual MUC1 glycopeptide.
In conclusion, glycopeptide epitopes expressed by
MUC1 may be predicted by bioinformatics tools. Glyco-epitopes
expressed on tumor cell surfaces may elicit antibody responses in
mice and cancer patients. Monoclonal antibodies can be generated
from mice immunized by tumor cells, and selected by ELISA
experiments using chemically synthesized glycopeptides predicted by
bioinformatics tools. Such monoclonal antibodies are valuable tools
for discovery of new biomarkers and targets for immunotherapy. This
approach was successful in generating a monoclonal antibody, 16A,
that was able to stain tissue sections. The nonglycosylated peptide
is being recognized, but 25-fold weaker than the glycopeptide.
Interestingly, 16A binds 3,4,6-tri-O-acetylated GalNAc glycopeptide
more strongly than the nonglycosylated peptide by a factor of
approximately 40, indicating that the absence of hydroxyl groups in
the sugar moiety does not abolish binding. One possible explanation
for this finding could be that the antibodies engage in
intermolecular contacts simultaneously with the peptide and those
parts of the Ac3GalNAc moiety that it has in common with
GalNAc, for example the acetamido group at position 2. Research
investigating this molecular recognition phenomenon is currently
underway.
Acknowledgements
We thank Long Vien and Laura Bover at
the University of Texas M.D. Anderson Cancer Center Monoclonal
Antibody Facility for technical support. This work was partially
funded by the grants from the University of Texas M.D. Anderson
Cancer Center (D.Z.), 1SC2CA148973-01 (KM), and R00CA133244
(E.A.M.). E.S.D. is supported by National Cancer Institute training
Grant T32CA009598. D.Z. is supported by NIAID grant AI079232. M.D.
Anderson Cancer Center is supported in part by NIH grant CA16672.
We thank the Biomolecule Analysis Core Facility at the Border
Biomedical Research Center/Biology/UTEP (NIH grants
2G12RR008124-16A1 and 2G12RR008124-16A1S1) for the access to the
Biacore T-100 and LC-MS instruments. D.Z. is President of
NanoCruise Pharmaceutical Suzhou Ltd., a consultant for BioTex,
Houston, TX, and an inventor involved in patents related to
technologies mentioned in this study, issued or in application.
References
|
1.
|
S GendlerJ Taylor-PapdimitriouT DuhigJ
RothbardJ BurchellA highly immunogenic region of a human
polymorphic epithelial mucin expressed by carcinoma is made up of
tandem repeatsJ Biol Chem263128201282319883417635
|
|
2.
|
JC ByrdRS BresalierMucins and mucin
binding proteins in colorectal cancerCancer Metastasis
Rev237799200410.1023/A:102581511359915000151
|
|
3.
|
SE BaldusK EngelmannFG HanischMUC1 and the
MUCs: a family of human mucins with impact in cancer biologyCrit
Rev Clin Lab Sci41189231200410.1080/1040836049045204015270554
|
|
4.
|
PK SinghMA HollingsworthCell
surface-associated mucins in signal transductionTrends Cell
Biol16467476200610.1016/j.tcb.2006.07.00616904320
|
|
5.
|
MA TarpH ClausenMucin-type O-glycosylation
and its potential use in drug and vaccine developmentBiochim
Biophys Acta1780546563200810.1016/j.bbagen.2007.09.01017988798
|
|
6.
|
RE BeatsonJ Taylor-PapadimitriouJM
BurchellMUC1
immunotherapyImmunotherapy2305327201010.2217/imt.10.17
|
|
7.
|
AM VladJC KettelNM AlajezCA CarlosOJ
FinnMUC1 immunobiology: from discovery to clinical applicationsAdv
Immunol82249293200410.1016/S0065-2776(04)82006-614975259
|
|
8.
|
E YangXF HuPX XingAdvances of MUC1 as a
target for breast cancer immunotherapyHistol
Histopathol22905922200717503348
|
|
9.
