Introduction
Metastasis is a complex process characterized by the
detachment of cells from the primary tumor followed by their
dissemination into surrounding tissues to form secondary tumors at
distant sites, which is the most common cause of death for cancer
patients (1). Several types of
molecules are involved in the metastatic process, including
adhesion molecules, such as cadherins and integrins, various
metalloproteases, as well as growth factors and their receptors
(2). Cancer cells need to be
motile in order to escape the tumor mass to form metastasis. They
move in different ways: as a group or as single cells, adopting an
elongated phenotype or moving in an ameboid shape (1), depending on the organization of actin
cytoskeleton. Among a variety of molecules involved in cell
migration, cortactin plays a pivotal role. Discovered as a Src
kinase substrate in RSV-transformed fibroblasts (3), cortactin is a multidomain
actin-binding protein (4,5). The N-terminal half of the molecule
includes the acidic domain (NTA), responsible for the interaction
with the Arp2/3 complex, and a tandem repeats containing the
F-actin binding site. The C-terminal half is composed by an
α-helical region, followed by a region rich in proline, serine and
threonine, which harbors critical tyrosine residues, and finally an
Src-homology 3 (SH3) domain (6).
Cortactin is enriched at membrane ruffles and lamellipodia of
several different cell types (4),
and plays a crucial role in regulating membrane dynamics and actin
assembly for the control of membrane movements (5). Furthermore, cortactin is reported to
be overexpressed in many different tumor types. The cortactin gene
EMS1, located on chromosome 11q13, is amplified in numerous human
carcinomas (7) and is usually
correlated with poor patient prognosis (8). Moreover, cortactin can be activated
by phosphorylation of tyrosines in position 421, 466 and 482 by Src
kinase after many different stimuli (9).
These properties of cortactin prompted us to examine
its role in cell migration promoted by insulin-like growth factor-1
(IGF-1), one of the component of the IGF system, and the epidermal
growth factor (EGF) in human breast cancer cell lines.
Previous studies have demostrated that IGF-1 is a
physiological peptide, whose biological function is carried out
following binding to its specific receptor (IGF-1R), a ubiquitous
and multifunctional tyrosine kinase receptor (2). Overexpression and activity of IGF-1R
have been associated with many different types of tumors, such as
breast, prostate, lung, colon and head and neck squamous cell
carcinoma (10). The IGF-1R is
overexpressed in estrogen receptor (ER)-positive cells. These data
are prominent because IGF-1R has been implicated in different
growth-related and growth-unrelated processes, critical for the
development and progression of malignant tumors, such as
proliferation, survival, and anchorage-independent growth, as well
as cell adhesion, migration and invasion (11,12).
The activation of the IGF-1R pathway promotes cell growth and
counteracts the anticancer-induced apoptosis. Nevertheless,
ER-negative tumors and cell lines, often exhibiting less
differentiated, mesenchymal-like phenotypes, express low levels of
IGF-1R (11,13,14).
Notably, these cells do not respond to IGF-1 with growth (11,15–18).
The lack of IGF-1R pathway activation impaired the metastatic
potential of ER-negative breast cancer cells by inhibiting both
mitogenic response and migration in vitro and tumorigenesis
in vivo (19–21). The epidermal growth factor receptor
(EGFR) is involved in various aspects of cell growth, survival,
differentiation, migration and invasion (22,23).
EGFR is overexpressed in MDA-MB-231 breast cancer cells, although
the event is not due to gene amplification (24), while MCF7 breast cancer cells lack
EGFR-overexpression (25).
Here we analyzed the effect of IGF-1 and EGF on the
epidermal growth factor receptor-2 (ERB-B2) negative human invasive
ductal breast carcinoma cell lines, MDA-MB-231 and MCF7 (26). The two cell lines differ in several
cytological and biochemical parameters: MCF7 cells possess a
functional ER, have an epithelial-like morphology and grow in
colonies, whereas MDA-MB-231 cells are (ER)-negative and
progesterone receptor (PgR)-negative and show a mesenchymal-like
morphology and grow as a monostrate. Furthermore, MDA-MB-231 cells
exhibit strong invasive and metastatic capacities, and are
representative of cells in late-stage breast cancer, while MCF7 are
a poorly invasive and non-metastatic breast cancer cell line
(27). MCF7 have a characteristic
luminal epithelial profile (27,28),
conversely MDA-MB-231 shows a gene expression pattern similar to
the claudin-low tumor subtype with the lowest expression of genes
involved in epithelial cell-cell adhesion (i.e., E-cadherin and
claudins 3, 4 and 7) vs the other breast cancer phenotypes, low
luminal differentiation (i.e., CD24, EpCAM), and high values for
the CD44/CD24 and CD49f/EpCAM mRNA ratios (29), and poor prognosis.
The present study investigates the mechanism
underlying the IGF-1 mediated migration of human HER2 negative
breast cancer cells, and all the experiments were performed
comparing the results obtained by using IGF-1 with those obtained
by EGF. We demonstrate that both growth factors mediate: i) the
motogenic effect on the two cell lines, with a stronger effect of
IGF-1 on the claudin-low cell line, ii) the actin cytoskeletal
organization in typical migratory structures, iii) the
translocation of the phospho-Y421-cortactin to the focal adhesions,
and we show that all these processes are Src-dependent.
Materials and methods
Materials
Human recombinant epidermal growth factor (EGF) and
human recombinant insulin-like growth factor type-1 (IGF-1),
TRITC-phalloidin, anti-tubulin monoclonal antibody, and anti-mouse
IgG peroxidase antibodies were purchased from Sigma (St. Louis, MO,
USA). Goat anti-rabbit IgG peroxidase antibody was from Amersham
Biosciences (Uppsala, Sweden). Anti-cortactin polyclonal antibodies
were from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and mouse
anti-paxillin monoclonal antibody was obtained from BD Pharmingen
(San Diego, CA, USA). Anti-phosphotyrosine monoclonal antibody
(clone 4G10) and anti-cortactin monoclonal antibody (clone 4F11)
were purchased from Upstate Biotechnology (Lake Placid, NY, USA).
Anti-phospho-cortactin pY421 polyclonal antibodies were purchased
from Invitrogen (Carlsband, CA, USA). FITC-conjugated goat
anti-rabbit IgG was obtained from Cappel Research Products (Durham,
NC, USA), Texas Red-conjugated goat anti-mouse IgG was from Jackson
Immunoresearch Laboratories (West Grove, PA, USA). Src inhibitor
SU6656 was obtained from Calbiochem (San Diego, CA, USA).
Cell lines and treatments
The human breast cancer cell lines MDA-MB-231 and
MCF7 were cultured in Dulbecco’s modified Eagle’s medium (DMEM,
Euroclone, Pero, Italy) supplemented with 10% fetal bovine serum
and antibiotics. For inhibition of Src specific family protein
tyrosine kinases, cells were preincubated with SU6656 (30), 5 μM for 1 h, before incubation with
IGF-1 or EGF in the presence of the same inhibitor at 37°C in
pre-warmed medium.
