Introduction
Hsp90 is a molecular chaperone that is responsible
for the stability and function of over 100 client proteins
(1). Hsp90 client proteins include
Akt, IKKα, B-Raf and GSK3β, which are critical for the cell
survival and proliferation (2).
Hsp90 represents 1–2% of total cellular protein, and is
overexpressed 2- to 10-fold in tumor cells compared to normal cells
in the same or related tissue (3).
In addition, tumor cells are more dependent on Hsp90 than normal
counterparts, suggesting that Hsp90 may be important for tumor cell
growth or survival (4). The
therapeutic efficacy of Hsp90 inhibitors probably relates to the
increased levels of active Hsp90 in tumors (5). The Hsp90 inhibitors have been shown
to accumulate in tumor tissue while being rapidly cleared in normal
tissue and shutdown multiple signaling pathways probably because of
the degradation of client proteins (6), and the inhibition of signaling
pathways, such as the IKKα/NF-κB signaling pathway, may induce
apoptosis.
Hsp90 is intimately associated with autophagy
(7) and the Hsp90 inhibitor
17-DMAG induces autophagy via the inhibition of mTOR signaling
(8). Autophagy is a physiological
process regulated by the Akt/mTOR and MAPK/Erk1/2 signaling
pathways, which are important in the induction of tumor cell death
(9,10). Autophagy is an important cellular
response to stress or starvation. Many studies have focused on the
importance of autophagy in cancer, however, it is still under
debate whether autophagy suppresses tumorigenesis or promotes cell
survival (11,12).
Currently, 14 unique chemical moieties are
undergoing clinical trials as potential cancer therapeutics
(1). 17-AAG, a derivative of
geldanamycin, is the first Hsp90 inhibitor to undergo clinical
testing and possesses encouraging biological and pharmacologic
properties (13). However, the
poor pharmaceutical properties such as aqueous solubility and
formulation difficulties are anticipated to limit its further
clinical development (14), and
this has catalyzed the efforts of identifying the novel scaffolds
with improved pharmacological and lower toxicity profiles. Thus,
novel synthetic Hsp90 inhibitors based on diverse chemical
scaffolds have been developed (15).
The novel Hsp90 inhibitor SNX-2112 can selectively
bind to the ATP/ADP binding pocket of Hsp90 and is more
pharmacologically effective than 17-AAG (16). Also, SNX-2112 is highly effective
against various cancer cells in vitro and in vivo
(17,18). We have previously reported that
SNX-2112 induces apoptosis and/or autophagy in K562, MCF-7 and A375
cell lines (16,19,20).
In addition, we also analyzed the pharmacokinetics of SNX-2112 in
rats with a sensitive and specific reversed-phase high-performance
liquid chromatography method (21,22).
BJ-B11 is a novel analog of SNX-2112, and has a
structure that differs in the cyclohexanol and inazolone moieties
(23). Previous research has shown
that BJ-B11 potently induces growth inhibition of a diverse range
of tumor cell lines in vitro. However, the molecular
mechanism by which BJ-B11 acts needs to be further elaborated.
In this study, we provide the first evidence that
BJ-B11 induces growth inhibition, G2/M cell cycle arrest, the
mitochondrial-mediated apoptosis, autophagy via the inhibition of
Akt/mTOR/p70S6K signaling and cytoskeleton polymerization in
Eca-109 cells.
Materials and methods
Cell culture and reagents
The Eca-109 cells (Cell Bank of the Chinese Academy
of Sciences, Shanghai, China) were cultured in RPMI-1640 containing
10% heat inactivated fetal bovine serum (FBS) and 100 U/ml
penicillin/streptomycin in a humidified incubator in a 5%
CO2 atmosphere at 37°C.
BJ-B11 was synthesized as previously described with
a purity of >98.0% (24), and
10 mmol/l BJ-B11 stock solutions in dimethylsulfoxide (DMSO) were
stored at −20°C. 17-AAG was purchased from Alexis Biochemicals (San
Diego, CA). The 3-(4,5-diethylthiazol-2-yl)-2,5-diphenyl
tetrazolium bromide (MTT) assay, 4′,6-diamidino-2-phenylindole
(DAPI), jasplakinolide (Jas), latrunculin A (Lat-A), paclitaxel
(PT) and vincristine (VCR) were purchased from Sigma (St. Louis,
MO, USA). The mitochondrial membrane potential assay kit with JC-1,
Annexin V-FITC/PI staining kit and 2′,7′-Dichlorofluorescin
diacetate (DCFH-DA) were purchased from Beyotime (Haimen, China).