|
T JuVI OttoRD CummingsThe Tn
antigen-structural simplicity and biological complexityAngew Chem
Int Ed5017701791201110.1002/anie.20100231321259410
|
|
10.
|
R SewellM BackstromM DalzielThe
ST6GalNAc-1-sialyltransferase localizes throughout the Golgi and is
responsible for the synthesis of the tumor-associated sialyl-Tn
O-glycan in human breast cancerJ Biol
Chem28135863594200610.1074/jbc.M51182620016319059
|
|
11.
|
H CaiZH HuangL ShiZY SunYF ZhaoH KunzYM
LiVariation of the glycosylation pattern in MUC1 glycopeptide BSA
vaccines and its influence on the immune responseAngew Chem Int Ed
Engl5117191723201210.1002/anie.20110639622247051
|
|
12.
|
A Hoffmann-RöderA KaiserS WagnerSynthetic
antitumor vaccines from Tetanus toxoid conjugates of MUC1
glycopeptides with the Thomsen-Friedenreich antigen and a
fluorinesubstituted analogueAngew Chem Int Ed
Engl4984988503201020878823
|
|
13.
|
S KracunE CloH ClausenSB LeveryKJ JensenO
BlixtRandom glycopeptide bead libraries for seromic biomarker
discoveryJ Proteom Res967056714201010.1021/pr100847720886906
|
|
14.
|
A SchietingerM PhilipBA YoshidaP AzadiH
LiuSC MeredithH SchreiberA mutant chaperone converts a wild-type
protein into a tumor-specific
antigenScience314304308200610.1126/science.112920017038624
|
|
15.
|
WHO1982The World Health Organization
histological typing of breast tumors. Second editionAm J Clin
Pathol7880681619827148748
|
|
16.
|
JQ ChenJ LittonL XiaoHZ ZhangCL WarnekeY
WuX ShenS WuA SahinR KatzM BondyG HortobagyiNL BerinsteinJL MurrayL
RadvanyiQuantitative immunohistochemical analysis and prognostic
significance of TRPS-1, a new GATA transcription factor family
member, in breast cancerHorm
Cancer12133201010.1007/s12672-010-0008-8
|
|
17.
|
MR PriceJA PughF HudeczW GriffithsE
JacobsIM SymondsAJ ClarkeWC ChanRW BaldwinC595 - a monoclonal
antibody against the protein core of human urinary epithelial mucin
commonly expressed in breast carcinomasBr J
Cancer61681686199010.1038/bjc.1990.1541692469
|
|
18.
|
G SafinaApplication of surface plasmon
resonance for the detection of carbohydrates, glycoconjugates, and
measurement of the carbohydrate-specific interactions: a comparison
with conventional analytical techniques. A critical reviewAnal Chim
Acta712929201210.1016/j.aca.2011.11.016
|
|
19.
|
C BrockeH KunzSynthesis of
tumor-associated glycopeptide antigensBioorg Med
Chem1030853112200210.1016/S0968-0896(02)00135-912150854
|
|
20.
|
S DziadekH KunzSynthesis of
tumor-associated glycopeptide antigens for the development of
tumor-selective vaccinesChem
Rec3308321200410.1002/tcr.1007414991920
|
|
21.
|
CL BrooksA SchietingerSN BorisovaP KuferM
OkonT HiramaCR MackenzieLX WangH SchreiberSV EvansAntibody
recognition of a unique tumor-specific glycopeptide antigenProc
Natl Acad Sci
USA1071005610061201010.1073/pnas.091517610720479270
|
|
22.
|
R KornfeldS KornfeldAssembly of
asparagine-linked oligosaccharidesAnnu Rev
Biochem54631664198510.1146/annurev.bi.54.070185.0032153896128
|
|
23.
|
S IngaleMA WolfertT BuskasGJ
BoonsIncreasing the antigenicity of synthetic tumor-associated
carbohydrate antigens by targeting Toll-like
receptorsChembiochem10455463200910.1002/cbic.20080059619145607
|