Scratch assay
MDA-MB-231 and MCF7 cells were seeded at
5×105 and 1.2×106 cells on 35-mm plates,
respectively, and grown until confluence. Confluent cells were
serum starved for 12 h and then a standardized cell-free area was
introduced by scraping the monolayer with a sterile tip. After
intensive washing, the remaining cells were incubated for 24 h in
the presence of IGF-1 (10 ng/ml) or EGF (50 ng/ml). After
incubation, the cells were fixed with 4% paraformaldehyde for 30
min at 25°C and photographs were taken using an AxioObserver
inverted microscope (Carl Zeiss Inc.). Some plates were fixed and
photographed immediately after scratching, representing a T0
control. For inhibition of Src family protein tyrosine kinases,
cells were preincubated with SU6656 (5 μM) for 1 h before
stimulation with the growth factors. The effects of growth factors
and inhibitor on cell migration was evaluated by measuring the
distance remaining between the two sides of the scratch area,
performed using Axio Vision software (Carl Zeiss Inc.). The data
presented are a mean of triplicate experiments ± SEM. Statistical
analysis was carried out using Student’s t-test to evaluate
significative differences with respect to control.
Immunofluorescence microscopy
MDA-MB-231 and MCF7 cells, grown on coverslips, were
treated with IGF-1 or EGF for 10, 30 or 60 min, or pre-incubated
with the Src inhibitor SU6656 for 1 h before adding the growth
factors, as above. Cells were subsequently fixed in 4%
paraformaldehyde in phosphate-buffered saline (PBS) for 30 min at
25°C, followed by treatment with 0.1 M glycine in PBS for 20 min at
25°C and with 0.1% Triton X-100 in PBS for additional 5 min at 25°C
to allow permeabilization. Cells were then incubated for 1 h at
25°C with the following primary antibodies: anti-cortactin
polyclonal antibodies (1:100 in PBS), anti-cortactin monoclonal
antiboby (1:100 in PBS), anti-paxillin monoclonal antibody (1:100
in PBS) and anti-phospho-cortactin pY421 polyclonal antibodies
(1:100 in PBS). The primary antibodies were visualized, after
appropriate washing in PBS, using FITC-conjugated goat anti-rabbit
IgG (1:100 in PBS) or Texas Red-conjugated goat anti-mouse IgG
(1:50 in PBS). Actin cytoskeleton was visualized using
TRITC-phalloidin (1:50) for 45 min at 25°C. Coverslips were finally
mounted with mowiol for observation. Fluorescence signal was
analyzed by recording stained images using an AxioObserver inverted
microscope, equipped with the ApoTome System (Carl Zeiss Inc.).
Immunoprecipitation and western blot
analysis
Subconfluent cultures of MDA-MB-231 and MCF7 cells,
treated with IGF-1 or EGF and SU6656, as above, were lysed in a
buffer containing 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 1
mM EDTA, supplemented with protease inhibitors (10 μg/ml aprotinin,
10 μg/ml leupeptin, 2 mM PMSF), and phosphatase inhibitors (1 mM
sodium orthovanadate, 20 mM sodium pyrophosphate, 50 mM sodium
fluoride). Total protein (50 µg) was resolved under reducing
conditions by 8% SDS-PAGE and transferred to reinforced
nitrocellulose (Protran, Schleider and Schuell, Keene, NH, USA).
The membrane was blocked with 5% non-fat dry milk in PBS 0.1%
Tween-20, and incubated with anti-cortactin polyclonal antibodies
diluted 1:1,000, followed by enhanced chemiluminescence detection
(ECL). For the equal loading, the membrane was probed with
anti-tubulin monoclonal antibody. For immunoprecipitation
experiments, 1 mg of total protein from cell lysates, prepared as
above, was incubated with 4 µg/ml anti-cortactin polyclonal
antibodies. Immunocomplexes, aggregated with 50 µl of γ-bind
protein-G sepharose (Amersham Biosciences), were washed four times
with 0.5 ml of buffer. The pellets were boiled in Laemmli buffer
for 5 min, and the protein resolved under reducing conditions by 8%
SDS-PAGE and transferred to reinforced nitrocellulose (Protran).
The membranes were blocked with 5% non-fat dry milk in PBS 0.1%
Tween-20 and incubated with anti-phosphotyrosine monoclonal
antibody diluted 1:1,000 for 1 h at 25°C, followed by goat
anti-mouse-HRP secondary antibody and enhanced chemiluminescence
detection (ECL, Amersham Biosciences, Arlington Heights, IL, USA).
To estimate the protein equal loading, the membranes were
rehydrated by being washed in PBS Tween-20, stripped with 100 mM
mercaptoethanol and 2% SDS for 30 min at 55°C, and then reprobed
with anti-cortactin polyclonal antibodies diluted 1:1,000.
Densitometric analysis was performed using Image J software. The
signal intensity for each band was calculated and the background
subtracted from experimental values. The resulting values were then
normalized, expressed as fold increase with respect to the control
value and visualized as a graph.
Results
IGF-1 induces migration of breast cancer
cells in a Src-dependent manner
In order to assess the motogenic effect of IGF-1 on
human breast cancer cells, we analyzed cell migration through
scratch assay. To this aim, MDA-MB-231 and MCF7 cells were seeded
onto Petri dishes and allowed to grow to confluence. Then, a
cell-free area was introduced in monolayers and cells were allowed
to migrate from the edge of the scratch for 24 h in the presence of
IGF-1. To better evaluate the effect of IGF-1, we used EGF as
positive control for migration, as this growth factor has been
shown to be strongly motogenic for cancer cell lines (31,32).
As shown in Fig. 1, IGF-1
displayed a stronger migratory effect on MDA-MB-231 cells compared
to MCF7. On the other hand, EGF treatment induced a more evident
migration on MCF7 with respect to MDA-MB-231 (Fig. 1). Moreover, both the growth factors
induced significant migration with respect to control untreated
cells (Fig. 1).
Since the protein tyrosine kinase Src has been
previously shown to be involved in cell motility (33), to evaluate the potential role of
Src in regulating the migratory effect of IGF-1 and EGF on
MDA-MB-231 and MCF7 cells, we performed the scratch assay, as
above, in the presence of the selective Src family protein tyrosine
kinase inhibitor, SU6656 (30).
Treatment with SU6656 was able to significantly prevent the
migration of both the cancer cell lines upon either IGF-1 and EGF
stimulation (Fig. 1), thus
suggesting the involvement of Src in this process. Taken together,
these data indicate that both IGF-1 and EGF stimulate migration of
human cancer cells and strongly suggest a crucial role of Src in
this process.