Z-VAD-fmk and the LC3 antibody were purchased from MBL (Japan).
Antibodies against cleaved caspase 3, PARP, β-actin, Akt, p-mTOR,
p-p70S6K, p-4EBP1, and p-S6 were purchased from Cell Signaling
Technology (Beverly, MA, USA). Antibodies against cytochrome
c were purchased from Santa Cruz Biotechnology (Santa Cruz,
CA, USA).
MTT
Cells (3×103/well) were plated in 96-well
plates in 100 μl media, cultured overnight and exposed to a
range of concentrations of BJ-B11 (or 17-AAG) for 24, 48 or 72 h.
After the addition of 20 μl of 5 mg/ml MTT solution/well,
the plates were incubated for 4 h, the media were removed, the
formazan crystals were solubilized in 100 μl DMSO/well and
the absorbance values were read at 570 nm.
Cell cycle analysis
Cells were exposed to the indicated concentrations
of BJ-B11 for 48 h, harvested in cold PBS, fixed in 70% ethanol,
stored overnight at 4°C, washed twice with PBS, resuspended in 50
μg/ml PI staining reagent containing 100 μg/ml RNase
and 0.1% Triton X-100 for 30 min in the dark. Cells were analyzed
by flow cytometry (Becton-Dickinson, CA) and the percentage of
cells in the different phases of the cell cycle was analyzed with
Becton-Dickinson software.
Annexin V-FITC/PI analysis
Cells were exposed to the indicated concentrations
of BJ-B11 for 48 h, harvested, washed twice with ice cold PBS,
resuspended in 500 μl incubation buffer containing Annexin
V-FITC and PI, incubated in the dark for 15 min and analyzed by
flow cytometry.
DAPI staining assay
Cells were exposed to 0.5 μM BJ-B11, washed
twice in ice-cold PBS, fixed in 4% paraformaldehyde for 15 min at
room temperature, washed with ice-cold PBS, stained with 5
μg/ml DAPI for 10–15 min and examined by fluorescence
microscopy.
Transmission electron microscopy
(TEM)
Cells were exposed to 0.5 μM BJ-B11 for 48 h,
harvested, washed twice in ice-cold PBS, and then fixed with
ice-cold 2.5% glutaraldehyde overnight at 4°C. The samples were
post-fixed with 1% OsO4 in the same buffer for 1 h and
then subjected to electron microscopic analysis. Representative
areas were chosen for ultra-thin sectioning and were observed with
a Philips Technai-10 transmission electron microscope (Philips
Eindhoven, The Netherlands).
Determination of intracellular ROS
Cells were exposed to 0.5 μM BJ-B11 for the
indicated time periods, harvested, incubated with 10 μM
DCFH-DA for 15 min at 37°C. The fluorescence intensity of cells was
measured by flow cytometry with an excitation wavelength of 488 nm
and an emission wavelength of 525 nm.
Evaluation of the MMP
Cells were exposed to 0.5 μM BJ-B11 for the
indicated time periods, harvested, resuspended in 500 μl PBS
containing 10 μg/ml of JC-1 dye, incubated for 15 min at
37°C, immediately centrifuged to remove the supernatant,
resuspended in PBS and analyzed by flow cytometry. The loss of MMP
was quantified as the percentage of cells expressing JC-1 monomer
fluorescence.
MDC staining
Cells were exposed to BJ-B11 with or without 3-MA
for 48 h, harvested, washed twice in ice-cold PBS, and cultured
with 0.05 mM MDC at 37°C for 60 min. The cellular fluorescence
intensity was analyzed by flow cytometry.
Immunofluorescence assay
Cells were cultured and treated on glass cover-slips
for indicated time periods, fixed in fresh 4% paraformaldehyde in
PBS for 15 min at room temperature, permeabilized with 0.1% Triton
X-100 for 20 min, blocked with 5% bovine serum albumin (BSA) for 1
h, incubated with β-tubulin antibody [Alexa Fluor (R) 647 Conj.]
(1:500) for 1 h or Phalloidin for 40 min, respectively, washed
three times in PBST. Nuclei were counterstained using 5
µg/ml DAPI for 15 min and the cells were washed, mounted and
examined by laser scanning confocal microscopy (LSM510 Meta Duo
Scan, Zeiss, Germany).