IGF-1 differentially affects migratory
phenotype and actin reorganization in MDA-MB-231 and MCF7
cells
Since the actin cytoskeleton reorganization in
lamellipodia and membrane ruffles is a crucial step for cells to
migrate (34), here we analyzed
the reorganization of actin cytoskeleton in human breast cancer
cells upon IGF-1 treatment. To this purpose, starved MDA-MB-231 and
MCF7 cells were stimulated with IGF-1 or EGF at different time
points, and stained with TRITC-phalloidin, that specifically
recognizes filamentous actin cytoskeleton. Immunofluorescence
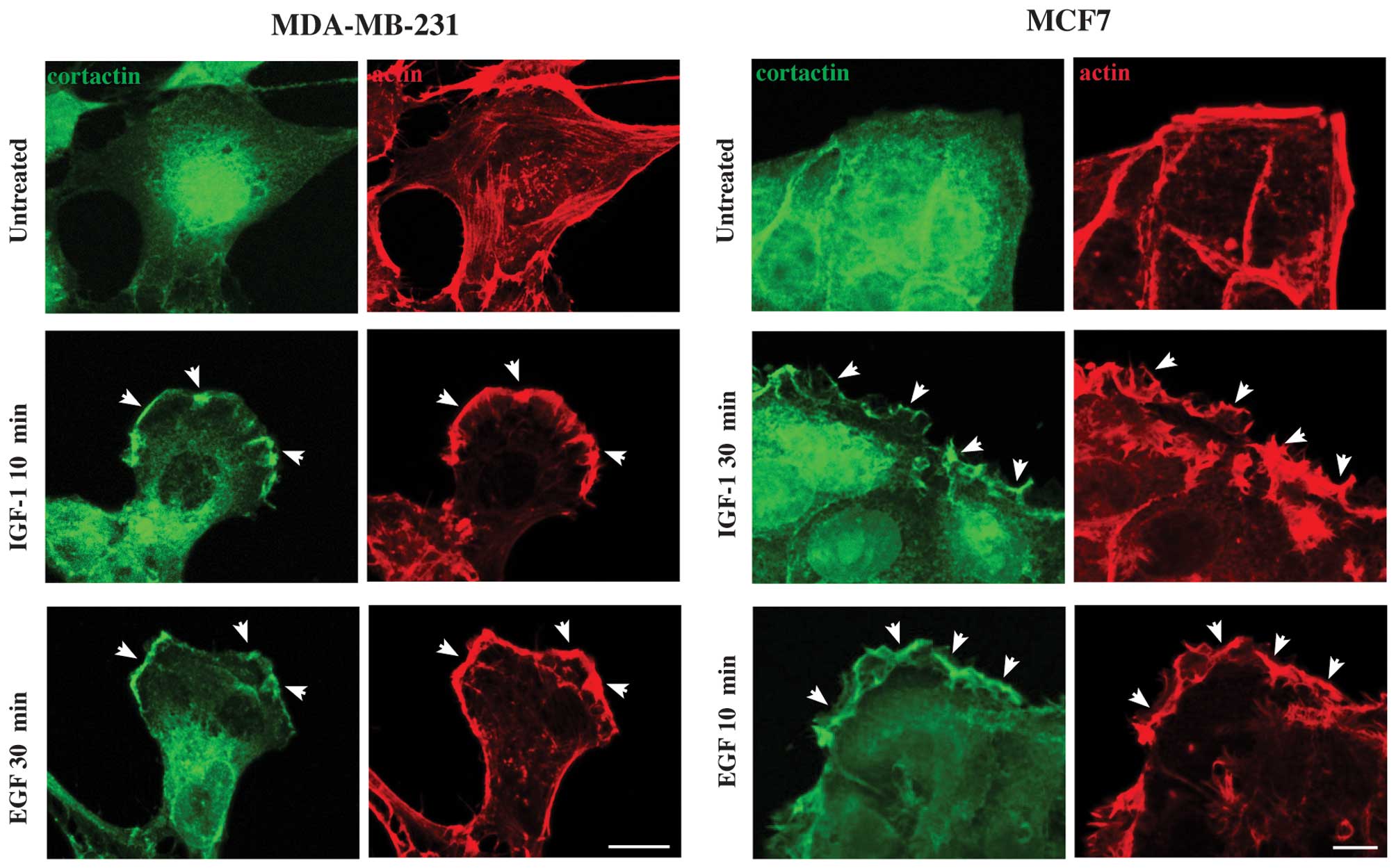
analysis revealed that untreated MDA-MB-231 cells exhibited a
polygonal, enlarged and flat shape, with actin cytoskeleton mainly
organized in stress fibers (Fig.
2, arrows, upper left panel), as previously described (35). Upon treatment with IGF-1,
MDA-MB-231 cells showed actin reorganization in membrane ruffles
and lamellipodia already after 10 min (Fig. 2, arrowheads, left panels), which
were still present at 30 and 60 min (Fig. 2, arrowheads, left panels),
althought after 60 min of treatment with IGF-1 some stress fibers
were also evident (Fig. 2, arrow,
left panels). Moreover, cells displayed an elongated shape, which
is the typical phenotype of migrating cells (Fig. 2, left panels). In contrast, the
actin cytoskeleton organization in membrane ruffles and
lamellipodia was evident after 30 and 60 min of stimulation with
EGF (Fig. 2, arrowheads, left
panels). whereas, after 10 min the actin cytoskeleton was organized
mainly in stress fibers (Fig. 2,
arrows, left panels) and in some small lamellipodia and ruffles
(Fig. 2, arrowheads, left panels).
Furthermore, cells displayed phenotype that changes depending on
time of treatment with EGF, being polygonal at 10 min, and
elongated at 30 and 60 min of stimulation, showing a clear leading
edge (Fig. 2, left panels). In the
case of MCF7, we focused our attention on cells that are localized
at the periphery of colonies, because they are the ones that
undergo migration. Untreated MCF7 showed clearly separated colonies
with regular edges, and the actin mainly organized in well defined
stress fibers (Fig. 2, arrows,
upper right panel). Upon IGF-1 treatment, MCF7 cells showed the
actin cytoskeleton organized in membrane ruffles and lamellipodia,
more evident at 30 and 60 min of stimulation, and only partially at
10 min (Fig. 2, arrowheads, left
panels). The treatment with EGF induced reorganization of actin
cytoskeleton in membrane ruffles and lamellipodia already after 10
min, which were less evident at 30 and 60 min of stimulation
(Fig. 2, arrowheads, left panels).
Interestingly, in MCF7 cells both IGF-1 and EGF induce
reorganization of actin in a large array of lamellipodia, which
remain confined throughout the cell cluster and do not extend
beyond the edges of the cell colonies (Fig. 2, left panels). Moreover, after
stimulation of both growth factors, cells at the periphery of MCF7
clusters showed a partially elongated cell shape, typical of cells
that are separating from the colony (Fig. 2, left panels). Thus, a migratory
phenotype was achieved by breast carcinoma cells after stimulation
with both growth factors, but at different time points. To evaluate
if the actin cytoskeleton reorganization in the cell lines that we
assayed is dependent on Src activation, we performed parallel
experiments in the presence of the Src inhibitor SU6656. Results
showed that SU6656 prevented both the migratory phenotype and the
formation of membrane ruffles and lamellipodia on MDA-MB-231 and
MCF7 cells (Fig. 2, right panels).
Taken together, these results suggest that IGF-1 and EGF are able
to induce actin reorganization in human carcinoma cell lines,
although the effect of IGF-1 was more rapid and consistent in
MDA-MB-231 than in MCF7 cells, and that this process is
Src-dependent.