Western blotting
Cells were exposed to 0.5 μM BJ-B11 for
indicated time periods, harvested, washed twice in ice-cold PBS,
lysed in RIPA buffer for 30 min on ice, centrifuged at 14,000 g for
15 min and the supernatants were collected. Equivalent amounts of
lysate (20–30 μg) were denatured in SDS sample buffer,
resolved on 6–15% SDS-PAGE gels, transferred to PVDF membranes,
blocked in 5% skimmed milk in Tris-buffered saline (TBS) containing
0.1% Tween-20 at room temperature for 1 h and probed with
appropriate dilutions (1:100–1:5,000) of primary antibody overnight
at 4°C. The membranes were washed three times in TBST for 5 min,
incubated with secondary antibody (1:3,000) for 1 h at room
temperature, washed and the bound antibodies were detected using an
enhanced chemiluminescence kit (Haimen) following the
manufacturer’s instructions.
Statistical analysis
Data are expressed as the means ± SD. Differences
between two groups were analyzed using the Student’s t-test and
groups of three or more were analyzed using One-way ANOVA multiple
comparisons. *P<0.05 and **P<0.01 were
considered statistically significant.
Results
BJ-B11 induces growth inhibition and cell
cycle arrest in Eca-109 cells
Initially, we assessed the effects of BJ-B11 and
17-AAG on the growth of Eca-109 cells using MTT assay. Eca-109
cells were cultured in the presence of increasing concentrations of
BJ-B11 (or 17-AAG) for 24, 48 or 72 h. Both BJ-B11 and 17-AAG
inhibited Eca-109 cell growth in a time- and
concentration-dependent manner with the IC50 values of
0.31±0.01 and 1.10±0.02 μM following 48-h incubation,
respectively (Fig. 1A and B).
To investigate the mechanism by which cell growth
was inhibited, the cell cycle progression was examined by flow
cytometry. BJ-B11 treatment resulted in the accumulation of cells
in the G2/M phase, with a concomitant reduction in the proportion
of cells in the G0/G1 and S phase in a concentration-dependent
manner (Fig. 1C). The percentage
of cells in the G2/M phase increased to 12.45±2.33, 44.20±0.71 and
57.30±3.96% at a concentration of 0.125, 0.5 and 2 μM,
respectively.
BJ-B11 induces caspase-dependent
apoptosis in Eca-109 cells
To confirm whether BJ-B11 induced cell death
occurred via apoptosis, we examined the ability of BJ-B11 to induce
the characteristic morphological changes of apoptosis with DAPI