 | Figure 2IGF-1 induces actin cytoskeleton
reorganization and migratory phenotype on the two breast cancer
cell lines in a different manner. MDA-MB-231 and MCF7 cells were
stimulated with IGF-1 or EGF at 10, 30 and 60 min, fixed,
permeabilized and stained with TRITC-phalloidin to visualize the
actin cytoskeleton organization. Untreated MDA-MB-231 cells show a
polygonal cell shape, with actin cytoskeleton mainly organized in
stress fibers (arrows). In MDA-MB-231, IGF-1 induces actin
organization in membrane ruffles and lamellipodia already after 10
min of stimulation (arrowheads), still present at 30 and 60 min,
during which some stress fibers are also evident (arrow). Moreover,
cells display a typical migratory phenotype. After 10 min of
stimulation with EGF, actin cytoskeleton is organized mainly in
stress fibers (arrows) and in membrane ruffles and lamellipodia
after 30 min (arrowheads), which were more evident at 60 min
(arrowheads). Furthermore, cells show a phenotype that changed
depending on the time of stimulation, being at 10 min polygonal,
and elongated at 30 and 60 min. Untreated MCF7 show clearly
separated colonies with regular edges, and the actin mainly
organized in well defined stress fibers (arrows). Upon IGF-1
stimulation, MCF7 cells display actin organized in membrane ruffles
and lamellipodia, especially evident after 30 and 60 min of
stimulation, and only partially at 10 min (arrowheads). The
treatment with EGF promotes formation of membrane ruffles and
lamellipodia already after 10 min, which are less evident at 30 and
60 min (arrowheads). After stimulation of both growth factors,
cells at the periphery of MCF7 clusters display a partially
elongated cell shape, typical of cells that are separating from the
colony. In addition, in the presence of the Src inhibitor SU6656
the formation of membrane ruffles and lamellipodia on either
MDA-MB-231 and MCF7 cells was prevented, and the cells do not
achieve a migratory phenotype. The images are representative of
three independent experiments. Bars, 10 μm. |
IGF-1 induces rapid tyrosine
phosphorylation of cortactin in breast cancer cells
Since one of the molecules involved in the
reorganization of actin cytoskeleton is cortactin, an actin-binding
protein, whose activity depends on tyrosine phosporylation mediated
by the tyrosine kinase Src (6), we
evaluated the possible involvement of cortactin in human breast
carcinoma cells upon IGF-1 stimulation. To this purpose, we first
determined the expression of cortactin in MDA-MB-231 and MCF7 cells
by western blot analysis. Cells were lysed and blotted with
anti-cortactin antibodies, and the membrane was subsequently
stripped and blotted with anti-α-tubulin antibody to assess the
equal loading. Results showed that both the human breast cancer
cell lines expressed the protein cortactin, although MCF7 cells
exhibited a greater amount of the protein with respect to that
expressed by MDA-MB-231 (Fig. 3A),
in keeping with previous reports (36). To analyze the activation of
cortactin, we performed a biochemical study of cortactin tyrosine
phosphorylation in serum starved MDA-MB-231 and MCF7 cells, treated
with IGF-1 or EGF at different intervals of time, as reported in
Materials and methods. Immunoprecipitation with anti-cortactin
antibodies and immunoblot with anti-phosphotyrosine antibody,
demonstrated that in MDA-MB-231 cells both IGF-1 and EGF induced
tyrosine phosphorylation of cortactin, although IGF-1 effect was
already evident at 10 min, and was still present at 30 and 60 min
of stimulation (Fig. 3B, left
panels). On the other hand, EGF induced a lower intensity of
tyrosine phosphorylation of cortactin, with a peak of
phosphorylation after 30 min of treatment (Fig. 3B, right panels). In MCF7 cells, the
highest point of phosphorylation of the protein is found after 30
min of treatment with IGF-1, still present at 60 min (Fig. 3C, left panels), whereas, the
treatment with EGF led to a good activation of cortactin already
after 10 min, which decreased at 30 min and was virtually absent
after 60 min of stimulation (Fig.
3C, right panels). Thus, both IGF-1 and EGF were able to induce
tyrosine phosphorylation of cortactin in breast cancer cells,
althoug with a different intensity and kinetics, being IGF-1
stronger, faster and lasting with respect to EGF in MDA-MB-231. To
analyze if the activation of cortactin in the cellular models that
we assayed is dependent on Src activity, we performed parallel
experiments in the presence of SU6656. The results showed that the
Src inhibitor was able to block tyrosine phosphorylation of
cortactin (Fig. 3B and C), as
expected (37), thus suggesting a
direct involvement of Src in the activation of cortactin also in
our cellular models.
 | Figure 3IGF-1 induces tyrosine
phosphorylation of cortactin. (A) Expression of cortactin in
MDA-MB-231 and MCF7 cells by western blot analysis. Cells were
lysed and blotted with anti-cortactin antibodies and the membrane
was subsequently stripped and blotted with anti-α-tubulin antibody
to assess the equal loading. A band corresponding to the protein
cortactin is present in both the cell lines, although the amount of
the protein expressed by MCF7 cells is greater than that exhibited
by MDA-MB-231. Biochemical study of cortactin tyrosine
phosphorylation in serum starved (B) MDA-MB-231 and (C) MCF7. Cells
were treated with IGF-1 or EGF at different time points,
immunoprecipitated with anti-cortactin antibodies and immunoblotted
with anti-phosphotyrosine antibody. In MDA-MB-231 cells (B), IGF-1
stimulation induces a strong intensity level of tyrosine
phosphorylation of cortactin already evident after 10 min and still
present at 30 and 60 min. Whereas, EGF stimulation promotes a lower
intensity of tyrosine phosphorylation of cortactin, with a peak
after 30 min of treatment. In the case of MCF7 cells (C), the
highest point of the phosphorylation of the protein is found after
30 min of treatment with IGF-1, still present at 60 min, whereas,
the treatment with EGF induces a good activation of cortactin
already after 10 min, which decreases at 30 min and is virtually
absent after 60 min of stimulation. The membranes were subsequently
stripped and blotted with anti-cortactin antibodies to assess the
equal loading. Parallel experiments performed in the presence of
SU6656, show that the Src inhibitor blocks tyrosine phosphorylation
of cortactin (B and C). The intensity of the bands was evaluated by
densitometric analysis; the values from a representative experiment
were normalized, expressed as fold increase with respect to the
control value and reported as a graph (A–C). |
IGF-1 induces translocation of tyrosine
phosphorylated cortactin to the plasma membrane at sites of focal
adhesion
Since we have previously shown that after KGF or
FGF10 treatment, cortactin translocates to the plasma membrane of
human keratinocytes in areas where actin is organized in ruffles
and lamellipodia (37), we
wondered whether upon IGF-1 treatment the activated phosphorylated
cortactin translocates to the plasma membrane also in breast cancer
cells. To this purpose, we first performed immunofluorescence
analysis of cortactin localization in the cellular models assayed
in the present study, using polyclonal antibodies directed against
cortactin and, subsequently, the TRITC-phalloidin to stain the
filamentous actin cytoskeleton. Cells were treated with either
IGF-1 or EGF and observed at time points in which cortactin
activation has been documented to be stronger, as described in
experiments shown in Fig. 3.
Results demonstrated that in untreated MDA-MB-231 and MCF7 cells
the cortactin staining appeared mainly localized in the cytoplasm,
with no localization at the plasma membrane (Fig. 4), as previously shown (37). After stimulation with IGF-1 for 10
min or EGF for 30 min, MDA-MB-231 cells showed translocation of
cortactin to the plasma membrane, in areas where actin is mainly
organized in ruffles and lamellipodia (Fig. 4, arrowheads, left panels). Similar
results were obtained in MCF7 cells after treatment with IGF-1 for
30 min or EGF for 10 min (Fig. 4,
arrowheads, right panels).
It has been shown that cortactin is phosphorylated
at tyrosine residues Y421, Y466 and Y482 (9,38)
upon various signals, including growth factors (39), and that this occurs in a sequential
manner, the tyrosine position 421 being the first to be
phosphorylated, and then tyrosine 466 and finally 482 (9). In addition, cortactin phosphorylated
on tyrosine 421 is found localized on lamellipodia (9). Based on these observations, we
wondered whether the cortactin form that translocates to the plasma
membrane upon IGF-1 and EGF stimulation, is the one phosphorylated
on tyrosine in position 421 in our cellular models. To this
purpose, we performed double immunofluorescence analysis using
anti-cortactin monoclonal antibody and anti-pY421 cortactin
polyclonal antibodies, that specifically recognize the cortactin
phosphorylated on tyrosine in position 421. Results obtained showed
that in MDA-MB-231 cells upon stimulation with IGF-1 for 10 min or
EGF for 30 min, phospho-cortactin colocalized with cortactin on the
plasma membrane, where it was translocated (Fig. 5A, arrowheads, upper panels),
whereas, in untreated cells no colocalization between
phosphorylated and normal cortactin was evident on the plasma
membrane (Fig. 5A, upper panels).