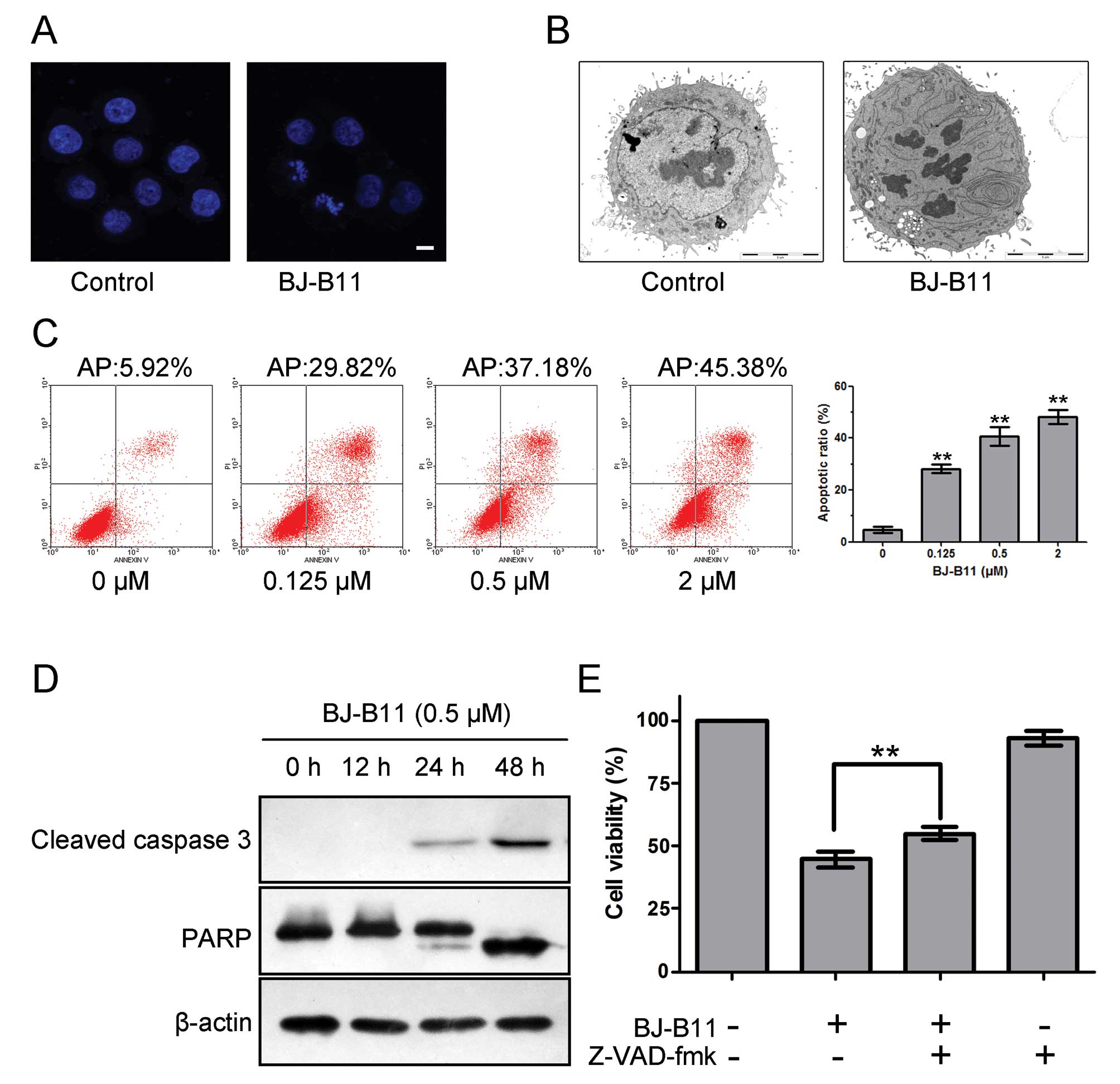
staining and TEM. As shown in Fig. 2A
and B, marked morphologic alterations indicative of apoptosis
such as nuclear condensation, chromatin marginalization, and DNA
fragmentation were observed in cells treated with 0.5 μM
BJ-B11 for 48 h.
To further investigate whether BJ-B11 induced growth
inhibition was due to apoptosis, we performed the Annexin-V FITC/PI
double staining analysis. As shown in Fig. 2C, BJ-B11 induced significant
apoptosis in a concentration-dependent manner, and the rate of
early apoptotic and late apoptotic cells and necrotic cell death in
Eca-109 cells increased markedly to 28.19±1.63, 40.68±3.50 and
48.14±2.76% at a concentration of 0.125, 0.5 and 2 μM,
respectively.
Next, we examined the effects of BJ-B11 on caspase
activity to determine whether caspase activation occurs in the
BJ-B11-induced apoptosis. Western blot analysis indicated that
treatment of Eca-109 cells with 0.5 μM BJ-B11 for 0, 12, 24
and 48 h resulted in cleavage of caspase 3 and PARP (Fig. 2D). The cleavage of caspases plays a
key role in the process of apoptotic cell death (25). We therefore wondered whether the
inhibition of caspases with the general caspase inhibitor Z-VAD-fmk
would prevent BJ-B11 induced cell death. As shown in Fig. 2E, pretreatment of cells with 25
μM Z-VAD-fmk significantly improved the cell viability as
assessed by MTT assay, indicating that the cell death induced by
BJ-B11 is caspase-dependent.
BJ-B11 induces mitochondrial dysfunction
in Eca-109 cells
To investigate whether mitochondria were involved in
the mechanism of BJ-B11-induced apoptosis, we evaluated the
production of ROS, the release of cytochrome c, and the
reduction of MMP. As shown in Fig.
3A, treatment of Eca-109 cells with 0.5 μM BJ-B11
resulted in significant production of ROS, which increased to
1.33±0.10-fold of control at 12 h. BJ-B11 induced the release of
cytochrome c into the cytosol of Eca-109 cells in a
time-dependent manner (Fig. 3B).
It is well established that apoptotic stimuli alter the MMP
(26). We examined the MMP by JC-1
staining. As shown in Fig. 3C,
BJ-B11 induced significant reduction of MMP after treatment of
Eca-109 cells for 48 h. These results suggested that mitochondrial
dysfunction was involved in the BJ-B11-induced apoptosis.