Also in MCF7 cells, after stimulation with IGF-1 for 30 min or EGF
for 10 min, cortactin colocalized with the phosphorylated form of
the protein on the plasma membrane (Fig. 5A, arrowheads, lower panels), as
previously demonstrated (40),
whereas no colocalization was observed in untreated cells (Fig. 5A, lower panels). The presence of
SU6656 showed that there were neither translocation of cortactin
nor staining of phospho-cortactin on the plasma membrane (Fig. 5A, upper and lower panels).
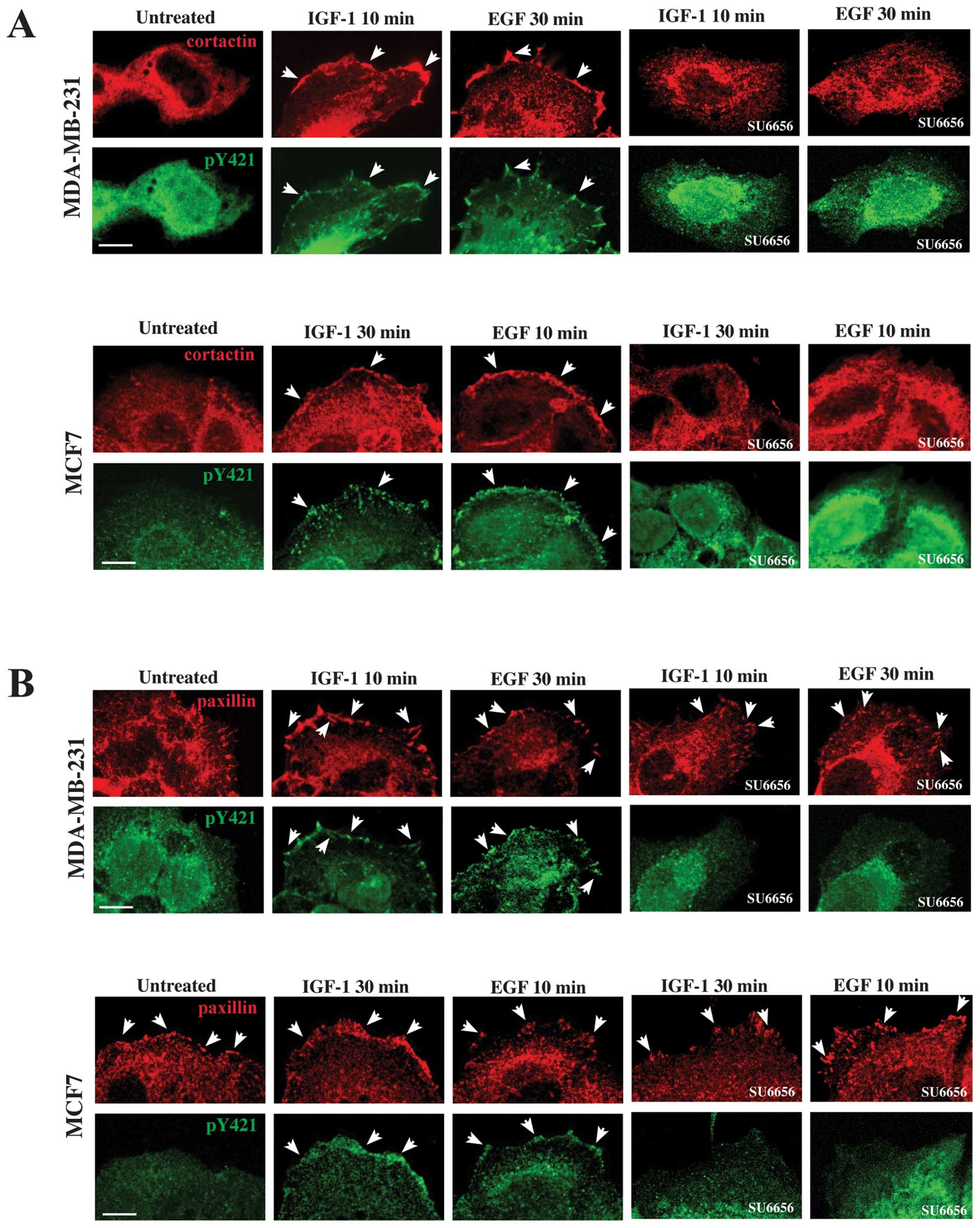 | Figure 5Phospo-cortactin localizes to the
focal adhesions. (A) Serum starved MDA-MB-231 and MCF7 cells were
treated with IGF-1 or EGF at different time points, fixed,
permeabilized and labeled with anti-cortactin monoclonal antibody
and anti-pY421 cortactin polyclonal antibodies, which specifically
recognize the cortactin phosphorylated on tyrosine in position 421.
Double immunofluorescence analysis show that in MDA-MB-231 cells
upon stimulation with IGF-1 for 10 min or EGF for 30 min
phospho-cortactin and cortactin colocalize on the plasma membrane
(arrowheads), whereas, in untreated cells no virtual colocalization
between them is evident. Upon treatment with IGF-1 for 30 min or
EGF for 10 min, even in the case of MCF7 cells, cortactin
colocalizes with the phosphorylated form of cortactin on the plasma
membrane (arrowheads), whereas no virtual colocalization is
observed in untreated cells. Parallel experiments performed in the
presence of SU6656 demonstrate that there are neither translocation
of cortactin nor staining of phospho-cortactin on the plasma
membrane. (B) Serum starved MDA-MB-231 and MCF7 cells were
stimulated with IGF-1 or EGF, fixed, permeabilized and incubated
with anti-paxillin monoclonal antibody to reveal paxillin, a
component of the focal adhesions and with anti-pY421-cortactin to
reveal the phospho-cortactin. In untreated MDA-MB-231 and MCF7
cells, paxillin staining is present on the plasma membrane
(arrowheads), whereas the labeling of phospho-cortactin is not.
After IGF-1 treatment for 10 min or EGF for 30 min, in MDA-MB-231
cells colocalization of phospho-cortactin with paxillin at the
plasma membrane is evident (arrowheads). Similar results are
obtained in MCF7 cells, upon stimulation with IGF-1 for 30 min or
EGF for 10 min (arrowheads). SU6656 is able to prevent the movement
of pY421 cortactin to the focal adhesions, whereas the staining of
paxillin is regular (arrowheads). The images are representative of
three independent experiments. Bars, 10 μm. |
Since the cortactin translocated to the plasma
membrane appeared at specific sites which resembled focal adhesion,
we focused our attention on paxillin, a component of focal
adhesions, with a role in cell migration (41). Serum starved MDA-MB-231 and MCF7
cells were stimulated with IGF-1 or EGF, fixed, permeabilized and
incubated with anti-paxillin monoclonal antibody to reveal
paxillin, and with anti-pY421-cortactin to reveal the
phospho-cortactin. In untreated MDA-MB-231 and MCF7 cells the
labeling of phosphocortactin was not present at the plasma membrane
(Fig. 5B, upper and lower panels),
whereas the staining of paxillin was evident at the periphery of
the cells (Fig. 5B, arrowheads,
upper and lower panels). After IGF-1 treatment for 10 min or EGF
for 30 min, MDA-MB-231 cells showed an evident colocalization of
phospho-cortactin with paxillin in correspondence of focal
adhesions (Fig. 5B, arrowheads,
upper panels). Also in the case of MCF7 cells stimulation with
IGF-1 for 30 min or EGF for 10 min induced the localization of
pY421-cortactin at the plasma membrane where paxillin was evident
(Fig. 5B, arrowheads, lower
panels). The presence of SU6656 inhibited the localization of pY421
cortactin to the focal adhesions, whereas the staining of paxillin
was regular (Fig. 5B, arrowheads,
upper and lower panels).
Taken together, these results indicate that IGF-1
and EGF were able to induce translocation of pY421-cortactin to the
plasma membrane at the focal adhesions, where it colocalizes with
paxillin. Moreover, the employment of SU6656 strongly suggest a
direct involvement of Src in this process.
Discussion
The rationale of our investigation on IGF-1 and
EGF-dependent migration and on the pathways involved in ER-negative
and ER-positive breast cancer cell lines, is that the migration of
cancer cells greatly contribute to their metastatic potential, that
is the major clinical threat in cancer patients. Overexpression and
activation of IGF-1R have been associated with some different
aspects of the progression in cancer (10). Several studies indicate that in
breast cancer, both IGF-1 and EGF are involved in the processes of
cell adhesion, invasion and anchorage-independent growth that
finally lead to the migration and metastatic spreading (19–23).
Both IGF-1R and EGFR activate common downstream signaling pathways,
meanly those involving MAP kinase and Akt. IGF-1R functions are
predominantly mediated through IGF-1-induced activation of the
phosphoinositide 3-kinase (PI3K)-AKT, RAS-RAF-MAPK and p38 MAPK
signaling cascades (42). The
cooperativity between Src and EGFR, as well as other motogenic
receptors, is well established (43), and a variety of docking proteins
that contain Src homology-2 (SH2) and phosphotyrosine binding (PTB)
domains, bind the intracellular domains of the activated receptors
(44). Our study demonstrates that
both IGF-1 and EGF induce a significant effect on migration of
human breast cancer cells. IGF-1 treatment of MDA-MB-231 cells
resulted in significantly enhanced cell migration. The effect is
earlier, stronger and steadier, when compared to the EGF and
untreated control cells under the same experimental conditions. The
effect of EGF, with respect to IGF-1 treatment, on ER-positive MCF7
cells in inducing cell migration is, on the contrary, greater. Our
results are consistent with those obtained by Bae et al
(45), who concluded that the
motogenic effect of IGF-1 does not induce a motile phenotype on
MCF7 cell line, and with those of Jackson et al (46), who showed that IGF-1 stimulated the
migration of MDA-MB-231 cells. Moreover, we are in agreement also
with Tong et al (47), who
observed that EGF produced a chemoattractant effect in estrogen
receptor (ER)-positive MCF-7 cells, whereas in ER-negative
MDA-MB-231 breast cancer cells, EGF produced very little invasive
activity.
All these observations are consistent with the view
that the expression level of IGF-1R, as well as that of EGFR, are
important in controlling cellular responses in terms of migration
to both the growth factors. This scenario has been strenghten by
the demonstration that the knockdown of the IGF-1R decreases the
migration stimulated by IGF-1 in MCF7 and MDA-MB-231 cells
(48).
These data strongly suggest a cell specific ability
of the motogenic receptor to modulate the migratory potential of
breast cancer cells. The modulation of cell motility and cell
adhesion, that is essential for metastasis, arises as a result of
changes in the cytoskeletal architecture (49,50).
Engagement or activation of cell surface growth factors and
adhesion receptors affects the assembly and arrangement of F-actin
networks (51,52), and the cell shape. In a previous
paper, DePasquale (53) showed
that the treatment of MCF7 cells with estrogen induces a
reorganization of the actin cytoskeleton in a complex arrangement
of lamellipodia that is actively motile. In line with this report
is our observation that in MCF7 cells both IGF-1 and EGF induce
reorganization of actin in a large array of lamellipodia, which
remains confined throughout the cell clusters and do not extend
beyond the edges of the cell colonies, suggesting that the cells at
the periphery of the colonies move all together along the direction
of migration. Moreover, we found that MDA-MB-231 cells show typical
migratory features and an evident motile phenotype upon both IGF-1
and EGF treatment, although the effect of IGF-1 is more rapid than
that of EGF, reflecting the behavior of cells in terms of
migration.
Cortactin has been shown to enhance lamellipodial
persistence and the rate of adhesion formation in lamellipodia,
consistent with an important function in regulating directed cell
motility (54). As reported by
Wang et al (55), we
demonstrate that upon IGF-1 stimulation, cortactin is tyrosine
phosphorylated at Y421 and translocates to the focal adhesions,
where it colocalizes with paxillin. This finding may explain the
observed phenotype and these changes in local F-actin dynamics may
regulate cell motility.
Our data show that the inhibition of Src abolishes
IGF-1- and EGF-mediated migration in both ER-positive and
ER-negative cells. We demonstrated that the motogenic effect and
the switch in the phosphorylation state of cortactin is Src
signaling-dependent, as shown by using the selective Src-inhibitor
SU6656. In our model system, MDA-MB-231 cells, which have a high
invasive potential, the translocation of phospho-cortactin to the
focal adhesion is IGF-1-dependent, and Src is necessary, because
tyrosine phosphorylation of cortactin is linked to Src activity, as
previously shown (3,56,57).
The initial accumulation of the post-translational activated
cortactin and actin, is potentially involved in the clinical
aggressiveness of both ER-positive and ER-negative breast
carcinoma, because actin polymerization at the leading edge
provides the protrusive force required for the extension of
lamellipodia observed during cell motility and spreading (58). In agreement with Liu and Feng
(59), our data suggest the Src
inhibition as a potential clinical therapeutic strategy for
Her2-negative breast cancer patients, but the treatment approach
and the combination with chemotherapy needs to be further
elucidated.
We also showed that the expression level of
cortactin is different in the two cell lines, being higher in MCF7,
according to their molecular subtype characteristic of luminal
epithelial phenotype (27,28), compared to MDA-MB-231. Our data are
in agreement with a previous report in which a profile of the level
of cortactin expression was assessed in a panel of breast cancer
cells related to the different degree of EMS1 gene amplification
(36), and with the reduced
epithelial properties of MDA-MB-231 cells according to their gene
expression pattern, similar to the claudin-low tumor subtype with a
strong tendency to mesenchymal transition, and a low expression of
genes involved in epithelial cell-cell adhesion (29). Epithelial cells are characterized
by the formation of intercellular junctions, which connect and
immobilize adjacent cells. Cortactin is reported to be involved in
the formation of adhesion junctions after recruitment to cell-cell
adhesive contacts, and it is recruited in response to homophilic
cadherin ligation (60). Recently,
it has been reported that inhibition of cortactin enhanced the
expression of fibronectin, a specific mesenchymal marker, and
accelerated TGF-β1-induced EMT, indicating that cortactin is
involved in maintaining the epithelial properties of cells
(61). Based on these observations
and on the results of this study, we speculate that also in our
cellular models the basal level of cortactin is related to a
potential function in maintaining epithelial properties in
Her2-negative breast cancer cells.