BJ-B11 induces autophagy via inhibition
of Akt/mTOR/p70S6K signaling in Eca-109 cells
As the pretreatment of Z-VAD-fmk could only increase
part of the cell viability, we next investigated whether BJ-B11
induced autophagy in Eca-109 cells. Treatment of Eca-109 cells with
0.5 μM BJ-B11 resulted in autophagic vacuoles (Fig. 4A and B), revealed by TEM and
monodansylcadaverine (MDC) staining. We also examined the levels of
LC3-II induced by BJ-B11 treatment, since this protein is a good
indicator of autophagosome formation (27). As shown in Fig. 4B and C, BJ-B11 induced punctate
redistribution and cleavage of LC3 in a time- and
concentration-dependent manner. Specifically, treatment of Eca-109
cells with 0.5 μM BJ-B11 resulted in the fluorescence
intensity of MDC-positive vacuoles increased to 1.37±0.15-fold than
that of control, which could be inhibited by the pretreatment of
the general autophagy inhibitor 3-MA (Fig. 4D). We next evaluated the ratio of
apoptotic cells induced by BJ-B11 with or without 3-MA. As shown in
Fig. 4E, pretreatment of Eca-109
cells with 3-MA could significantly reduce the ratio of apoptotic
cells induced by BJ-B11, indicating that autophagy promoted
apoptosis.
Akt/mTOR/p70S6K signaling pathway plays a key role
in regulating autophagy (28). We
next examined the role of Akt/mTOR/p70S6K in BJ-B11-induced
autophagy. As shown in Fig. 4F,
treatment with BJ-B11 reduced the expression of Akt, p-mTOR,
p-p70S6K, p-4EBP1 and p-S6. These results suggest that BJ-B11 may
induce autophagy via Akt/mTOR/p70S6K signaling inhibition in
Eca-109 cells.
BJ-B11 induces β-tubulin and F-actin
polymerization in Eca-109 cells
As reported, the polymerization or depolymerization
of tubulin and F-actin are related with the induction of G2/M cell
cycle arrest, apoptosis, and autophagy (29–32).
To evaluate whether BJ-B11 interferes with cytoskeleton network, we
examined its effects on β-tubulin and F-actin by laser scanning
confocal microscopy. Paclitaxel (microtubule polymerizing agent)
and vincristine (microtubule depolymerizing agent) was used as
positive or negative control, respectively. BJ-B11 induced
β-tubulin polymerization at 12, 24 and 48 h, respectively, with an
increased density of cellular microtubules and formation of long
thick microtubule bundles surrounding the nucleus, which was
similar to the effects with paclitaxel treatment (Fig. 5A). Fig. 5B shows that BJ-B11 induced
polymerization of F-actin at 12, 24 and 48 h, respectively. Jas
inducing the polymerization and stabilization of actin filaments
and Lat-A preventing actin polymerization, were used as controls.
Furthermore, to quantify the polymerization, we next analyzed the
fluorescence intensity by flow cytometry. As shown in Fig. 5C, BJ-B11 increased β-tubulin to
1.34±0.19-fold of the control at 24 h, whereas, BJ-B11 increased
F-actin to 1.47±0.19-fold of the control at 24 h (Fig. 5D). These results indicate that the
growth inhibition of Eca-109 cells induced by BJ-B11 may also be
associated with the cytoskeleton polymerization.
Discussion
Previous studies demonstrated that BJ-B11 displayed
less toxicity on normal human cells and more potent anti-tumor
activity than the positive control 17-AAG (23). In this study, we showed that BJ-B11
induced Eca-109 cell cycle arrest, apoptosis, autophagy and
cytoskeleton polymerization, resulting in cell death. BJ-B11
induced growth inhibition in Eca-109 cells in a more potent manner
than 17-AAG (Fig. 1A and B).
BJ-B11 induced G2/M cell cycle arrest in Eca-109 cells, different
from the G0/G1 cell cycle arrest in K562 cells (23), suggesting that BJ-B11 might affect
the mitosis of Eca-109 cells (Fig.
1C). Thus, the G2/M cell cycle arrest may be responsible for
the growth inhibition of Eca-109 cells induced by BJ-B11.
BJ-B11 induced apoptosis in Eca-109 cells,
characterized by altered cell morphology, DNA fragment, caspase
activation and PARP cleavage (Fig.
2). It is known that apoptosis is regulated by two major
pathways: the mitochondrial pathway and death receptor pathway
(33). Mitochondria play a central
role in determining cell survival or death in response to diverse
stimuli (34). The mitochondrial
dysfunction is characterized by the production of ROS, release of
cytochrome c, and disruption of MMP, and the mitochondrial
apoptotic pathway is associated with caspase activation (35). The generation of ROS damages the
mitochondria which may result in apoptosis or autophagy (36,37).