In addition, we observed that cortactin is tyrosine
phosphorylated by IGF-1 and EGF with different amplitude and
kinetics, IGF-1 is stronger and faster than EGF in activating this
protein in MDA-MB-231 cells. Whereas, in MCF7 cells, EGF-induced
activation of cortactin appears to be more intense than that of
IGF-1. We hypothesize that these differences in the expression,
kinetics, magnitude and duration of cortactin tyrosine
phosphorylation can account for the discrepancies between IGF-1 and
EGF in promoting cell migration.
Collectively, our study underlines the
multipotential properties of the actin-binding protein cortactin in
tumor cell migration, and, in conclusion, suggests that the
IGF-1/EGF-Src-cortactin pathway dependent enhancement of cell
motility leads to a motile phenotype, and determines the formation
of cancer cells which are migratory and invasive, hallmarks of
cells that have the potential to generate metastases.
Acknowledgements
This study was funded by grant from
MIUR 2009 (Ministero dell’ Istruzione, dell’ Università e della
Ricerca).
References
|
1.
|
Brooks SA, Lomax-Browne HJ, Carter TM,
Kinch CE and Hall DM: Molecular interactions in cancer cell
metastasis. Acta Histochem. 112:3–25. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Bahr C and Groner B: The IGF-1 receptor
and its contributions to metastatic tumor growth-novel approaches
to the inhibition of IGF-1R function. Growth Factors. 23:1–14.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Wu H, Reynolds AB, Kanner SB, Vines RR and
Parsons JT: Identification and characterization of a novel
cytoskeleton-associated pp60src substrate. Mol Cell Biol.
11:5113–5124. 1991.PubMed/NCBI
|
|
4.
|
Wu H and Parsons JT: Cortactin, an
80/85-kilodalton pp60src substrate, is a filamentous actin-binding
protein enriched in the cell cortex. J Cell Biol. 120:1417–1426.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Weed SA and Parsons JT: Cortactin:
coupling membrane dynamics to cortical actin assembly. Oncogene.
20:6418–6434. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Daly RJ: Cortactin signalling and dynamic
actin networks. Biochem J. 382:13–25. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Schuuring E, Verhoeven E, Litvinov S and
Michalides RJ: The product of the EMS1 gene, amplified and
overexpressed in human carcinoma, is homologous to a v-src
substrate and is located in cell-substratum contact sites. Mol Cell
Biol. 13:2891–2898. 1993.PubMed/NCBI
|
|
8.
|
Ormandy CJ, Musgrove EA, Hui R, Daly RJ
and Sutherland RL: Cyclin D1, EMS1 and 11q13 amplification in
breast cancer. Breast Cancer Res Treat. 78:323–335. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Head JA, Jiang D, Li M, Zorn LJ, Schaefer
EM, Parsons JT and Weed SA: Cortactin tyrosine phosphorylation
requires Rac1 activity and association with the cortical actin
cytoskeleton. Mol Biol Cell. 14:3216–3229. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Rosenzweig SA and Atreya HS: Defining the
pathway to insulin-like growth factor system targeting in cancer.
Biochem Pharmacol. 80:1115–1124. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Surmacz E: Function of the IGF-I receptor
in breast cancer. J Mammary Gland Biol Neoplasia. 5:95–105. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Baserga R: The IGF-I receptor in cancer
research. Exp Cell Res. 253:1–6. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Jackson JG, White MF and Yee D: Insulin
receptor substrate-1 is the predominant signaling molecule
activated by insulin-like growth factor I, insulin, and
interleukin-4 in estrogen receptor-positive human breast cancer
cells. J Biol Chem. 273:9994–10003. 1998. View Article : Google Scholar
|
|
14.
|
Schnarr B, Strunz K, Ohsam J, Benner A,
Wacker J and Mayer D: Downregulation of insulin-like growth
factor-I receptor and insulin receptor substrate-1 expression in
advanced human breast cancer. Int J Cancer. 89:506–513. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Peyrat JP, Bonneterre J, Dusanter-Fourt I,
Leroy-Martin B, Dijane J and Demaille A: Characterization of
insulin-like growth factor 1 receptors (IGF-IR) in human breast
cancer cell lines. Bull Cancer. 76:311–309. 1989.PubMed/NCBI
|
|
16.
|
Sepp-Lorenzino L, Rosen N and Lebwohl DE:
Insulin and insulin-like growth factor signaling are defective in
the MDA-MB-468 human breast cancer cell line. Cell Growth Differ.
5:1077–1083. 1994.PubMed/NCBI
|
|
17.
|
Jackson JG and Yee D: IRS-1 expression and
activation are not sufficient to activate downstream pathways and
enable IGF-I growth response in estrogen receptor negative breast
cancer cells. Growth Horm IGF Res. 9:280–289. 1999. View Article : Google Scholar
|
|
18.
|
Godden J, Leake R and Kerr DJ: The
response of breast cancer cells to steroid and peptide growth
factors. Anticancer Res. 12:1683–1688. 1992.PubMed/NCBI
|
|
19.
|
Dunn SE, Ehrlich M, Sharp NJ, Reiss K,
Solomon G, Hawkins R, Baserga R and Barrett JC: A dominant negative
mutant of the insulin-like growth factor-I receptor inhibits the
adhesion, invasion and metastasis of breast cancer. Cancer Res.
58:3353–3361. 1998.PubMed/NCBI
|
|
20.
|
Arteaga CL, Kitten LJ, Coronado EB, Jacobs
S, Kull FC Jr, Allred DC and Osborne CK: Blockade of the type I
somatomedin receptor inhibits growth of human breast cancer cells
in athymic mice. J Clin Investig. 84:1418–1423. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Doerr ME and Jones Jl: The roles of
integrins and extracellular matrix proteins in the insulin-like
growth factor I-stimulated chemotaxis of human breast cancer cells.
J Biol Chem. 271:2443–2447. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Schlessinger J: Cell signaling by receptor
tyrosine kinases. Cell. 103:211–225. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Arteaga CL: Epidermal growth factor
receptor dependence in human tumors: more than just expression?
Oncologist. 4:31–39. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Davidson NE, Gelmann EP, Lippman ME and
Dickson RB: Epidermal growth factor receptor gene expression in
estrogen receptor-positive and negative human breast cancer cell
lines. Mol Endocrinol. 1:216–223. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Mamot C, Drummond DC, Greiser U, Hong K,
Kirpotin DB, Marks JD and Park JW: Epidermal growth factor receptor
(EGFR)-targeted immunoliposomes mediate specific and efficient drug
delivery to EGFR- and EGFRvIII-overexpressing tumor cells. Cancer
Res. 63:3154–3161. 2003.
|
|
26.
|
Callahan R and Hurvitz S: Human epidermal
growth factor receptor-2-positive breast cancer: current management
of early, advanced, and recurrent disease. Curr Opin Obstet
Gynecol. 23:37–43. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Fillmore CM and Kuperwasser C: Human
breast cancer cell lines contain stem-like cells that self-renew,
give rise to phenotypically diverse progeny and survive
chemotherapy. Breast Cancer Res. 10:R252008. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Charafe-Jauffret E, Ginestier C, Monville
F, Finetti P, Adélaïde J, Cervera N, Fekairi S, Xerri L, Jacquemier
J, Birnbaum D and Bertucci F: Gene expression profiling of breast
cell lines identifies potential new basal markers. Oncogene.