Cytochrome c release activates caspase-mediated apoptosis
pathway. In the present study, the time-dependent production of
ROS, release of cytochrome c into the cytosol and depletion
of MMP were observed in BJ-B11-treated Eca-109 cells (Fig. 3). Thus, BJ-B11 may induce
mitochondrial-mediated apoptosis in Eca-109 cells.
We provide evidence that BJ-B11 also induced
autophagy in Eca-109 cells. At present, the precise molecular
mechanism that switches between apoptosis and autophagy needs to be
further investigated. Autophagy and apoptosis can be induced in
response to different cellular stresses, and they can act in
synergy or in a mutually exclusive manner (38). In our study, pretreatment of
Eca-109 cells with 3-MA decreased the fluorescence intensity of
MDC-positive vacuoles and the ratio of apoptotic cells induced by
BJ-B11, indicating that autophagy promoted apoptosis induced by
BJ-B11 (Fig. 4D and E).
It has been suggested that Akt/mTOR/p70S6K pathway
negatively regulate autophagy in cancer cells (39). Our results suggest that BJ-B11 may
induce autophagy via the inhibition of Akt/mTOR/p70S6K pathway, and
that BJ-B11-induced autophagy may be associated with Akt
degradation in a mechanism dependent on Hsp90 inhibition and
Akt-mediated inhibition of mTOR activity. This is consistent with
several other reports (39–41).
It has been suggested that microtubules are crucial
in the development and maintenance of cell shape, the
transportation of vesicles and mitochondria throughout the cells,
and in the cell signaling, division and mitosis (42). Actin is the major component of the
cytoskeleton and also plays many important roles in cell growth,
division, motility, signal transduction, cell- cell adhesion, and
wound-healing processes (43). In
recent years, increasing numbers of compounds have been reported to
interact with the microtubule and actin cytoskeleton, including
paclitaxel, vincristine, colchicines, latrunculin A, jasplakinolide
and azadirachtin A (44–46). Paclitaxel is reported as an inducer
of G2/M arrest, and causes caspase-8-induced apoptosis (47). Paclitaxel can also induce apoptosis
and autophagy in v-Ha-ras-transformed fibroblasts (48). In our study, BJ-B11 induced
β-tubulin and F-actin polymerization at 12, 24 and 48 h, which
might be responsible for the G2/M cell cycle arrest, apoptosis, and
autophagy (31,32).
In conclusion, this study provides the first
evidence that BJ-B11 induces G2/M cell cycle arrest, apoptosis,
autophagy and polymerization of cytoskeleton in Eca-109 cells.
These findings may facilitate future investigations on the
potential of BJ-B11 as a targeted therapy agent for the treatment
of human esophageal squamous carcinoma.
Acknowledgements
This study was supported by Grants
from the Industry-Academia-Research Demonstration Base of Guangdong
Higher Education Institutes (Innovative Culturing Base of
Graduates) (no. 2010B091000013), the Twelfth Five-Year National
Science and Technology Support Program (no. 2012BAI29B06), the
National Natural Science Foundation of China (no. 81001449), and
the Natural Science Foundation of Guangdong Province of China
(10451063201005506).
References
|
1.
|
Porter JR, Fritz CC and Depew KM:
Discovery and development of Hsp90 inhibitors: a promising pathway
for cancer therapy. Curr Opin Chem Biol. 14:412–420. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Mahalingam D, Swords R, Carew JS, Nawrocki
ST, Bhalla K and Giles FJ: Targeting HSP90 for cancer therapy. Br J
Cancer. 100:1523–1529. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Lu X, Xiao L, Wang L and Ruden DM: Hsp90
inhibitors and drug resistance in cancer: The potential benefits of
combination therapies of Hsp90 inhibitors and other anti-cancer
drugs. Biochem Pharmacol. 83:995–1004. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Oude Munnink TH, Korte MA, Nagengast WB,
et al: (89Zr-trastuzumab PET visualises HER2 downregulation by the
HSP90 inhibitor NVP-AUY922 in a human tumour xenograft. Eur J
Cancer. 46:678–684. 2010.PubMed/NCBI
|
|
5.
|
Eccles SA, Massey A, Raynaud FI, et al:
NVP-AUY922: a novel heat shock protein 90 inhibitor active against
xenograft tumor growth, angiogenesis, and metastasis. Cancer Res.
68:2850–2860. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Whitesell L and Lindquist SL: HSP90 and
the chaperoning of cancer. Nat Rev Cancer. 5:761–772. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Xu C, Liu J, Hsu LC, Luo Y, Xiang R and
Chuang TH: Functional interaction of heat shock protein 90 and
Beclin 1 modulates Toll-like receptor-mediated autophagy. FASEB J.
25:2700–2710. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Palacios C, Martin-Perez R, Lopez-Perez
AI, Pandiella A and Lopez-Rivas A: Autophagy inhibition sensitizes
multiple myeloma cells to
17-dimethylaminoethylamino-17-demethoxygeldanamycin-induced
apoptosis. Leuk Res. 34:1533–1538. 2010. View Article : Google Scholar
|
|
9.
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Gou X, Ru Q, Zhang H, et al: HAb18G/CD147
inhibits starvation-induced autophagy in human hepatoma cell
SMMC7721 with an involvement of Beclin 1 down-regulation. Cancer
Sci. 100:837–843. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Rami A and Kogel D: Apoptosis meets
autophagy-like cell death in the ischemic penumbra: two sides of
the same coin? Autophagy. 4:422–426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Solit DB and Chiosis G: Development and
application of Hsp90 inhibitors. Drug Discov Today. 13:38–43. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Li Y, Zhang T, Schwartz SJ and Sun D: New
developments in Hsp90 inhibitors as anti-cancer therapeutics:
mechanisms, clinical perspective and more potential. Drug Resist
Updat. 12:17–27. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Taldone T, Sun W and Chiosis G: Discovery
and development of heat shock protein 90 inhibitors. Bioorg Med
Chem. 17:2225–2235. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Wang SX, Ju HQ, Liu KS, et al: SNX-2112, a
novel Hsp90 inhibitor, induces G2/M cell cycle arrest and apoptosis
in MCF-7 cells. Biosci Biotechnol Biochem. 75:1540–1545. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Liu KS, Ding WC, Wang SX, et al: The heat
shock protein 90 inhibitor SNX-2112 inhibits B16 melanoma cell
growth in vitro and in vivo. Oncol Rep. 27:1904–1910.
2012.PubMed/NCBI
|
|
18.
|
Okawa Y, Hideshima T, Steed P, et al:
SNX-2112, a selective Hsp90 inhibitor, potently inhibits tumor cell
growth, angiogenesis, and osteoclastogenesis in multiple myeloma
and other hematologic tumors by abrogating signaling via Akt and
ERK. Blood. 113:846–855. 2009. View Article : Google Scholar
|
|
19.
|
Jin L, Xiao CL, Lu CH, et al:
Transcriptomic and proteomic approach to studying SNX-2112-induced
K562 cells apoptosis and anti-leukemia activity in K562-NOD/SCID
mice. FEBS Lett. 583:1859–1866. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Liu KS, Liu H, Qi JH, et al: SNX-2112, an
Hsp90 inhibitor, induces apoptosis and autophagy via degradation of
Hsp90 client proteins in human melanoma A-375 cells. Cancer Lett.
318:180–188. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Zhai QQ, Gong GQ, Liu Z, et al:
Preclinical pharmacokinetic analysis of SNX-2112, a novel Hsp90
inhibitor, in rats. Biomed Pharmacother. 65:132–136. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Zhai QQ, Gong GQ, Luo Y, et al:
Determination of SNX-2112, a selective Hsp90 inhibitor, in plasma
samples by high-performance liquid chromatography and its
application to pharmacokinetics in rats. J Pharm Biomed Anal.
53:1048–1052. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Ju HQ, Wang SX, Xiang YF, et al: BJ-B11, a
novel Hsp90 inhibitor, induces apoptosis in human chronic myeloid
leukemia K562 cells through the mitochondria-dependent pathway. Eur
J Pharmacol. 666:26–34. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Ju HQ, Xiang YF, Xin BJ, et al: Synthesis
and in vitro anti-HSV-1 activity of a novel Hsp90 inhibitor BJ-B11.