25:2273–2284. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Prat A, Parker JS, Karginova O, Fan C,
Livasy C, Herschkowitz JI, He X and Perou CM: Phenotypic and
molecular characterization of the claudin-low intrinsic subtype of
breast cancer. Breast Cancer Res. 12:R682010. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Blake RA, Broome MA, Liu X, Wu J, Gishizky
M, Sun L and Courtneidge SA: SU6656, a selective Src family kinase
inhibitor, used to probe growth factor signaling. Mol Cell Biol.
20:9018–9027. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Dittmar T, Husemann A, Schewe Y, Nofer JR,
Niggemann B, Zanker KS and Brandt BH: Induction of cancer cell
migration by epidermal growth factor is initiated by specific
phosphorylation of tyrosine 1248 of c-erbB-2 receptor via EGFR.
FASEB J. 16:1823–1825. 2002.PubMed/NCBI
|
|
32.
|
Hirsch DS, Shen Y and Wu WJ: Growth and
motility inhibition of breast cancer cells by epidermal growth
factor receptor degradation is correlated with inactivation of
Cdc42. Cancer Res. 66:3523–3530. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Playford MP and Schaller MD: The interplay
between Src and integrins in normal and tumor biology. Oncogene.
23:7928–7946. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Ridley AJ, Schwartz MA, Burridge K, Firtel
RA, Ginsberg MH, Borisy G, Parsons JT and Horwitz AR: Cell
migration: integrating signals from front to back. Science.
302:1704–1709. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Azios NG and Dharmawardhane SF:
Resveratrol and estradiol exert disparate effects on cell
migration, call surface actin structures, and focal adhesion
assembly in MDA-MB-231 human breast cancer cells. Neoplasia.
7:128–140. 2005. View Article : Google Scholar
|
|
36.
|
Campbell DH, deFazio A, Sutherland RL and
Daly RJ: Expression and tyrosine phoshorylation of EMS1 in human
breast cancer cell lines. Int J Cancer. 68:485–492. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Ceccarelli S, Cardinali G, Aspite N,
Picardo M, Marchese C, Torrisi MR and Mancini P: Cortactin
involvement in the keratinocyte growth factor and fibroblast growth
factor 10 promotion of migration and cortical actin assembly in
human keratinocytes. Exp Cell Res. 313:1758–1777. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Huang C, Liu J, Haudenschild CC and Zhan
X: The role of tyrosine phosphorylation of cortactin in the
locomotion of endothelial cells. J Biol Chem. 273:25770–25776.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Weed SA, Du Y and Parsons JT:
Translocation of cortactin to the cell periphery is mediated by the
small GTPase Rac1. J Cell Sci. 111:2433–2443. 1998.PubMed/NCBI
|
|
40.
|
Ren G, Helwani FM, Verma S, McLachlan RW,
Weed SA and Yap AS: Cortactin is a functional target of
E-cadherin-activated Src family kinases in MCF7 epithelial
monolayers. J Biol Chem. 284:18913–18922. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Brown MC and Turner CE: Paxillin: adapting
to change. Physiol Rev. 84:1315–1339. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Vincent AM and Feldman EL: Control of cell
survival by IGF signaling pathways. Growth Horm IGF Res.
12:193–197. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Biscardi JS, Tice DA and Parsons SJ:
c-Src, receptor tyrosine kinases, and human cancer. Adv Cancer Res.
76:61–119. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Ravichandran KS: Signaling via Shc family
adapter proteins. Oncogene. 20:6322–30. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Bae SN, Arand G, Azzam H, Pavasant P,
Torri J, Frandsen TL and Thompson EW: Molecular and cellular
analysis of basement membrane invasion by human breast cancer cells
in matrigel-based in vitro assays. Breast Cancer Res Treat.
24:241–255. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Jackson JG, Zhang X, Yoneda T and Yee D:
Regulation of breast cancer cell motility by insulin receptor
substrate-2 (IRS-2) in metastatic variants of human breast cancer
cell lines. Oncogene. 20:7318–7325. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Tong GM, Rajah TT and Pento JT: The
differential influence of EGF, IGF-I and TGF-beta on the
invasiveness of human breast cancer cells. In Vitro Cell Dev Biol
Anim. 36:493–494. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
de Blaquiere GE, May FE and Westley BR:
Increased expression of both insulin receptor substrates 1 and 2
confers increased sensitivity to IGF-1 stimulated cell migration.
Endocr Relat Cancer. 16:635–647. 2009.PubMed/NCBI
|
|
49.
|
Lester BR and McCarthy JB: Tumor cell
adhesion to the extracellular matrix and signal transduction
mechanisms implicated in tumor cell motility, invasion and
metastasis. Cancer Metastasis Rev. 11:31–44. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Yamaguchi H, Pixley F and Condeelis J:
Invadopodia and podosomes in tumor invasion. Eur J Cell Biol.
85:213–218. 2005. View Article : Google Scholar
|
|
51.
|
Zigmond SH: Signal transduction and actin
filament organization. Curr Opin Cell Biol. 8:66–73. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Schoenwaelder SM and Burridge K:
Bidirectional signaling between the cytosckeleton and integrins.
Curr Opin Cell Biol. 11:274–286. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
DePasquale JA: Rearrangement of the
F-actin cytoskeleton in estradiol-treated MCF7 breast carcinoma
cells. Histochem Cell Biol. 112:341–350. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Bryce NS, Clark ES, Leysath JL, Currie JD,
Webb DJ and Weaver AM: Cortactin promotes cell motility by
enhancing lamellipodial persistence. Curr Biol. 15:1276–1285. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Wang W, Liu Y and Liao K: Tyrosine
phosphorylation of cortactin by the FAK-Src complex at focal
adhesions regulates cell motility. BMC Cell Biol. 13:12–49.
2011.PubMed/NCBI
|
|
56.
|
Mader CC, Oser M, Magalhaes MA,
Bravo-Cordero JJ, Condeelis J, Koleske AJ and Gil-Henn H: An
EGFR-Src-Arg-Cortactin pathway mediates functional maturation of
invadopodia and breast cancer cell invasion. Cancer Res.
71:1730–1741. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Artym VV, Zhang Y, Seillier-Moiseiwitsch
F, Yamada KM and Mueller SC: Dynamic interactions of cortactin and
membrane type 1 matrix metalloproteinase at invadopodia: defining
the stages of invadopodia formation and function. Cancer Res.
66:3034–3043. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Small JV and Resch GP: The comings and
goings of actin: coupling protrusion and retraction in cell
motility. Curr Opin Cell Biol. 17:517–523. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
59.
|
Liu X and Feng R: Inhibition of epithelial
to mesenchymal transition in metastatic breast carcinoma cells by
c-Src suppression. Acta Biochim Biophys Sin (Shanghai). 42:496–501.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Helwani FM, Kovacs EM, Paterson AD, Verma
S, Ali RG, Fanning AS, Weed SA and Yap AS: Cortactin is necessary
for E-cadherin-mediated contact formation and actin reorganization.
J Cell Biol. 164:899–910. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
61.
|
Zhang K, Wang D and Song J: Cortactin is
involved in tran-forming growth factor-beta1-induced
epithelial-mesenchymal transition in AML-12 cells. Acta Biochim
Biophys Sin (Shanghai). 41:839–845. 2009. View Article : Google Scholar : PubMed/NCBI
|