Bioorg Med Chem Lett. 21:1675–1677. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Kumar S: Caspase function in programmed
cell death. Cell Death Differ. 14:32–43. 2007. View Article : Google Scholar
|
|
26.
|
Green DR and Kroemer G: The
pathophysiology of mitochondrial cell death. Science. 305:626–629.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Shinojima N, Yokoyama T, Kondo Y and Kondo
S: Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in
curcumin-induced autophagy. Autophagy. 3:635–637. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Viola G, Bortolozzi R, Hamel E, et al:
MG-2477, a new tubulin inhibitor, induces autophagy through
inhibition of the Akt/mTOR pathway and delayed apoptosis in A549
cells. Biochem Pharmacol. 83:16–26. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
McElligott AM, Maginn EN, Greene LM, et
al: The novel tubulin-targeting agent pyrrolo-1,5-benzoxazepine-15
induces apoptosis in poor prognostic subgroups of chronic
lymphocytic leukemia. Cancer Res. 69:8366–8375. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Tang YA, Wen WL, Chang JW, et al: A novel
histone deacetylase inhibitor exhibits antitumor activity via
apoptosis induction, F-actin disruption and gene acetylation in
lung cancer. PLoS One. 5:e124172010. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Hwang JH, Takagi M, Murakami H, Sekido Y
and Shin-ya K: Induction of tubulin polymerization and apoptosis in
malignant mesothelioma cells by a new compound JBIR-23. Cancer
Lett. 300:189–196. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Hogstrand K, Hejll E, Sander B, Rozell B,
Larsson LG and Grandien A: Inhibition of the intrinsic but not the
extrinsic apoptosis pathway accelerates and drives MYC-driven
tumorigenesis towards acute myeloid leukemia. PLoS One.
7:e313662012. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Adams JM and Cory S: Life-or-death
decisions by the Bcl-2 protein family. Trends Biochem Sci.
26:61–66. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Wang YC, Lee CM, Lee LC, et al:
Mitochondrial dysfunction and oxidative stress contribute to the
pathogenesis of spinocerebellar ataxia type 12 (SCA12). J Biol
Chem. 286:21742–21754. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Nicolau-Galmes F, Asumendi A,
Alonso-Tejerina E, et al: Terfenadine induces apoptosis and
autophagy in melanoma cells through ROS-dependent and -independent
mechanisms. Apoptosis. 16:1253–1267. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Liu B, Cheng Y, Zhang B, Bian HJ and Bao
JK: Polygonatum cyrtonema lectin induces apoptosis and autophagy in
human melanoma A375 cells through a mitochondria-mediated
ROS-p38-p53 pathway. Cancer Lett. 275:54–60. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Saiki S, Sasazawa Y, Imamichi Y, et al:
Caffeine induces apoptosis by enhancement of autophagy via
PI3K/Akt/mTOR/p70S6K inhibition. Autophagy. 7:176–187. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Takeuchi H, Kondo Y, Fujiwara K, et al:
Synergistic augmentation of rapamycin-induced autophagy in
malignant glioma cells by phosphatidylinositol 3-kinase/protein
kinase B inhibitors. Cancer Res. 65:3336–3346. 2005.PubMed/NCBI
|
|
41.
|
Degtyarev M, De Maziere A, Orr C, et al:
Akt inhibition promotes autophagy and sensitizes PTEN-null tumors
to lysosomotropic agents. J Cell Biol. 183:101–116. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Shen L, Xu W, Li A, Ye J and Zhou J: JWA
enhances AsO-induced tubulin polymerization and apoptosis via p38
in HeLa and MCF-7 cells. Apoptosis. 16:1177–1193. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Moon SS, Rahman AA, Kim JY and Kee SH:
Hanultarin, a cytotoxic lignan as an inhibitor of actin
cytoskeleton polymerization from the seeds of Trichosanthes
kirilowii. Bioorg Med Chem. 16:7264–7269. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Canta A, Chiorazzi A and Cavaletti G:
Tubulin: a target for antineoplastic drugs into the cancer cells
but also in the peripheral nervous system. Curr Med Chem.
16:1315–1324. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Allingham JS, Klenchin VA and Rayment I:
Actin-targeting natural products: structures, properties and
mechanisms of action. Cell Mol Life Sci. 63:2119–2134. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Fenteany G and Zhu S: Small-molecule
inhibitors of actin dynamics and cell motility. Curr Top Med Chem.
3:593–616. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Mielgo A, Torres VA, Clair K, Barbero S
and Stupack DG: Paclitaxel promotes a caspase 8-mediated apoptosis
through death effector domain association with microtubules.
Oncogene. 28:3551–3562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Eum KH and Lee M: Crosstalk between
autophagy and apoptosis in the regulation of paclitaxel-induced
cell death in v-Ha-ras-transformed fibroblasts. Mol Cell Biochem.
348:61–68. 2011. View Article : Google Scholar : PubMed/NCBI
|